
Abstract
Phase equilibria in systems polystyrene–block and gradient n-butyl acrylate–styrene copolymers are studied for the first time using optical interferometry for polystyrene of various molecular weight and copolymers of different architecture. Phase diagrams, which are characterized by the upper critical solution temperatures, are constructed. The pair interaction parameters for these systems are calculated, and the thermodynamics of mixing of the components is evaluated. The data on the interdiffusion of polystyrene into the copolymer and the copolymer into polystyrene are obtained. The effect of the composition of copolymers on their interaction with polystyrene is analyzed. The friction coefficients of macromolecules and the diffusion coefficients of monomer units are calculated. It is shown that these values are independent of the molecular weight of the diffusant and the architecture of the copolymer.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
This paper is an extension of our previous studies on interdiffusion and phase equilibria in the systems PS–poly(butyl acrylate) (PBA) and PS–butyl acrylate–styrene random copolymers [1–4]. Phase diagrams were constructed and analyzed. It was shown that most of these systems are characterized by amorphous separation diagrams with the upper critical solution temperatures. In terms of the classical Flory–Huggins–Scott theory [5, 6] pair interaction parameters were determined, and their dependence on the molecular weight of PS, the composition of random copolymers, and temperature was studied. The distribution profiles of components in interdiffusion zones were obtained, and the partial diffusion coefficients of PS into the copolymers and the copolymers into PS were estimated. The activation energies of diffusion were calculated. It was demonstrated that the structural heterogeneity of random copolymer macromolecules exerts a lower impact on the phase and diffusion parameters of the systems than the difference in their composition.
In this study, the block and gradient n-butyl acrylate–styrene (BASC) copolymers of different composition are used as blend components, and PS and PBA homopolymers are used as macromolecular probes. This group of the copolymers was chosen owing to the fact that, compared to the previously studied random copolymers, they provide an opportunity to unveil specific features of the solubility and translational mobility of macromolecules of complex architecture in evidently compositionally heterogeneous, alternating, block, and graft copolymers.
Our thermochemical and structural morphological studies made it possible to identify substantial differences between block and gradient BASC [4]. The presence of extended random fragments in gradient-BASCs strongly affects the conformation of a macromolecular chain in general, thereby allowing PS blocks to form small structural heterogeneities that do not cause the isolation of PS as a separate phase. The same behavior is exhibited by block-BASC containing a small (up to 40%) amount of styrene, although the packing density of domains grows, and the glass transition temperature of these copolymers deviates from the Flory–Fox additivity rule. With an increase in the content of styrene in block-BASC (above 40%) the phase transition and, as a consequence, isolation of individual phases enriched in PS block fragments occur; this leads to the emergence of two glass transition temperatures.
One of the key issues in the field of polymer blends concerns the level of thermodynamic miscibility of components, because this parameter crucially influences the morphology and mechanical behavior of the blend. In this study, we present our data on the phase equilibria in the systems PS–BASC block copolymers and PS–BASC gradient copolymer in a wide range of MW values of both components. Owing to a similar structure of random, block, and gradient copolymers, the PS–BASC blends are suitable and interesting model systems for gaining insight into the general features of polymer miscibility.
This study addresses the experimental investigation of phase equilibria and interdiffusion in the systems PS–BASC block and gradient copolymers in a wide range of temperatures and MW of both components.
EXPERIMENTAL
The objects of research were PBA, PS, and n-butyl acrylate–styrene block and gradient copolymers of different microstructure which were synthesized by reversible addition-fragmentation chain transfer copolymerization. The content of styrene in the copolymers was varied in the range of 7–46 mol % (Table 1). The conditions of synthesis and the technique of studying the composition of copolymers were described in [7, 8]. The random copolymer of n-butyl acrylate and styrene containing 17 mol % styrene units was used for comparison.
The block copolymer has the block-PS–block-PBA–block-PS triblock structure [4, 8], in which the RAFT agent is situated within the PBA block. The gradient copolymer has a more complex triblock structure, in which small fragments of the random copolymer are present at the boundary of blocks. It is expected that this structure will provide a smoother transition between blocks in the copolymer. Thus, this gradient copolymer can partially be considered as a five-block copolymer block-PS–block-random–block-PBA–block-random–block-PS.
The chosen block and gradient copolymers are distinguished by the presence of a single glass transition temperature for all compositions of the copolymers, except block-BASC-46. The average MW of the copolymers is (47–67) × 103, except block-BASC-46, whose MW is two times smaller. The MW of the random copolymers of PS and PBA (random-BASC) [2] is close to that of block-BASC; it is in the range of (47–52) × 103. The molecular weight of PS is in the range of (0.8–30) × 103, which is comparable with the sizes of blocks in the copolymers.
The solubility of polymers was studied by laser microinterferometry [9, 10]. All measurements were conducted using polystyrene and copolymer films with a thickness of 120–150 µm which were obtained by pressing [2]. The interference patterns were measured and treated as described in [1, 2]. Measurements were performed under the step-by-step increase/decrease in temperature from 280 to 500 K; however, phase diagrams were constructed using the data obtained under the step-by-step rise in temperature to 440 K, which is associated with the thermal degradation of the copolymers [3].
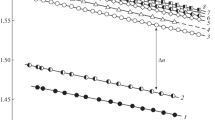
According to preliminary experiments, the temperature dependences of the refractive index were determined for the copolymers and their blends (Fig. 1). Measurements were carried out on an ATAGO NAR-2T refractometer (Japan) in the above-mentioned temperature and composition ranges. For all the studied components, both linear temperature dependences of the refractive index and additivity dependences of this parameter on composition were observed; therefore, using the obtained interference patterns, the concentration profiles and their evolution with time and temperature may be estimated. It was shown that, at temperatures above Тg, for example, at 380 K for PS-4, the difference between the refractive indexes of PS and PBA is 0.13; between the refractive indices of PS and block-BASC-17 or gradient-BASC-18, it is 0.11; and between the refractive indexes of PS and block-BASC-46, it is 0.07. These results indicate that from 42 to 23 interference bands, respectively, are formed in the interdiffusion zone. This means that, when moving from one band to another, the composition (concentration) increment is registered with an accuracy of ∼2.4 vol %. This information is of crucial value for calculating the compositions of coexisting phases in amorphous phase-separated systems.

