
Abstract
A number of bifunctional composite cobalt-based catalysts were prepared with various types of molecular sieves in the H-form (zeolites ZSM-5, Y, Beta, and Mordernite, and silicoaluminophosphate SAPO-11) as an acid component. The catalytic performance of these catalysts was comparatively assessed in integrated Fischer-Tropsch synthesis of fuels. The catalysts were found to exhibit high activity and high C5+ selectivity at 2.0 MPa, 240–250°C, and a gas hourly space velocity (WHSV) of 1000 h–1. With the CO conversion reaching 80.9–93.7%, the productivity was in the range of 146.4 kg/(m3cat h). The study further revealed the effects of the crystalline and porous structures of the molecular sieves on the hydrocarbon and fractional compositions of the fuel products and on the selectivity towards branched hydrocarbons. It was shown that catalysts based on HZSM-5 and HBeta zeolites are preferable for high-performance and selective synthesis of fuels. In the presence of the HZSM-5-based catalyst, an effect atypical of Fischer–Tropsch synthesis was observed for the first time: an elevation of the process temperature leads to an enhancement of C5+ selectivity.

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Of late, the well-known catalytic Fischer–Tropsch (FT) synthesis has been in demand in the green technology market [1, 2]. This process converts a CO/H2 mixture (referred to as syngas) to environmentally friendly low-sulfur engine fuels (e.g., gasoline and diesel fuels) and various chemicals (e.g., alkenes, oils, aromatics, and oxygenates) [3, 4]. In recent attempts to effectively intensify this process, researchers have increasingly considered a process solution that involves two consecutive steps carried out in one reactor unit: synthesizing linear hydrocarbons (on metal catalytic sites) followed by isomerizing them into branched compounds (generally on active acid sites). Integrated technologies based on this approach are mostly intended for the fuel market, and are considered promising as industrially proven processes. Among them, GTL (gas-to-liquid) technologies are of particular interest.
To implement GTL processes, novel catalyst types and modifications have been created (in commercial practice, these are generally represented by iron-based and cobalt-based catalysts). Provided that the chemical composition of the fuel products is optimized, these catalysts largely resolve the challenge of overcoming the Anderson–Schulz–Flory distribution [5–7]. According to this distribution, the maximum selectivity towards gasoline (C5–C10) and diesel (C11−C18) hydrocarbons is limited to 45 and 30%, respectively. In FT synthesis, iron catalysts promote the formation of mostly alkenes (when under elevated pressure and temperature) or alcohols (at increased syngas hourly space velocity) [8, 9]. Chemicals synthesized over conventional cobalt FT catalysts such as Co/SiO2, Co/Al2O3, and Co/TiO2 consist mostly of n-alkanes, the yield and composition of which are directly determined by process conditions, while the catalysts themselves are distinguished by extended durability [10–12]. High selectivity towards straight-chain alkanes causes the production of liquid fuels with low octane numbers [13, 14]. To enhance the octane number, it has been proposed to use aromatics or FT processes designed to selectively produce a portion of chemicals in the branched form [15]. On the other hand, the implementation of integrated FT synthesis over bifunctional metal–zeolite catalysts allows gasoline and diesel fuels to be produced in one step with high yields and high iso-alkane concentrations [16]. Unlike the conventional FT method, this approach allows significant quantities of fuels to be produced without the need to have a downstream hydrocracking or hydrotreating step or to install several reactor units. As a consequence, this approach reduces the production costs and improves the overall synthesis performance. However, such a design requires that effective techniques must be implemented to combine conventional metal FT catalysts and acid components in one reactor [17].
Molecular sieves have been deemed promising as acid components in FT catalysts because they contain acid sites that promote secondary synthesis reactions such as the commonly known hydroprocessing (including hydrocracking and hydroisomerization) [18]. Due to their molecular-sieve effect and well-developed microporous structure, molecular sieves have been in high demand as catalysts and catalyst supports for FT synthesis [19]. Due to their high activity, low cost, and good stability, they have been extensively applied in industry [20]. Hybrid catalysts for integrated FT synthesis, which are prepared by mixing an FT catalyst and a molecular sieve, have two types of active sites: one type to synthesize hydrocarbons (mostly unbranched alkanes and α-alkenes), and the other for the hydrocracking and isomerization thereof. Thus, proper variation in the properties of the molecular sieves makes it possible to largely control the properties of related hybrid multifunctional systems, including the composition of FT fuels [21, 22].
Many reports have been published on bifunctional (hybrid) zeolite-based catalysts for integrated FT synthesis [23–28]. Among these catalysts, of particular interest are composite catalytic systems prepared by mixing two components: a cobalt-based FT catalyst [26, 27], and a molecular sieve (e.g., ZSM-5, Y, Beta, Mordernite, SAPO-11, etc.) as an acid component. There have been relevant publications on the implementation, as a cobalt-based component, of promoted catalysts containing compounds of noble metals (e.g., Pt, Pd, Re, etc.) [27, 28]. Composite catalysts with oxides (Al2O3, MnOx, etc.) as promoters have been underexplored.
