
Abstract
The rheological properties and the liquid crystalline phase transitions of cellulose ethers solutions are studied. The concentration dependences of the enthalpy of the activation of a viscous flow are described by the curves with extremes (maxima and minima) caused by liquid crystalline transitions. These dependences are compared with the phase diagrams of studied systems.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Liquid crystals (LC) play an enormous role in science and engineering [1‒8]). The high capability of these compounds for self-organization is of considerable interest for the development of new materials. Macromolecules of cellulose and its derivatives have a rigid helical conformation and are capable of ordering and forming cholesteric-type liquid crystals in concentrated solutions [1, 4].
The solutions of rigid-chain polymers have a special concentration dependence of viscosity: this dependence is described by a curve with a sharp maximum. For the first time, this was shown by Hermans [9] for PBG solutions in DMF, Yang [10] for PBG solutions in m-cresol, and Iizuka [11] for poly(alkyl glutamate)s. Later, such dependence was found for solutions of other polymers [12‒15]. The concentration at the maximum point corresponds to the onset of the formation of the anisotropic phase, which leads to a decrease in the viscosity due to the presence of macromolecules which undergo orientation under flow. Robinson [16] assumed that the decrease of viscosity is caused by the layer-by-layer flow of anisotropic solutions. The viscosity minimum is due to the completion of the liquid crystalline phase formation throughout the solution volume. Further increase in polymer concentration leads to an increase in the viscosity, caused by enhancement of the intermolecular interaction. The same dependences were observed for the solutions of hydroxypropyl cellulose (HPC)–poly(ethylene glycol), HPC–water, HPC–dimethylsulfoxide (DMSO) [17], and hydroxypropyl cellulose-based nanocomposites [18]. Thus, the increase in viscosity with the increase in polymer concentration is typical of single-phase (isotropic or anisotropic) solutions, while the viscosity decreases with the increase in polymer concentration in a two-phase region where the isotropic and liquid crystalline phases coexist.
It is known [19, 20] that the viscosity is related to the enthalpy of activation of a viscous flow ∆H via the ratio
where A is a constant, R is the gas constant, and T is the absolute temperature. So, if an extremum appears on the concentration dependence of viscosity, then this may be due to a sharp change in the activation enthalpy of a viscous flow. However, the data on the concentration dependence of the activation enthalpy of viscous flow for rigid-chain polymer solutions in the isotropic and anisotropic regions are practically absent [15].
The goal of this work was to determine the concentration dependences of the enthalpy of activation of a viscous flow for the cellulose ether–solvent systems and to compare the obtained data with the phase diagrams of these systems.
EXPERIMENTAL
Experiments were carried out with samples of HPC supplied by Acros Organics (Belgium) with Mw = 1 × 105 and a molar substitution of 3.2, ethyl cellulose (EC) (Acros Organics, Belgium) with Мη = 4.7 × 104 and a degree of substitution DS = 2.6, cyanoethyl cellulose (CEC) with Мw = 9 × 104 and DS = 2.6, and methylhydroxyethyl cellulose (MHEC) (6000R, Hercules Culminal, Aqulon) with Mη = 1 × 106 and DS(‒OCH3) = 1.8 and DS(‒OCH2CH2OH) = 0.3. Cyanoethyl cellulose was prepared according to the following procedure. Cotton linter was treated with an 18% NaOH solution at 20°С for 2 h, squeezed until 3- to 4-fold weight was achieved, and loosened. The prepared product was treated under stirring with acrylonitrile (20 mol per glucose unit) at 35°С for 3 h. The reaction mass was cooled to 5°С, diluted with acetone, neutralized with a 15% alcohol solution of acetic acid, repeatedly washed with water, and finally extracted in a Soxhlet extractor with alcohol for 10 h. The resulting ester was dried at 50°С in a vacuum chamber. The molar substitution of HPC and DS of EC and MHEC were calculated from elemental analysis data. The DS value of CEC was estimated from the percentage of nitrogen determined by the Kjeldahl method. DMSO, dimethylacetamide (DMAA), ethanol, and distilled water were used as solvents. The purity of the solvents was judged from their refractive indices [21]. The solutions were prepared for several weeks at 330–350 K.