Temperature dependence of the refractive index for (1) PBA, (2) block-BASC-7, (3) block-BASC-12, (4) block-BASC-17, (5) gradient-BASC-18, (6) block-BASC-46, (7) PS-1, (8) PS-2, (9) PS-3, (10) PS-4, (11) PS-5, and (12) PS-6.
RESULTS AND DISCUSSION
Interdiffusion Zones
Figures 2 and 3 show the typical interference patterns of interdiffusion zones that spontaneously appear upon the conjugation of block-BASC and gradient-BASC phases with the phase of PA at different temperatures and MW of the homopolymer. It is seen that, in the case of the low molecular weight PS (Мn < 1.2 × 103), the interdiffusion zones are the classical example of completely mutually soluble components (Figs. 2a, 2e). In these systems for both block and gradient copolymers, the continuous concentration distribution profile is detected in the self-diffusion zone when moving from PS to the copolymer. Both branches of the concentration distribution profile are symmetric with respect to the middle point of the ordinate (Fig. 4а).

Effect of temperature and molecular weight of PS on the nature of interference patterns for the systems PS–block-BASC-17 at a molecular weight of PS of (a) 1140, (b, f) 4100, (c, g) 9000, and (d, h) 30 000 and (e) for the system PS-2–block-BASC-12. Т = (a–e) 383 and (f–h) 433 K.

Interference patterns for the interaction of PS-4 with (a) PBA and copolymers (b) random-BASC-17, (c) gradient-BASC-18, (d) block-BASC-17, and (e) block-BASC-46 at 393 K.