We have developed and field-tested a bifunctional composite catalytic system for direct production of hydrocarbon fuels [29–31]. To ensure stable, high-performance, and selective FT synthesis of long-chain hydrocarbons, a Co–Al2O3/SiO2 catalyst was used as a metal component [32]. The acid component was represented by a pentasil-group ZSM-5 zeolite (with 10 T-atoms) in the H-form with a medium-porous structure and molecular selectivity that suppressed the formation of multibranched isomers [33]. However, catalysts that would contain other types of molecular sieves as an acid component, particularly when compared to ZSM-5 have yet to be characterized. Further research is needed into the processes that take place on the catalyst surface under known diffusion limitations caused by the composition of hydrocarbons being formed, including the impact of these processes on the selectivity of individual reactions and entire FT synthesis, and on the fractional and group compositions of the products. This research would provide new relevant data that would make it possible to control the selectivity towards the hydrocarbon products. New efforts should be dedicated to optimize the previously developed catalyst, to search for other promising catalyst compositions, and to properly choose the type, chemical composition, and framework of the molecular sieve, taking into account its effects on the physicochemical properties and structure of the active component of the composite system. These efforts could afterwards serve as a matrix for the compilation of a list of catalysts on which targeted chemical reactions could be carried out.
The purpose of this study was to investigate the effects of the type and structure of industrial molecular sieves on the catalytic performance of bifunctional FT catalysts prepared by mixing a metal component (Co–Al2O3/SiO2), an acid component (H-form ZSM-5, Y, Beta, Mordernite, or SAPO-11), and a binder in the composite mixture form.
EXPERIMENTAL
Samples of cobalt-based bifunctional FT catalysts were prepared as a composite mixture. We used a Co–Al2O3/SiO2 catalyst for selective FT synthesis of long-chain hydrocarbons (20.0 wt % Co and 1.0 wt % Al2O3) on a KSKG grade silica gel support (manufactured by the Salavat Catalyst Plant, Republic of Bashkortostan, Russia) [31] as a metal component; zeolites and silicoaluminophosphate SAPO-11 in the H-form as an acid component; and Pural Sasol SB Al(OH)O pseudoboehmite TH 80 (manufactured by Sasol) as a binder. The following commercial zeolites were used for the study: HZSM-5 and HY (manufactured by the Ishimbay Specialized Chemical Catalyst Plant, Republic of Bashkortostan, Russia), Beta and Mordernite (Zeolyst International), and SAPO-11 (synthesized by IPC RAS). Before testing, the HZSM-5 and HY zeolites were dried at 120–150°C for 2 h; the Beta and Mordernite in ammonium form were calcined at 550°C for 4 h; and the SAPO-11 was calcined at 600°C for 6 h. The H-forms of ZSM-5, Y, Beta, Mordernite, and SAPO-11 were labeled HZ, HY, HB, HM, and HS, respectively. The related catalysts were designated as HZ+, HY+, HB+, HM+, and HS+, respectively.
The catalyst samples were prepared by mixing powders (<100 μm) of the components [32]. To plasticize the powder mixture, an aqueous–alcoholic solution of triethylene glycol with nitric acid was used. A nitric acid solution was prepared by introducing 1–2 mL of 65 wt % nitric acid into 90–100 mL of distilled water per 100 g of the powder mixture. Triethylene glycol was then added based on a nitric acid to triethylene glycol volumetric ratio of 1 : 3. Catalyst pellets were successively dried at 80°C for 4 h and at 100–150°C for 2–4 h, and calcined at 400°C for 4 h. The pellets were then ground to particles 1–2 mm in size. The catalysts had the following compositions: Co–Al2O3/SiO2 catalyst, 35 wt %; H-form molecular sieve (ZSM-5, Y, Beta, Mordernite, or SAPO-11), 30 wt %; boehmite, 35 wt %.
The catalysts were analyzed by X-ray diffraction (XRD) on an ARL X'TRA powder diffractometer (Thermo Fisher Scientific, Switzerland) with monochromated CuKα radiation using point-to-point scanning (step 0.01°, counting time 2 s) in the 2θ range of 10°–90°. The phase composition was identified using Crystallographica software and the PDF-2 database [34]. The X-ray patterns were processed using FullProf software. The average size of Co3O4 particles for the characteristic line with 2θ = 36.8° was derived from the Scherrer equation [35], and the average size and dispersion of Co0 particles were evaluated as described in [36, 37].
The BET specific surface area and the micropore and mesopore volumes were measured by the low-temperature nitrogen adsorption/desorption on a Nova 1200e sorption analyzer (Quantachrome, USA). The BET specific surface area was calculated at a relative partial pressure (P/P0) of 0.05 to 0.2. The micropore volume in the presence of mesopores was evaluated by the t-Plot method. The pore size distribution was calculated using BJH (Barrett–Joyner–Halenda) desorption branch analysis. Before testing, the samples were evacuated at 350°C for 5 h.
The cobalt content in the catalysts was identified by X-ray fluorescence analysis (XRF) on an ARL QUANT'X energy-dispersive spectrometer (Thermo Scientific, USA) under the following conditions: air environment, Teflon support, effective irradiation area 48.9 mm2.