The solution phase state was studied using an OLYMPUS BX-51 polarization microscope and a polarization photoelectric installation [22]. An ampoule with a solution was placed in the gap between the crossed polarizer and analyzer and cooled using a temperature control jacket. The polarized light of a He–Ne laser was transmitted through the polarizer and analyzer in the direction normal to the ampoule. When the solution was transparent (isotropic), the intensity of the transmitted light was zero. As the system became turbid upon variation in temperature or increase in the concentration of solution, the intensity of transmitted polarized light increased. This indicated the formation of the anisotropic phase. The temperature at which a stable opalescence appeared was taken as the phase transition point. The solution cooling rate was 0.2 K/min. The solution viscosity was investigated using a Rheotest RN 4.1 rheometer equipped with a coaxial cylindrical working unit.
RESULTS AND DISCUSSION
The phase diagrams of cellulose derivative–solvent systems (Fig. 1) were constructed earlier and discussed in [4, 13‒15]. The boundary curves separate the isotropic regions from the anisotropic ones. The effect of polymer molecular weight and chemical structure of components on phase transitions is analyzed in detail in [4].

Boundary curves of the systems: (a) CEC–DMAA; (b) HPC–DMSO (1) and HPC–ethanol (2); (c) EC–ethanol (1) and EC–DMAA (2); (d) MHEC–water. (I) The region of isotropic solutions; (II) the region of anisotropic solutions. ω2 is the mass fraction of polymer.
Figure 2 shows the micrographs of HPC solutions in ethanol and DMSO. The rainbow coloration suggests the anisotropic phase state of solutions. Similar data were obtained for other studied systems.

Micrographs of solutions under crossed polaroids for systems: (a) HPC–ethanol, ω2 = 0.55; (b) HPC–DMSO, ω2 = 0.50 (dark spots depict air bubbles). T = 298 K.
Figure 3 shows the typical dependences of the viscosity on the shear rate at different temperatures for the MHEC solution in water.

Viscosity vs. shear rate for MHEC solution in water. ω2 = 0.01 at T = (1) 338, (2) 298, and (3) 288 K.
The curves have a non-Newtonian character caused by the destruction of the initial structure of the polymer solution and orientation of the molecules in the flow, leading to the decrease in the viscosity. Such dependences were discovered for the solutions of the all studied polymers. Similar results are known for the systems Na-carboxymethyl cellulose–water [24], HPC–ethanol, HPC–DMSO [25], HPC–ethylene glycol, EC–DMF [26, 27]), HPC–m-cresol [28], CEC–DMF, CEC–DMAA, HPC–DMF [15], diacetate cellulose–trifluoroacetic acid [29], CEC–DMF [30], and HPC–acetic acid [31]. As the temperature increases the viscosity decreases.
These data were used to calculate the enthalpy of activation of a viscous flow. For this purpose, the viscosity values at a low shear rate (2 s–1) were chosen because it was shown in [9, 10, 12] that the concentration dependence of the viscosity determined at a low shear rate is typical of anisotropic solutions.
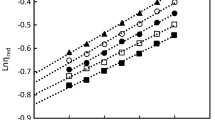
Figure 4 shows the dependences of viscosity on 1/T for aqueous solutions of MHEC at different polymer concentrations.

Dependences of the logarithm of viscosity on 1/T value for the MHEC–water system. ω2 = (1) 0.05, (2) 0.025, and (3) 0.01.
Table 1 shows the enthalpy ∆H of activation of a viscous flow of the solvents both experimental [32, 33] and calculated via the equation ∆H ≈ ∆H*/4 [19], where ∆H* is the enthalpy of solvent evaporation [34].
Figure 5 shows the concentration dependences of the enthalpy of activation of the viscous flow for the studied systems.

Concentration dependence of ∆H for the systems: (a) CEC–DMAA; (b) CEC–DMSO; (c) HPC–ethanol; (d) EC–DMAA; (e) EC–ethanol; (f) MHEC–water.
The concentration dependences of the enthalpy of activation of viscous flow are described by curves with extrema (maxima and minima) caused by liquid crystalline transitions. The calculated ∆Н values of isotropic polymer solutions correlate with similar data for other polymer systems [34]. The initial increase in the enthalpy of activation of viscous flow with the increase in polymer concentration is indicative of an increase in the interaction between macromolecules at approach to the concentration of liquid crystalline transitions. The region of the decrease in ∆Н corresponds to the appearance of an anisotropic phase (Fig. 1). This follows from the comparison to the phase diagrams.