Concentration distribution profile in the interdiffusion zone for the systems (a) PS-2–block-BASC-7 at 383 K and (b) PS-5–block-BASC-46 at 433 K.
As the molecular weight of PS is increased ((4 × 103) < Мn < (10 × 103)), the nature of the transition region of conjugated phases changes. In the middle composition range, an interface is observed which separates the region of dissolution (the diffusion region) of PS macromolecules in the copolymer phase (on the right of the interface) from the region of dissolution of copolymer macromolecules in the homopolymer phase (on the left of the interface). Under isothermal conditions, the position of the interface on the interference patterns does change with time and the concentration jump on the interface remains invariable and only temperature dependent. In heating/cooling cycles, the jump of concentrations φ' and φ'' is reproduced quantitatively. This fact makes it possible to state that equilibrium is established on the interface between the compositions of coexisting phases; i.e., at a given temperature, the copolymers are soluble in PS and PS is soluble in the copolymers, respectively.
As temperature is increased, the solubility of PS in the phase of block and gradient copolymers increases, which provides evidence that the UCST exists in the systems PS–block-BASC and PS–gradient-BASC.
Upon further increase in the molecular weight of PS, the mutual solubility of the polymers worsens (Fig. 2). When the molecular weight of PS is above 30 × 103, the solubility of the components is almost zero. Under these conditions, the zone of phase conjugation coincides with the interface (Figs. 2d, 2h). The same effect of the molecular weight of PS on the mixing of PS with PBA and random-BASC was described in [2].
The specific behavior of block and gradient copolymers manifests itself in the high-temperature range. For example, starting from 415 K in the interdiffusion zone of systems PS-4–block-BASC-17 (Figs. 2b, 2f) and PS-5–block-BASC-17 (Figs. 2c, 2g), the system undergoes swelling and the second optical boundary is formed in the region of dilute solutions of the block copolymer in PS. As temperature is increased to 433 K, the optical boundary degenerates аnd the second concentration wave appears on the concentration distribution profile (Fig. 4b). Note that the concentration distribution curve becomes asymmetric relative to the middle point of the ordinate. In our opinion, this finding may be attributed to different solubilities of the components.
In Fig. 3, the interference patterns measured for the copolymers of different structure, including random, block, and gradient, and PBA are compared. It is clear that the nature of the copolymer slightly affects the nature of diffusion zones. A marked difference is detected only in the case of block-BASC-46, whose rapid swelling under heating was described above.
Interestingly, for the copolymer containing up to 40% styrene, the position of the interface and the composition of coexisting phases are reproduced completely under heating/cooling regimes beyond the mentioned temperature range. For the copolymer block-BASC-46 upon swelling, no phase separation is observed with decreasing temperature.
In our opinion, these effects may be explained by the fact that the matrix of the block copolymers contains the network of three-dimensional bonds, which is related to the segregation of PS domains and fragmented PBA chains to the pseudophase. Under the influence of the diffusant, this phase (domain) network is broken (melts) at elevated temperatures and the system becomes single phase. It may be assumed that, for the original supramolecular structure of block and gradient copolymers to be recovered, the procedure of copolymer processing used previously (at the first step) should be implemented.
Phase Diagrams and Pair Interaction Parameters
The phase diagrams of the systems PS−block-BASC and PS−gradient-BASC (Figs. 5, 6) were constructed by the common method using the temperature dependences of compositions of coexisting phases. Only the experimental data that were reproduced in heating/cooling cycles were taken into account. On the diagrams, the regions of high temperatures (above 433 K) are marked off, in which the thermal degradation of the copolymers takes place during the long-term processes of contacting the components [3, 11].

Phase diagram of block-BASC-7 with (1) PS-4, (3) PS-5, and (4) PS-6 and (2) the calculation data on the spinodal for block-BASC-7 with PS-4. I is the region of true solutions, II is the region of homogeneous state, and III is the region of metastable states. The region of the thermal instability of BASC is marked in gray [3, 11].