The catalyst surface processes were examined by hydrogen temperature-programmed reduction (H2–TPR) using a ChemiSorb 2750 analyzer (Micromeritics, USA) equipped with a thermal conductivity detector (TCD). Prior to starting the H2–TPR procedure, the samples were degassed by holding in a helium flow at 200°C for 1 h. They were then reduced with a 10% H2/90% N2 mixture (20 mL/min) in the range of 20–800°C at a heating rate of 20°C/min.
The acidity of the samples was measured by ammonia temperature-programmed desorption (NH3-TPD) on a Quantachrome Autosorb analyzer (Quantachrome, USA). Prior to starting the adsorption, the samples were annealed in helium at 600°C for 5 h, then treated with a 10% NH3/90% He (v/v) mixture at 100°C for 2 h, and purged with helium to remove physically adsorbed ammonia. The NH3 desorption was carried out in the range of 100–600°C at a heating rate of 10°C/min.
The concentrations of weak, medium, and strong acid sites were derived from the amount (μmol/g) of NH3 desorbed in the ranges of 100–250°C, 250–400°C, and 400–600°C, respectively.
The FT catalytic performance was investigated in a 16 mm dia. isothermal reactor with a fixed catalyst bed. The catalyst (1–2 mm particles) in an amount of 10 cm3 mixed with 30 cm3 of quartz (with an identical particle size distribution) was introduced into the reactor. The catalyst was reduced with hydrogen for 1 h at 400°C and a gas hourly space velocity (WHSV) of 3000 h–1. The activation of the samples with syngas (H2/CO = 2) and the catalytic tests were carried out at 2.0 MPa and WHSV 1000 h–1 by heating from 180°C to 240–250°C at a rate of 2.5°C/h. The balance tests were run for 50–90 h, with the gas composition and flow rate at the test unit outlet being analyzed every 2 h. The measurement error was within 2.5%. The catalyst activity was assessed by measuring the CO conversion, the selectivity and productivity of the catalyst, and the fractional and group compositions of the synthetic products.
The syngas feed and gaseous products were analyzed on a Crystal 5000 gas chromatograph (Chromatec, Russia) equipped with a thermal conductivity detector and two columns (Haysep R active phase and NaX molecular sieves). The heating was programmed at a rate of 8°C/min. The condensed synthetic products were separated, by distillation at atmospheric pressure, into a C5–C10 gasoline fuel (<180°C), a C11–C18 diesel fuel (180–330°C), and a C19+ bottom residue (>330°C). The C5+ hydrocarbon composition was identified by chromatography-mass spectrometry using an Agilent 7890A instrument (Agilent Techno1ogies, USA) equipped with a 5975C mass-selective detector (MSD) and an HP-5MS capillary column.
RESULTS AND DISCUSSION
The commercial samples of MFI, FAU, BEA, and MOR type zeolites (ZSM-5, Y, Beta, and Mordernite, respectively) and the H-form SAPO-11 were investigated as acid components of bifunctional cobalt-based FT catalysts (Table 1).
Among the wide variety of silicoaluminophosphates with crystals composed of AlO4, PO4, and SiO4 tetrahedra, SAPO-11 is of particular interest due to its one-dimensional channel porous structure [38].
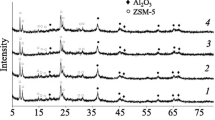
The XRF data show that the cobalt concentration in the catalysts was within 7.3–7.4 wt %. The phases of the catalyst components [34] were identified by XRD (Figs. 1a, 1b). The precursor of the metal component was an oxide phase (Co3O4) with a cubic spinel structure (Fd3m), which was detected as a series of reflections in the 2θ range of 18°–68°. SiO2 is X-ray amorphous, as indicated by a halo in the XRD pattern of Co–Al2O3/SiO2. The narrow spreads of the Scherrer-derived particle sizes of metallic cobalt and of their dispersion (10.8–11.4 nm and 8.4–8.9%, respectively, see Table 1) resulted from the fixation of Co3O4 crystals on the catalyst surface during the heat treatment. The phase composition of the molecular sieves was confirmed by the XRD examination (Fig. 1a). The acid component in the composite catalysts is indicated by reflections in the following 2θ ranges [34]: 7°–30° for HZ+, 5°–35° for HY+ and HB+, 5°–50° for HM+, and 5°–35° for HS+ (Fig. 1b). The low-crystallinity alumina phase formed during the thermal decomposition of the pseudoboehmite binder is represented by reflections between 47° and 70°.

X-ray patterns. (a) Molecular sieves: (1) HZ; (2) HY; (3) HB; (4) HM, and (5) HS. (b) Catalysts: (1) HZ+; (2) HY+; (3) HB+; (4) HM+; (5) HS+; and (6) Co–Al2O3/SiO2.