It should be noted that the temperature of the phase transition point Tph determined by the viscometric method corresponds to the phase inversion and not to the appearance of the first drop of the new phase. For this reason, the values of Tph determined by the viscometric method and cloud point technique can differ even by several degrees. The difference of Tph may be also caused by the influence of a hydrodynamic field on the phase equilibrium [13]. The results of comparison of the concentration dependence ∆H and phase diagrams at 298 K are presented in Table 2. It is shown that there is satisfactory matching of the \(\omega _{2}^{*}\) and \(\omega _{2}^{{**}}\) values corresponding to the maximum on concentration dependence ∆H = f(ω2) and to the onset of the formation of the anisotropic phase on the phase diagram at 298 K.
Thus, the decrease in ∆Н is caused by the change in the flow mechanism. In an isotropic phase, there is the flow of disordered macromolecules that demands a bigger shear stress; in an anisotropic phase, macromolecules form domains that are easily oriented in the direction of the flow. Therefore, the decrease in the value of ∆Н is caused by the presence of oriented domains in the system and by the layer-by-layer flow of anisotropic solutions. This part of the curves corresponds to the coexistence of two phases: isotropic and anisotropic. The appearance of liquid crystal domains in solutions leads to a decrease in the energy of activation of viscous flow by 1.5‒2.0 times. The minima of curves correspond to the formation of single-phase anisotropic systems.
Similar results were obtained earlier in [35] for the HPC–DMSO system. A sharp drop in activation energy from 50 to 30 kJ/mol was found upon the transition from isotropic to liquid crystalline solutions. This is due to the easy orientation of the liquid crystalline domains in the direction of flow.
Such data correlate well with the results of the concentration dependences of sizes of light scattering particles D in cellulose ether solutions [27, 36]. The concentration dependences of D are described by curves with maxima. The concentrations of solutions with the maximum particle size coincide with the concentrations of the transition from an isotropic solution to an anisotropic solution. In isotropic solutions, macromolecules and their associates are not oriented relative to each other. With an increase in polymer concentration, they form large particles as a result of the intensification of the interchain interaction. The formed large particles do not have a dense packing; i.e., they may contain abundant solvent. During the transition to the LC state with a further increase in the polymer concentration, the emerging orientation of macromolecules and supramolecular particles toward each other leads to the increase in interchain interaction. This phenomenon may result in the squeezing out of the solvent from supramolecular particles, an event that is manifested in a decrease in their size. Therefore, the decrease in viscosity in this concentration region is caused both by easy orientation of macromolecules and by the decrease in the sizes of their associates.
The third part of these curves (Figs. 5d–5f) characterizes single-phase anisotropic solutions. As the polymer concentration increases, the enthalpy of activation of the viscous flow increases as well. This is caused by the increase in an interchain interaction. Thus, the concentration dependence of ∆Н makes it possible to determine the borders of the biphasic region of equilibria in which the isotropic and anisotropic phases coexist.
CONCLUSIONS
The rheological properties and liquid crystalline phase transitions of the methylhydroxyethyl cellulose–water, hydroxypropyl cellulose–ethanol, HPC–dimethylsulfoxide, cyanoethyl cellulose–dimethyl-acetamide, ethyl cellulose–dimethylacetamide, and ethyl cellulose–ethanol systems have been investigated. The concentration dependences of the enthalpy of activation of viscous flow ∆Н are described by curves with maxima and minima caused by the liquid crystalline phase transitions. The concentration dependences of ∆Н are compared with the phase diagrams of the studied systems. The dependences of ∆Н = f(ω2) make it possible to determine the borders of the biphasic region of equilibria in which the isotropic and anisotropic phases coexist.
REFERENCES
V. G. Kulichikhin and L. K. Golova, Khim. Drev., No. 3, 9 (1985).
Orientation Phenomena in Polymer Solutions and Melts, Ed. by A. Ya. Malkin and S. P. Papkov (Khimiya, Moscow, 1980) [in Russian].
S. P. Papkov and V. G. Kulichikhin, Liquid Crystalline State of Polymers (Khimiya, Moscow, 1977) [in Russian].
S. A. Vshivkov and E. V. Rusinova, Polym. Sci., A 60, 65 (2018).
Liquid Crystal Polymers, Ed. by N. A. Platé (Plenum, New York, 1993).