It is clear that all the studied systems feature the UCST. Like most polymer systems in which the components exhibit noticeably different molecular weights, the diagrams are asymmetric [12] and critical concentrations are shifted to a lower MW component. Note that the data obtained are in good agreement with the phase diagrams reported in [2]. It is shown that all the above-mentioned phase states are reproduced in the heating/cooling cycles.
The solubility of the components increases with temperature. This implies that all the studied systems, including PBA–PS and PS–random-BASC, are characterized by amorphous separation phase diagrams with the UCST situated in the region of the thermal degradation of the copolymers.
There is a fairly pronounced asymmetry in the position of binodal curves on the temperature–concentration field of the diagrams. The right branch of the binodal, which characterizes the solubility of PS in block-BASC, is situated in the region of dilute solutions of BASC in the PS phase (less than 5 vol %). As the molecular weight of PS is increased from 4 × 103 to 30 × 103, its position on the temperature–concentration field of the diagram remains almost unchanged. The solubility of PS in block-BASC (the left branch of the binodal) decreases abruptly with the molecular weight of PS. The minimum values of solubility in this composition region are attained at Мn ≥ 30 × 103.
Using PS-4 as an example, Fig. 6 demonstrates the effect of composition and structure of the copolymer on the phase diagram. It is seen that the solubility of block-BASCs in PS is slightly different from the solubility of PBA in PS (the right branches of the binodals). A noticeable difference in the positions of boundary curves begins only at temperatures above 440 K.
The greater the content of styrene units in the copolymer, the longer the region of solubility of the components. For block-BASC-7, the solubility of PS-4 in the copolymer is in the range of 0.1–0.2 volume fractions; for block-BASC-17, it is in the range of 0.1–0.3 volume fractions; for block-BASC-46, it is in the range of 0.3–0.7 volume fractions. It is interesting that the values of solubility of PS-4 in gradient-BASC-18 (curve 3) and block-BASC-17 (curve 4) are similar.
The influence of the structure and topology of the macromolecular chains of copolymers is the most distinct when examining the isothermal cross sections of the phase diagrams. Figure 7 presents the generalized data for systems PS–PBA, PS–random-BASCs, PS–block-BASCs, and PS–gradient-BASCs. Under such a representation of the bimodal curves, the region of the heterogeneous state is situated between them, and the point of their intersection is equivalent to the critical point (the critical composition) at a given temperature. The dome of the binodal corresponds to the content of styrene in block-BASC on the order of 48–50% (for random-BASC, this value is 50–55%), at which complete miscibility of the components commences. It is seen that, for systems PS–block-BASC, the experimental compositions of coexisting phases fall on the common binodal curves at different temperatures (curve 1 at 393 K and curve 2 at 433 K). It should be emphasized that the data obtained for gradient-BASC (symbols 5, 6) coincide with those for block-BASC at the respective temperatures. For random-BASC, the dependences are analogous to those plotted for block-BASC, but the numerical values of PS solubility in it are much smaller (curves 3 and 4, respectively). Similar differences may be attributed to different molecular weights of the copolymers.