The porous properties of the molecular sieves and catalysts are presented in Table 2. The metal component (Co–Al2O3/SiO2) of the catalysts, with average pore size of below 11 nm, had a BET specific surface area of 216 m2/g and a pore volume of 0.564 cm3/g, with no micropores detected. The BET specific surface area of the boehmite binder was 180 m2/g. In terms of the pore size, the molecular sieves are classified as small-pore (0.30–0.45 nm), medium-pore (0.45–0.60 nm), and large-pore (0.6–0.8 nm) [33]. HY and HB had a well-developed surface (about 600 m2/g), mostly consisting of micropores, and a total pore volume of about 0.3 and about 0.4 cm3/g, respectively. HZ exhibited somewhat lower values: a surface area of 345 m2/g (with micropores accounting for up to 70%) and a pore volume 1.2-fold and 1.5-fold smaller than that of HY and HB, respectively. The lowest surface area was observed for the silicoaluminophosphate (HS). In the composite systems, with their developed porous structure, both the number and surface area of micropores declined when compared to the zeolites and the silicoaluminophosphate (by more than 90% for HZ+ and HS+, by 30% for HY+, and by 45% for HB+). Nonetheless, the total surface area and total pore volume (values being determined by the combination of the porous structures of all components and the system of newly-generated transport pores of the catalysts) remained fairly high, thus providing access to the catalyst’s active sites.
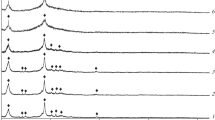
The H2–TPR spectra of the catalyst samples are largely similar (Fig. 2), especially in terms of the curve shapes and temperature maxima. The reduction of the cobalt-containing component follows a two-step sequential mechanism to ensure the Co3+→Co0 transformation. The ratio of the corresponding peak areas differs from the theoretical value (3) calculated in accordance with the Co3O4 reduction stoichiometry: it varies between 2.4 and 2.8.

H2–TPR curves: (1) HZ+; (2) HY+; (3) HB+; (4) HZ+; (5) HS+; and (6) Co-Al2O3/SiO2.
In general, the zeolites proved to have only a minor effect on Co3O4 reduction. This effect, presumably caused by the formation of small amounts of difficult-to-reduce mixed compounds of cobalt oxides and catalyst components [39, 40], is indicated by slight shifts or rises in the temperature and area for some of the peak maxima.
The catalytic performance characteristics of the bifunctional FT catalysts at 2.0 MPa, WHSV 1000 h–1, H2/CO = 2, and temperatures of 240 and 250°C are presented in Table 3. The relevant characteristics for the Co–Al2O3/SiO2 sample at 225°C, shown for comparison purposes, confirm the high activity and C5+ selectivity of the metal component of FT catalysts.
The bifunctional catalysts exhibited high activity in FT synthesis. The crystal structure and shape selectivity of the molecular sieve used as an acid component proved to be critical factors in the process performance. At 240°C, the CO conversion over the tested catalysts ranged between 72.6 and 85.1%, and the highest activity was achieved on HB+ and HS+. The highest selectivity towards target C5+ hydrocarbons (73.2%) was achieved on HB+, with productivity of 135.6 kg/(m3cat h). For the other catalyst samples, this selectivity parameter was between 65.5 and 67.5%. When the synthesis temperature was elevated to 250°C, the process intensification enhanced, predictably, the CO conversion to about 80–90%––the highest values again being observed for HB+ and HS+.
The highest productivity was achieved on HB+ and HZ+: 146.4 and 130.3 kg/(m3cat h), respectively. Given the increasing formation of C1–C4 and CO2 (as a result of the accelerating water–gas shift reaction [41, 42]), the C5+ selectivity declines for most catalysts regardless of their type [43]. The increasing concentration of low-molecular-weight hydrocarbons in the synthetic products is likely caused by a declining probability of the chain growth and/or by chain breaking [44] to form lower-molecular-weight products [45]. In the case of HZ+, however, the C5+ selectivity response to temperature elevation was atypical of FT synthesis: the C5+ selectivity was enhanced from 67.1 to 72.8%. We posit that, in the presence of a catalyst, the chain growth sites are blocked by long-chain C19+ hydrocarbons (waxes). With the HZ+ catalyst, the heating speeded up the hydrocracking of these hydrocarbons, thus unblocking the chain growth sites and enhancing the C5+ selectivity.
In the cases of HY and HM zeolites, the more intensive carbonaceous depositions in the presence of large amounts of high-molecular-weight condensed products may be caused by the porous structures of these catalyst components. In particular, HY has a 3D porous structure, and its 0.74 nm 12-membered rings limit the entrance to cavities about 1.3 nm in size [46]. HM zeolites are generally regarded as a 1D pore system, which leads to certain steric hindrances for most bulk molecular compounds and limits mass transfer [47].
Figure 3 shows the time trends of CO conversion at 240 and 250°C for the catalysts running continuously for 40–50 h. The CO conversion decline over time observed with all samples can likely be explained by carbonaceous depositions [31] that blocked active metal and acid sites on the catalyst surface during the operation. At 240°C, the most rapid catalyst deactivation was observed for HY+ and HM+, while the slowest deactivation was achieved on HZ+. Moreover, at 250°C, HZ+ even increased in activity.