P. G. De Gennes, The Physics of Liquid Crystals (Cambridge Univ. Press, London, 1974).
N. A. Platé and V. P. Shibaev, Comb-Shaped Polymers and Liquid Crystals (Plenum, New York, 1988).
A. P. Filippov, Polym. Sci., Ser. B 46, 66 (2004).
J. Hermans, Colloid Sci. 17, 638 (1962).
J. T. Yang, J. Am. Chem. Soc. 81, 3902 (1959).
E. Iizuka, Mol. Cryst. Liq. Cryst. 25, 287 (1974).
V. G. Kulichikhin, A. Ya. Malkin, and S. P. Papkov, Vysokomol. Soedin., Ser. A 26, 499 (1984).
S. A. Vshivkov, Phase Transitions and Structure of Polymer Systems in External Fields (Cambridge Scholars Publ., Newcastle, 2019).
S. A. Vshivkov and A. G. Galyas, Polym. Sci., Ser. A 53, 1032 (2011).
S. A. Vshivkov and E. V. Rusinova, Russ. J. Appl. Chem. 84, 1830 (2011).
J. C. Ward and R. B. Beevers, Discuss. Faraday Soc. 25, 29 (1958).
V. G. Kulichikhin, V. V. Makarova, M. Yu. Tolstykh, and G. V. Vasil’ev, Polym. Sci., Ser.A 52, 1196 (2010).
V. G. Kulichikhin, V. V. Makarova, M. Yu. Tolstykh, S. J. Picken, and E. Mendes, Polym. Sci., Ser. A 53, 748 (2011).
A. A. Tager, Physics and Chemistry of Polymers (Khimiya, Moscow, 1978) [in Russian].
A. Ya. Malkin, Fundamentals of Rheology (Professiya, St. Petersburg, 2018) [in Russian].
I. V. Ioffe, Refractometric Methods in Chemistry (Khimiya, Leningrad, 1974) [in Russian].
S. A. Vshivkov, E. V. Rusinova, N. V. Kudrevatykh, A. G. Galyas, M. S. Alekseeva, and D. K. Kuznetsov, Polym. Sci., Ser. A 48, 1115 (2006).
S. A. Vshivkov, E. V. Rusinova, and A. G. Galyas, Eur. Polym. J. 59, 326 (2014).
S. A. Vshivkov and A. A. Byzov, Polym. Sci., Ser. A 55, 102 (2013).
S. A. Vshivkov and T. S. Soliman, Polym. Sci., Ser. A 58, 307 (2016).
S. A. Vshivkov and T. S. Soliman, Polym. Sci., Ser. A 58, 499 (2016).
S. A. Vshivkov and E. V. Rusinova, Polymer Rheology (TechOpen, London, 2018).
T. Asada, S. Hayaahida, and S. Onogi, Rep. Prog. Polym. Phys. Jpn. 23, 145 (1980).
S. Deyan, M. Gilli, and P. Sixou, J. Appl. Polym. Sci. 28, 1527 (1983).
N. G. Bel’nikevich, L. S. Bolotnikova, L. I. Kutsenko, Yu. N. Panov, and S. Frenkel’, Vysokomol. Soedin., Ser. B 27, 332 (1985).
P. J. Navard and M. Haudin, J. Polym. Sci., Polym. Phys. Ed. 24, 189 (1986).
I. A. Rennsky, T. A. Kamenskaya, and A. A. Rudnitskaya, Compensation Effect in Thermodynamics of Activation of a Viscous Flow of N-alkanes (Natl. Tech. Univ. Ukraine “KPI”, Kiev, 2010).
E. A. Maximov, B. G. Pashayev, G. Sh. Gasanov, and N. G. Gasanov, Adv. Mod. Nat. Sci. 10, 32 (2015).
D. R. Lide, CRC Handbook of Chemistry and Physics (CRC Press, Boca Raton, 2004).
V. G. Kulichikhin, V. A. Platonov, L. P. Braverman, T. A. Rozhdestvenskaya, E. G. Kogan, N. V. Vasil’eva, and A. V. Volokhina, Vysokomol. Soedin., Ser. A 29, 2537 (1987).
A. A. Panfilova, V. A. Platonov, V. G. Kulichikhin, V. D. Kalmikova, and S. P. Papkov, Colloid J. 37, 210 (1975).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Vshivkov, S.A., Rusinova, E.V. & Saleh, A.S. Rheological Properties of Liquid Crystalline Solutions of Cellulose Derivatives. Polym. Sci. Ser. A 63, 363–368 (2021). https://doi.org/10.1134/S0965545X21040088
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0965545X21040088