Solubility of PS-4 in (1, 2) block-BASC, (3, 4) random-BASC [2], and (5, 6) gradient-BASC. Т = (1, 3, 5) 393 and (2, 4, 6) 433 K.
The thermodynamic analysis of the experimental data on phase equilibria was performed using the Flory–Huggins theory of polymer solutions [5, 6]. Let us recall that the condition at which phases may coexist is the equality of chemical potentials of components (Δμi) in them:
where \(\Delta \mu _{i}^{'}\) and \(\Delta \mu _{i}^{{''}}\) are the chemical potentials of the ith component in the first and second phases, respectively.
The expressions for the chemical potentials of components for the amorphous phase-separated system are as follows [12]:
where φ1 and φ2 are the volume fractions of the first (PS) and second (BASC) components, respectively; r1 and r2 are the degrees of polymerization of the homopolymer and copolymers expressed in comparative volume units (for copolymers, the weight-average molecular weight was used in calculations); and χ is the pair interaction parameter.
Then as applied to the amorphous phase separation, we may write
and
The combined solution of these equations yields the mathematical expressions
Under the assumption of concentration independence, we arrive at
The relations presented above were used for the analysis of phase diagrams of amorphous equilibrium. The pair interaction parameters χ12 and χ21 derived for systems PS–block-BASC, PS–random-BASC, and PS–gradient-BASC show that they are similar quantitatively. This finding indicates that there is no concentration dependence of these parameters. Therefore, the data calculated using Eq. (8) are presented in Figs. 8 and 9 together with the inset which demonstrates the composition dependences of χcr, which were calculated in terms of the Flory–Huggins theory [5]. It is seen that the values of χ are in the narrow range from 0.01 to 0.05 and all the temperature dependences χ–1/Т are straight lines. It should be noted that all χ–1/Т plots have similar slopes. Within the framework of the refined Flory–Huggins theory [5, 6] the slope of the χ–1/Т dependence is related to the enthalpy constituent of the pair interaction parameter χH, that is, is proportional to the energy of interaction between segments of the mixed polymers. As the molecular weight of the oligomeric component increases, χ naturally decreases and asymptotically approaches a certain limiting value χ ≅ 0.02, which characterizes the interaction of two high molecular weight BASCs and PS. The free term of the χ–1/Т dependence characterizes the entropy constituent χS. The temperature dependences of χ and their extrapolation to χcr were used to calculate the UCST and the domes of the binodal curves.

Temperature dependence of the pair interaction parameter: PS-4 with (1) PBA, (2) block-BASC-7, (3) block-BASC-17, (4) block-BASC-46, (5) gradient-BASC-18, and (6) random-BASC-17; (7) PS-5 with block-BASC-17 and (8) PS-6 with block-BASC-17.

Dependence of the pair interaction parameter on copolymer composition at 400 K in the systems (1) random-BASC–PS-4 [2] and block-BASC with (2) PS-4, (3) PS-5, and (4) PS-6. The open triangle refers to the gradient-BASC with PS-4, the full squares refer to PBA with PS-4, PS-5, and PS-6, and the open square refers to PS–PS. The dashed line refers to the limiting value of the pair interaction parameter for the system PS–BASC. The inset shows the dependence of χcr on composition for block-BASC with (5) PS-4, (6) PS-5, and (7) PS-6.
Thus, the constant temperature coefficient of the enthalpy component, which varies in the limited interval 6.8 ± 2.0, confirms the assumption that the intersegment interaction energy is independent of molecular weight.
The effect of the molecular weight of PS on the position of boundary curves may be traced using block-BASC-17 as an example (Fig. 8, curves 2, 7, 8). When moving from PS-4 (2) through PS-5 (7) and to PS-6 (8), the numerical values of χ decrease, but the slope does not change. This indicates that the molecular weight of PS influences only the entropy constituent of the pair interaction parameter.
The effect of copolymer structure on the thermodynamics of mixing is the most evident in Fig. 9, in which the pair interaction parameter is plotted as a function of the composition of copolymers of different nature. For all systems, regardless of the architecture of macromolecules, there is the common trend of a change in χ and χcr with the composition of copolymers—the pair interaction parameter gradually decreases with an increase in the content of polystyrene units in the copolymer chain and asymptotically approaches the limiting value χ = 0.02, which is typical of the system oligo-PS–high molecular weight PS. In our opinion, the seemingly extremal pattern of dependences 2–4 in Fig. 9 may be explained by a much lower molecular weight of block-BASC-46 compared with the other block-BAS copolymers. This hypothesis is proved by the calculated composition dependences of χcr (Fig. 9, curves 6–8), which are influenced only by the MW of the components. Accordingly, it should be expected that the high molecular weight block-BASC with the content of styrene on the order of 50% should have lower values of χ comparable with the limiting one.
The Diffusion of PS in the Copolymers
Figure 10 shows the kinetics of diffusion front propagation of PS macromolecules into the matrix of block-BASC and gradient-BASC (curves 1–8) and the copolymer macromolecules into the matrix of PS (curves 1'−8 '). The linear character of the dependences plotted in coordinates Х–t1/2 (Х is the depth of penetration of the diffusing component, and t is the observation time) provides evidence for the diffusion mechanism of spontaneous mixing of the components in the region of solutions (I) on the phase diagrams. The limiting partial diffusion coefficients of PS and the copolymers were calculated according to the Boltzmann–Matano method and from the slope of the straight lines (Table 2) using both the simple formula of the mean square displacement of front D = X2/2t and the Boltzmann–Matano method [13, 14]. The partial self-diffusion coefficients of polystyrene \(D_{{12}}^{*}\) into the block and gradient copolymers and the copolymers \(D_{{21}}^{*}\) into the PS matrix were calculated by the equation
where DV is the interdiffusion coefficient, D* is the partial self-diffusion coefficient, φ1 is the solution composition corresponding to the position of the figurative point on the concentration distribution profile front, \(\frac{{{{M}_{2}}}}{{{{M}_{1}}}}\) is the geometry parameter equal to the ratio of molar volumes of polymers, and χ is the pair interaction parameter of components (in accordance with Fig. 8). The calculation results are summarized in Table 2.