CO conversion vs. FT synthesis time at 240°C (a) and 250°C (b) for catalyst samples: (1) HZ+; (2) HY+; (3) HB+; (4) HM+; (5) HS+.
It should be noted that the surface structure and acidity of the molecular sieves and related catalysts largely govern their activity in acid-catalyzed reactions. The protonic (decationized) form of molecular sieves utilized for the catalyst preparation exhibited Brønsted acidity; the particular concentration of acid sites depends on aluminum content, aluminum distribution over the crystals, SiO2/Al2O3 ratio, presence of compensating cations, and other characteristics [48].
The potential contribution of Lewis acid sites reveals itself through their interaction with Brønsted sites. In the presence of trace amounts of water, Lewis acid sites transform into Brønsted acid sites with high proton reactivity. Similar transformations are also possible in the presence of transition metals and hydrogen [33].
Catalyst samples generally have weak, medium, and strong acid sites, as clearly demonstrated for the cases of HZ and HB zeolites and related HZ+ and HB+ catalysts, which achieved the best FT synthesis performance (Table 4). However, weak acid sites made the dominant contribution (about 70%) to the surface acidity of the most active catalyst samples, while the presence of strong acid sites was almost negligible. On the other hand, both the composition and concentration of acid sites may vary depending both on the zeolite type and the component ratio in the bifunctional catalyst. In contrast, the activity and acidity of catalysts are not always directly correlated [33]. For example, the catalyst surface acidity has a major effect on the distribution of the hydroprocessing products of high-molecular-weight alkanes. However, it is not yet clear which type of acid sites is responsible for skeletal rearrangements of alkanes.
The data summarized in Table 5 reflect the significant differences in the fractional and group compositions of condensed C5+ hydrocarbons produced from FT synthesis using bifunctional catalysts at 240 and 250°C. For comparison purposes, the table additionally shows relevant values for the metal component (Co–Al2O3/SiO2). These values clearly indicate high selectivity towards C5+ hydrocarbons, in which normal compounds account for 97.4% and long-chain C19+ hydrocarbons represent 46.7%.
At 240°C, normal C5+ hydrocarbons were predominant in the products synthesized over all catalysts except HZ+. The HY+ and HS+ samples exhibited the highest concentrations of these hydrocarbons, as well as C19+ hydrocarbons, and the lowest concentrations of C5–C10 and C11–C18 fuels.
The concentration of iso-alkanes in the synthetic products mostly ranged between 18 and 22%, except for the 8.8% in the case of HS+. The HZ+ sample exhibited the highest total content of isomers and the highest iso/n and O/P ratios. Heating to 250°C stimulated the degradation of newly-formed alkanes and decreased the content of C5+ linear hydrocarbons (to 48–86%) and C19+. The highest total content of branched hydrocarbons (above 51%), as well as the highest iso/n and O/P ratios, were observed on HZ+. The amount of branched alkanes, taking into account the catalyst productivity, increased in the following order: HS+ < HY+ < HM+ < HB+ < HZ+. Thus, HZ and HB zeolites were found to be optimal acid components of bifunctional FT catalysts from the viewpoint of the catalyst activity, productivity, stability, and selectivity towards branched hydrocarbons in the fuel range. Although HB+ and HZ+ achieved relatively similar FT synthesis performance, the key factors lying behind this performance were different: the high activity (CO conversion) for HB+, and the selectivity towards fuel hydrocarbons in the case of HZ+.
The isomerization/cracking ratio is commonly assumed to depend on the metal type, acid site strength, and pore size of the catalyst, on the diffusivity of the reactants and intermediate carbenium ions, and on the reaction conditions (temperature, pressure, etc.) [33]. Isomerization is considerably affected by the characteristics of acid and metal sites, in particular by the balance between them [49]. As shown in Table 4, HB+ was superior to HZ+ in the surface concentration of acid sites. Bearing in mind that metal sites were located only on the surface of the cobalt catalyst, and that the cobalt concentration was equal in all bifunctional catalytic systems under study, it would be reasonable to assume that the concentration of metal sites was roughly equal for all samples. Therefore, given the almost equal amounts of cobalt in all catalyst samples and the presumably equal concentrations of catalytically active sites, the HZ+ catalyst had the higher ratio of metal to acid sites. This can serve as a potential rationale for the higher isomerization performance of this catalyst. In addition, the size of the zeolite channel cross-sections and the presence of a transport pore system remain important factors [49, 50]. In this regard, the better development of the pore system in HZ+, specifically the higher proportion of large and medium pores in this catalyst, is clearly indicated by the data in Table 2.
As far as hydrocracking reactions on zeolite acid sites are concerned [51], the following variations in fractional concentrations can be reported: an increase in C5–C10 (gasoline), a drop in C19+ (bottom residue), and almost no variation in C11–C18 (diesel). This suggests that high-molecular-weight hydrocarbons (C19+) is the group that undergoes cracking into C5–C10. The amount of gasoline hydrocarbons as the main hydrocracking product, taking into account the catalyst productivity, increases in the following order: HY+ < HM+ < HS+ < HZ+ < HB+. The process intensity affects the performance of FT synthesis during its operation.