The kinetics of diffusion front propagation at 393 K in the systems (1, 1 ') block-BASC-7–PS-2, (2, 2 ') block-BASC-7–PS-4, (3, 3 ') block-BASC-17–PS-4, (4, 4 ') block-BASC-46–PS-4, (5, 5 ') block-BASC-7–PS-5, (6, 6 ') block-BASC-17–PS-5, (7, 7 ') block-BASC-46–PS-5, and (8, 8 ') gradient-BASC-18–PS-4. (1–8) The kinetics of propagation in the copolymer matrix, and (1 '–8 ') the kinetics of propagation in the PS matrix.
The dependences of the diffusion coefficients of PS into the copolymers and the copolymers into the PS melt on the molecular weight of PS are presented in Fig. 11. It is seen that, for block-BASC-7 and block-BASC-17, the exponent of the dependence D = kM–b is almost two, while for block-BASC-46 it is much higher. We believed that this is associated the contribution of thermodynamic nonideality to the diffusion coefficient.

Dependence of the diffusion coefficient on the molecular weight of PS at 393 K in the phase of copolymers for the diffusion of PS-4 into the matrix of (1) block-BASC-7, (2) block-BASC-17, (3) block-BASC-46, and (4) gradient-BASC-18.
An analysis of the temperature dependence of the diffusion coefficients made it possible to calculate the activation energy of diffusion; its numerical values are listed in Table 3.
Figure 12 shows the interdiffusion coefficients of PS into the matrix of the copolymer and of the copolymers into the matrix of PS as a function of the composition of the copolymers. For comparison, the same figure presents the data on translational diffusion coefficients for the random copolymers (curve 1). For all the systems, there is a common tendency—reduction in the translational mobility of PS macromolecules in the matrix of the copolymers of different architecture with an increase in the amount of PS units in the chain. The specific nature of each system makes itself evident as the intensity of decline in the rate of the translational motion of PS macromolecules in the copolymers. For example, when the composition of block-BASC and gradient-BASC changes by 20%, the self-diffusion coefficient of PS changes by an order of magnitude for the systems based on PS-4 and two orders of magnitude for the systems based on PS-5. Note that the self-diffusion coefficient of the copolymers into PS changes by half an order of magnitude and one-and-a-half orders of magnitude for the systems based on PS-4 and PS-5, respectively. A similar effect is observed in the case of random-BASC. The exception is block-BASC-46, whose molecular weight is two times lower than the molecular weight of the remaining copolymers.