Finally, the surface carbonaceous deposits that block the active (metal) and acid sites [11, 31] should also be considered as a factor affecting the catalyst performance. For example, carbon compounds on the surface of ZSM-5 are mostly linear, while considerable concentrations of less cracking-prone polyaromatics have been detected on the surface of HBeta [51]. It is also known that a longer hydrocarbon chain corresponds to a higher viscosity of FT products, which condense in the pores [11, 31] and can enter into cracking reactions to form lighter hydrocarbons. These lighter hydrocarbons can be removed from the catalyst’s pore space, thereby unblocking the surface. Thus, the cracking capacity of HZ+ also makes a definite contribution to its superior stability.
CONCLUSIONS
A number of bifunctional composite cobalt-based catalysts were prepared with various types of molecular sieves in the H-form (zeolites ZSM-5, Y, Beta, and Mordernite, and silicoaluminophosphate SAPO-11) as an acid component. The catalytic performance of these catalysts was comparatively assessed in integrated Fischer–Tropsch synthesis of fuels. The effects of the structure and properties of the acid component on the hydrocarbon and fractional compositions of the fuel products were discussed. The crystal structure and shape selectivity of the molecular sieve were demonstrated to play a key role in the selectivity of fuel synthesis, particularly towards branched hydrocarbons.
The composite systems were found to exhibit high activity and high C5+ selectivity at 2.0 MPa, 240–250°C, and WHSV 1000 h–1. At a synthesis temperature of 250°C, the CO conversion reached 80.9–93.7%, and the productivity was in the range of 100.7–146.4 kg/(m3cat h). It was further shown that, from the viewpoint of catalyst performance, and in particular the stability and selectivity towards branched hydrocarbons, catalysts based on HZSM-5 and HBeta zeolites are preferable. In the presence of the HZSM-5-based catalyst, an effect atypical of Fischer–Tropsch synthesis was observed for the first time: an elevation of the process temperature leads to an enhancement of C5+ selectivity. A temperature increase in the presence of this catalyst speeds up the hydrocracking of the hydrocarbons that have blocked the catalyst surface, thus unblocking chain growth sites and enhancing the C5+ selectivity.
REFERENCES
Wang, C., Fang, W., Wang, L., and Xiao, F.S., J. Energy Chem., 2021, vol. 54, pp. 429–433. https://doi.org/10.1016/j.jechem.2020.06.006
Escorihuela, S., Toldra-Reig, F., Escolástico, S., Murciano, R., Martínez, A., Serra, J.M., J. Membr. Sci., 2021, vol. 619, p. 118516. https://doi.org/10.1016/j.memsci.2020.118516
Sulima, S.I., Bakun, V.G., Chistyakova, N.S., Larina, M.V., Yakovenko, R.E., Savost’yanov, A.P., Petrol. Chem., 2021, vol. 61, no. 11, pp. 1178–1189. https://doi.org/10.1134/S0965544121110013
Pedrolo, D.R.S., Ordomsky, V.V., Schwaab, M., Marcilio, N.R., and Khodakov, A.Y., J. Mater. Sci., 2021, vol. 56, pp. 18019–18030. https://doi.org/10.1007/s10853-021-06457-1
Guo, L., Sun, S., Li, J., Gao, W., Zhao, H., Zhang, B., He, Y., Zhang, P., Yang, G., and Tsubaki, N., Fuel, 2021, vol. 306, p. 121684. https://doi.org/10.1016/j.fuel.2021.121684
Sartipi, S., Makkee, M., Kapteijn, F., and Gascon, J., Catal. Sci. Technol., 2014, vol. 4, pp. 893–907. https://doi.org/10.1039/C3CY01021J
Chen, Y., Zhang, J., Jiang, X., Wei, L., Li, Z., and Liu, C., J. Taiwan Inst. Chem. Eng., 2020, vol. 116, pp. 153–159. https://doi.org/10.1016/j.jtice.2020.11.007
Yakovenko, R.E., Zubkov, I.N., Bakun, V.G., and Savostyanov, A.P., Petrol. Chem., 2021, vol. 61, no. 4, pp. 516–526. https://doi.org/10.1134/S096554412105008X
Khodakov, A.Y., Chu, W., and Fongarland, P., Chem. Rev., 2007, vol. 107, pp. 1692–1744. https://doi.org/10.1021/cr050972v
Sartipi, S., Parashar, K., Valero-Romero, M.J., Santos, V.P., van der Linden, B., Makkee, M., Kapteijn, F., and Gascon, J., J. Catal., 2013, vol. 305, pp. 179–190. https://doi.org/10.1016/j.jcat.2013.05.012
Savost’yanov, A.P., Yakovenko, R.E., Narochnyi, G.B., Zubkov, I.N., Sulima, S.I., Soromotin, V.N., Mitchenko, S.A., Petrol. Chem., 2020, vol. 60, no. 1, pp. 81–91. https://doi.org/10.1134/S0965544120010120