Dependence of the diffusion coefficient on copolymer composition at 393 K (a) in the phase of copolymers for the diffusion of PS-4 into the matrix of (1) random-BASC [2] and (2) block-BASC and (3) PS-5 into the matrix of block-BASC and (b) in the phase of PS for the diffusion of (1) random-BASC and (2) block-BASC into the PS-4 matrix and (3) block-BASC into the PS-5 matrix. The open triangle refers to the gradient-BASC, the full square refers to PBA, and the open square refers to PS.
It is known that, for binary systems, two limiting regions of compositions φ1 → 0 and φ1 → 1, in which a diffusant and a diffusion medium are diverse in nature, are of most interest. Obviously, in the former case (φ1 → 0), the case in point is the translational mobility of homopolymers in the medium of block and gradient copolymers; in the latter case (φ1 → 1), the translational mobility of the copolymers in the PS matrix. Within the framework of the reptation model [15], this change in the nature of the diffusion medium and diffusant implies that, in the first case, information on the predominant effect of copolymer macromolecules forming the wall of the tube on the translational mobility of PS is derived; in the second case, information on the effect of the structure of copolymer macromolecules moving along the tube formed by PS macromolecules on the diffusion coefficients. The schematic of motion of macromolecules illustrating the above considerations is shown in Fig. 13. It could be expected that the numerical values of the friction coefficients of macromolecules (or chains) characterizing their motion in the entanglement tube would be different. Following the data from [15], let us assume that the translational motion of macromolecules is described by the reptation model, according to which the partial self-diffusion coefficient may be expressed through the equation
where D is the self-diffusion coefficient; Z is the friction coefficient of a chain per unit, which characterizes its motion in the entanglement tube; and Ne is the number of units in a chain between entanglements (taken to be 100).

Schematic of motion of macromolecules in the tube formed in the matrix of (a) block-BASC and (b) PS. Bold straight lines refer to block-PS, and thin straight lines refer to block-PBA.
In our case, the self-diffusion coefficients of the homopolymer PS in the copolymer and the copolymer in PS depend on the local composition of the medium:
where
where ρ1 is the molar fraction of component units (in this case, PS) and z is the friction coefficient per monomer unit.
Figure 14 shows the calculated friction coefficients of chains for the translational motion of PS in the copolymers and the copolymers in PS. It is seen that, firstly, within the measurement error, the friction coefficients of chains depend almost equally on the composition of copolymers of different architectures. A certain scatter in these values may be attributed to the proximity of the frontal concentrations to the binodal curves, that is, to the contribution of thermodynamic nonideality to the values of diffusion coefficients. Secondly, in calculations of average friction coefficients (Eqs. (11)–(12)), the additivity rule is valid only in a narrow range of styrene contents in the copolymer (up to 30–40%). In general, when the composition is changed from zero to unity, the change in the friction coefficient is described by a third order polynomial. Thus, it may be stated that, in calculations of translational mobility coefficients, it is advisable to use local friction coefficients, which characterize the architecture of both the diffusant and the diffusion medium.

Dependences of the friction coefficient on copolymer composition at 393 K (а) in the phase of copolymers for the diffusion of PS-4 into the matrix of (1) PBA, (2) random-BASC, (3) block-BASC, (5) gradient-BASC, and (6) PS and (4) PS-5 into the matrix of block-BASC and (b) in the phase of PS for the diffusion of (1) PBA, (2) random-BASC, (3) block-BASC, and (5) gradient-BASC into the PS-4 matrix and (4) block-BASC into the PS-5 matrix.
Finally, it can be mentioned that a similar conclusion about the contribution to the diffusion coefficient can be made using our data on D0 = kT/Z. This expression has a meaning of the self-diffusion coefficient for the system of broken units, in which monomer units are not joined together into a chain (Fig. 15).