Eliseev, O.L., Savost’yanov, A.P., Sulima, S.I., and Lapidus, A.L., Mendeleev Commun., 2018, vol. 28, no. 4, pp. 345–351. https://doi.org/10.1016/j.mencom.2018.07.001
Wang, Y., Gao, W., Kazumi, S., Fang, Y., Shi, L., Yoneyama, Y., Yang, G., and Tsubaki, N., Fuel, 2019, vol. 253, pp. 249–256. https://doi.org/10.1016/j.fuel.2019.05.022
Weber, J.L., Martínez del Monte, D., Beerthuis, R., Dufour, J., Martos, C., de Jong, K.P., and de Jongh, P.E., Catal. Today, 2021, vol. 369, pp. 175–183. https://doi.org/10.1016/j.cattod.2020.05.016
Xing, C., Li, M., Zhang, G., Noreen, A., Fu, Y., Yao, M., Lu, C., Gao, X., Yang, R., and Amoo, C.C., Fuel, 2021, vol. 285, pp. 119233. https://doi.org/10.1016/j.fuel.2020.119233
Lu, P., Sun, J., Zhu, P., Abe, T., Yang, R., Taguchi, A., Vitidsant, T., and Tsubaki, N., J. Energy Chem., 2015, vol. 24, pp. 637–641. https://doi.org/10.1016/j.jechem.2015.08.004
Martínez del Monte, D., Vizcaíno, A.J., Dufour, J., and Martos, C., Int. J. Energy Res., 2021, vol. 46, pp. 5280–5287. https://doi.org/10.1002/er.7460
Sadek, R., Chalupka, K.A., Mierczynski, P., Rynkowski, J., Millot, Y., Valentin, L., Casale, S., and Dzwigaj, S., Catal. Today, 2020, vol. 354, pp. 109–122. https://doi.org/10.1016/j.cattod.2019.05.004
Xing, C., Yang, G., Wu, M., Yang, R., Tan, L., Zhu, P., Wei, Q., Li, J., Mao, J., Yoneyama, Y., and Tsubaki, N., Fuel, 2015, vol. 148, pp. 48-57. https://doi.org/10.1016/j.fuel.2015.01.040
Zhang, M., He, Z., Zhang, M., Wang, L., Wang, Q., Zhang, X., and Li, G., Fuel, 2022, vol. 316, p. 123166. https://doi.org/10.1016/j.fuel.2022.123166
Park, G., Ahn, C., Park, S., Lee, Y., Kwak, G., and Kim, S.K., Appl. Catal. A: Gen., 2020, vol. 607, p. 117840. https://doi.org/10.1016/j.apcata.2020.117840
Chalupka, K.A., Sadek, R., Szkudlarek, L., Mierczynski, P., Maniukiewicz, W., Rynkowski, J., Gurgul, J., Casale, S., Brouri, D., and Dzwigaj, S., Res. Chem. Intermed., 2021, vol. 47, pp. 397–418. https://doi.org/10.1007/s11164-020-04343-0
Yao, M., Yao, N., Liu, B., Li, S., Xu, L., and Li, X., Catal. Sci. Technol., 2015, vol. 5, pp. 2821–2828. https://doi.org/10.1039/C5CY00017C
Kang, J., Wang, X., Peng, X., Yang, Y., Cheng, K., Zhang, Q., and Wang, Y., Ind. Eng. Chem. Res., 2016, vol. 55, pp. 13008–13019. https://doi.org/10.1021/acs.iecr.6b03810
Sartipi, S., Makkee, M., Kapteijn, F., and Gascon, J., Catal. Sci. Technol., 2014, vol. 4, pp. 893–907. https://doi.org/10.1039/C3CY01021J
Kriventseva, E.V., Gryaznov, K.O., Khat’kova, E.Yu., Sineva, L.V., and Mordkovich, V.Z., Vestn. MITKhT im. M.V. Lomonosova, 2013, vol. 8, no. 6, pp. 9–16.
Sineva, L.V., Asalieva, E.Y., and Mordkovich, V.Z., Russ. Chem. Rev., 2015, vol. 84, p. 1176. https://doi.org/10.1070/RCR4464
Adeleke, A.A., Liu, X., Lu, X., Moyo, M., and Hildebrandt, D., Rev. Chem. Eng., 2020, vol. 36, no. 4, pp. 437–457. https://doi.org/10.1515/revce-2018-0012
Savost’yanov, A.P., Narochnyi, G.B., Yakovenko, R.E., Saliev, A.N., Sulima, S.I., Zubkov, I.N., Nekroenko, S.V., and Mitchenko, S.A., Petrol. Chem., 2017, vol. 57, pp. 1186–1189. https://doi.org/10.1134/S0965544117060251
Yakovenko, R.E., Zubkov, I.N., Bakun, V.G., Agliullin, M.R., Saliev, A.N., and Savost’yanov, A.P., Catal. Ind., 2021, vol. 13, pp. 230–238. https://doi.org/10.1134/S2070050421030120
Yakovenko, R.E., Savost’yanov, A.P., Narochniy, G.B., Soromotin, V.N., Zubkov, I.N., Papeta, O.P., Mit-chenko, S.A., and Svetogorov, R.D., Catal. Sci. Technol., 2020, vol. 10, no. 22, pp. 7613–7629. https://doi.org/10.1039/D0CY00975J
Sulima, S.I., Bakun, V.G., Yakovenko, R.E. Shabel’skaya, N.P., Saliev, A.N., Narochnyi, G.B., and Savost’yanov, A.P., Kinet. Catal., 2018, vol. 59, pp. 218–228 https://doi.org/10.1134/S0023158418020131
Akhmedov, V.M. and Al-Khowaiter, S.H., Cat. Rev.-Sci. Eng., 2007, vol. 49, no. 1, pp. 33–139. https://doi.org/10.1080/01614940601128427
PDF-2. The powder diffraction file TM. International Center for Diffraction Data (ICDD), 2014. https://www.icdd.com/
Young, R.A., The Rietveld Method, Oxford University Press, 1995, p. 298.
Schanke, D., Vada, S., Blekkan, E.A., Hilmen, A.M., Hoff, A., and Holmen, A., J. Catal., 1995, vol. 156, no. 1, pp. 85–95. https://doi.org/10.1006/jcat.1995.1234
Xu, D., Li, W., Duan, H., Ge, Q., and Xu, H., Catal. Lett., 2005, vol. 102, nos. 3–4, pp. 229–235. https://doi.org/10.1007/s10562-005-5861-7
Agliullin, M.R., Kutepov, B.I., Ostroumova, V.A., and Maximov, A.L., Petrol. Chem., 2021, vol. 61, no. 8, pp. 836–851. https://doi.org/10.1134/S096554412108003X
Okoye-Chine, C.G., Moyo, M., and Hildebrandt, D., Fuel, 2021, vol. 296, pp. 120667. https://doi.org/10.1016/j.fuel.2021.120667
van Steen, E., Sewell, G.S., Makhothe, R.A., Micklethwaite, C., Manstein, H., de Lange, M., and O’Connor, C.T., J. Catal., 1996, vol. 162, pp. 220–229. https://doi.org/10.1006/jcat.1996.0279
Lualdi, M., Lögdberg, S., Regali, F., Boutonnet, M., and Järås, S., Top. Catal., 2011, vol. 54, pp. 977–985. https://doi.org/10.1007/s11244-011-9719-5
Gnanamani, M.K., Shafer, W.D., Sparks, D.E., and Davis, B.H., Catal. Commun., 2011, vol. 12, pp. 936–939. https://doi.org/10.1016/j.catcom.2011.03.002
Yang, J., Ma, W., Chen, D., Holmen, A., and Davis, B.H., Appl. Catal. A: Gen., 2014, vol. 470, pp. 250–260. https://doi.org/10.1016/j.apcata.2013.10.061
Lögdberg, S., Yang, J., Lualdi, M., Walmsley, J.C., Järås, S., Boutonnet, M., Blekkan, E., Rytter, E., and Holmen, A., J. Catal., 2017, vol. 352, pp. 515–531.
Dry, M.E., Catal. Today, 2002, vol. 71, nos. 3–4, pp. 227–241. https://doi.org/10.1016/S0920-5861(01)00453-9
Pavlov, M.L. Basimova, R.A., Alyab’ev, A.S., and Makeeva, E.A., Neftegaz. Delo, 2012, vol. 10, no. 1, pp. 169–173.
Jin, Y., Li, Y., Zhao, S., Lv, Z., Wang, Q., Liu, X., and Wang, L., Micropor. Mesopor. Mater., 2012, vol. 147, no. 1, pp. 259–266. https://doi.org/10.1016/j.micromeso.2011.06.023
Cejka, J., Corma, A., and Zones, S., Zeolites and Catalysis: Synthesis Reactions and Applications; Weinheim: Wiley–VCH Verlag, GmbH & Co, 2010, p. 881.
Khairullina, Z.R., Agliullin, M.R., Alekhina, I.E., and Kutepov, B.I., Vestn. Bashkir. Univ., 2020, vol. 25, pp. 495–505. https://doi.org/10.33184/bulletin-bsu-2020.3.6
Weitkamp, J., Solid State Ionics, 2000, vol. 131, pp. 175–188. https://doi.org/10.1016/S0167-2738(00)00632-9
Martínez, A., Rollán, J., Arribas, M.A., Cerqueira, H.S., Costa, A.F., and Aguiar, E.F.S., J. Catal., 2007, vol. 249, pp. 162–173. https://doi.org/10.1016/j.jcat.2007.04.012
Funding
This study was carried out with financial support from the Ministry of Sciences and Higher Education of the Russian Federation within the state assignment (project no. 2019-0990), using equipment of the Nanotechnology Center for Collective Use, South Russian State Polytechnic University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflict of interest requiring disclosure in this article.
Rights and permissions
About this article

Cite this article
Yakovenko, R.E., Bakun, V.G., Agliullin, M.R. et al. Effects of Zeolite Type on Integrated Fischer–Tropsch Synthesis and Hydroprocessing. Pet. Chem. 62, 950–961 (2022). https://doi.org/10.1134/S0965544122070209
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0965544122070209