The reptation constant D0 as a function of copolymer composition at 393 K in the copolymer phase for the diffusion of PS-4 into the matrix of (1) PBA, (2) random-BASC, (3) block-BASC, and (5) gradient-BASC and (4) PS-5 into the matrix of block-BASC.
Thus, complex research into the processes of diffusion and solubility of polystyrenes of various molecular weight in the block and gradient n-butyl acrylate–styrene copolymers of different composition is performed and the results obtained are compared with the data available for PBA and random copolymers. The phase diagrams featuring the UCST are constructed. The data on the interdiffusion of polystyrene into the copolymer and the copolymer into polystyrene are obtained. The friction coefficients of macromolecules and the diffusion coefficients of monomer units are calculated. It is shown that the effect of copolymer structure is small compared with the effect of diffusant molecular weight.
REFERENCES
A. E. Chalykh, U. V. Nikulova, and A. A. Shcherbina, Polym. Sci., Ser. A 57, 445 (2015).
A. E. Chalykh, U. V. Nikulova, A. A. Shcherbina, and E. V. Chernikova, Polym. Sci., Ser. A 61, 175 (2019).
U. V. Nikulova and A. E. Chalykh, in Structure and Dynamics of Molecular Systems. Collected Papers, Ed. by A. A. Shcherbina (IFKhE RAN, Moscow, 2017), p. 177 [in Russian].
A. E. Chalykh, U. V. Nikulova, V. K. Gerasimov, and R. R. Khasbiullin, Polym. Sci., Ser. A 62, 85 (2020).
Polymer Blends, Ed. by D. R. Paul and S. Newman (Acad. Press; A Subsidiary of Harcout Brace Jovanovich Publ., New York; San Francisco; London, 1978).
Polymer Blends, Ed. by D. R. Paul and C. B. Bucknall (A Wiley-Intersci. Publ. John Wiley & Sons, Inc., New York; Chichester; Weinheim; Brisbane; Singapore; Toronto, 2000).
E. V. Chernikova, V. V. Yulusov, E. S. Garina, Yu. V. Kostina, G. N. Bondarenko, and A. Yu. Nikolaev, Polym. Sci., Ser. B 55, 176 (2013).
D. V. Vishnevetskii, Candidate’s Dissertation in Chemistry (MGU, Moscow, 2015).
A. Malkin, A. Ascadsky, V. Kovriga, and A. E. Chalykh, Experimental Methods of Polymer Physics (Mir Publ., Moscow, 1983).
A. E. Chalykh, A. I. Zagaitov, V. V. Gromov, and D. P. Korotchenko, Optical Diffusiometer “ODA-2”. Guideline (Inst. Fiz. Khim. RAN, Moscow, 1996) [in Russian].
E. V. Chernikova, A. V. Plutalova, E. S. Garina, and D. V. Vishnevetsky, Polym. Chem. 7, 3622 (2016).
A. E. Chalykh, V. K. Gerasimov, and Yu. M. Mikhailov, Phase State Diagrams of Polymer Systems (Yanus-K, Moscow, 1998) [in Russian].
A. E. Chalykh, Water Diffusion in Polymer Systems (Khimiya, Moscow, 1987) [in Russian].
I. N. Bekman, Diffusion Mathematics: Tutorial (OntoPrint, Moscow, 2016) [in Russian].
N. A. Plate, A. D. Litmanovich, and Ya. V. Kudryavtsev, Macromolecular Reactions in Polymer Melts and Blends: Theory and Experiment. A. V. Topchiev Inst. Petrochem. Synth. RAS (Nauka, Moscow, 2008) [in Russian].
ACKNOWLEDGMENTS
We are grateful to E.V. Chernikova (Moscow State University) for providing us with the copolymer samples.
Our studies were carried out using equipment of the Shared Research Center of the Frumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences.
Funding
This work was supported by the Ministry of Science and Higher Education of the Russian Federation (theme registration number АААА-А19-119011790097-9).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by T. Soboleva
Rights and permissions
About this article

Cite this article
Chalykh, A.E., Nikulova, U.V. & Shcherbina, A.A. Diffusion and Thermodynamics of Polystyrene Mixing with Block and Gradient Copolymers of Butyl Acrylate and Styrene. Polym. Sci. Ser. A 62, 368–382 (2020). https://doi.org/10.1134/S0965545X20040033
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0965545X20040033