
Abstract
The paper presents the first experimental results on the chemical interdiffusion of major components (SiO2, Al2O3, Na2O, CaO, MgO, and FeO) and the \(\text{CO}_{3}^{2-}\) anion at interaction between basalt and kimberlite melts under moderate pressures. The research was carried out using a high gas pressure apparatus of original design at Ar or CO2 pressures of 100 MPa and a temperature of 1300°C, with the use of the method of diffusion pairs. It is established that the rate of the oncoming chemical diffusion of all major components of melts (SiO2, Al2O3, Na2O, CaO, and MgO) and \({\text{CO}}_{3}^{{2 - }}\) anion is almost identical at the interaction of model basalt and kimberlite carbonate-containing melts and is approximately one order of magnitude higher than the diffusion rate of these components at the interaction of melts in the more polymerized andesite–basalt model system. The latter is explained by the significantly lower viscosity of the boundary melt (Montana boundary), which is formed during the interaction of model basalt and kimberlite melts. The equal diffusion rates of CaO and the \({\text{CO}}_{3}^{{2 - }}\) anion indicate that the CaCO3 carbonate diffuses from kimberlite to basalt (both model and natural) melts by means of the diffusion of the end members. The pattern of the diffusion processes significantly changes when melt of natural magnesian basalt interacts with model kimberlite. Thereby calcite diffuses into magnesian basalt also by means of diffusion of the end members. The diffusion rates of all other components of the melts (SiO2, MgO, and FeO) significantly increase. A weak exponential concentration dependence of all diffusing components is determined, with this dependence close to D(i) = constant.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Diffusion of components in silicate melts has long been proved and agreed to play a decisive role in many magmatic processes (Bowen, 1921). Various silicate melts of geological interest consist of three or more oxide components (e.g., SiO2, Al2O3, TiO2, FeO, MgO, CaO, Na2O, K2O, CO2, and H2O). Chemical diffusion in such systems is referred to as multicomponent diffusion. Multicomponent diffusion involves mass flows controlled by chemical potential gradients and is an important mass-transfer mechanism for many transport processes. Although diffusion is a microscopic effect, it can lead to macroscopic effect.
For example, the initial phases of explosive volcanic eruptions (or more widespread champagne-type eruptions) depend on the growth rates of the gas bubbles, which in turn, are controlled by diffusion that brings gas molecules into the bubbles. Inasmuch as diffusion plays a decisive role in many geological processes, petrology, and geochemistry, these data are often used in developing models, including interpretations of rock ages, thermal events, parameters of the origin and preservation of the chemical composition and isotope fractionation of minerals, control of the size of bubbles in volcanic rocks, and processes affecting the character of volcanic eruptions.
One of the main problems associated with many applications of diffusion data is to obtain reliable experimental data, because diffusion properties defined at various laboratories may differ by orders of magnitude (e.g. Liang, 2010).Characteristics of multicomponent chemical diffusion of major and volatile components at interaction between magmatic melts are understood still inadequately poorly (e.g., Watson and Baker, 1991; Watson, 1994; Kress and Ghiorso, 1995). Most literature data on the chemical diffusion of major components in magmatic melts were obtained by dissolving minerals (Watson and Baker, 1991; and others), whereas studies of the interdiffusion of these components in melts per se are still very scarce (Yoder, 1973; Watson and Baker, 1991). Practically no data are available so far on the multicomponent chemical diffusion at interaction between basalt and ultramafic carbonate-bearing magmas. This publication presents experimental data newly obtained on the interdiffusion of major components (SiO2, Al2O3, Na2O, CaО, MgO, and FeO) and the carbonate ion \({\text{CO}}_{3}^{{2 - }}\) at interaction between kimberlite and basalt melts at a pressure of 100 MPa and temperature of 1300°C. The experiments were carried out using the method of diffusion pairs and an IHPV of unique original design.
EXPERIMENTAL AND ANALYTICAL DATA
The experiments were carried out in a unique internally heated pressure vessel (IHPV) equipped with an original internal device with a high-pressure equalizer–separator.
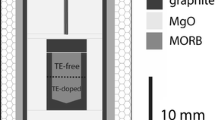
The diffusion experimental runs were made in platinum capsules, whose geometry has not been changed in the course of the runs at high pressures and temperatures (Fig. 1). The internal volume of the reactor (5) with a welded platinum capsule containing a starting diffusion pair (6) and equalizer–separator (9) under a piston (10) was filled with Ar or CO2 under a pressure of 10 MPa, using a specialized system.

Internal equalizer–separator of original design in the internally heated pressure vessel (IHPV). (1, 3) Insulators; (2) two-unit heater; (4) measurement thermocouples; (5) molybdenum reactor; (6) platinum capsule with the diffusion-pair melt; (7) adjustment thermocouples; (8) stopper plug; (9) equalizer–separator casing; (10) piston; (11) screw valve; (12) detector of piston position; (13) lid; (14) platinum capsule with an oxygen buffer (Persikov et al., 2018).
The assembled device with an internal heater (2) was mounted within IHPV in such a way that the capsule with a sample (6) occurred in the gradient-free temperature zone of the heater. Piston movements in the course of the runs always maintained Ar pressure in the vessel equal to the Ar or CO2 pressure in the inner volume of the reactor (5). Early in the course of the runs, Ar pressure in the vessel and the reactor (5) was increased for 1 h to the desired value of 100 MPa. Then the temperature was raised to 1100°C, and the run was held for 2 h in an automated regime. At these parameters, i.e., before the melting of the samples, the interdiffusion rates were very low. Then the temperature was rapidly increased for 1–2 min to the target value (1300°C), and the parameters were maintained during the required time of the diffusion runs (180 and 360 s in the first set of the runs and 80 and 120 s in the second one). After this, the experiments were isobarically quenched at the switched off internal heater. The quenching rate was thereby sufficiently high (~300°C/min) for the glasses to be preserved without crystallization of the melt. The temperature measurements were accurate to ±5°C, and the Ar or CO2 pressure was measured accurate to ±0.1 rel. %. Upon isobaric quenching, the device was taken from the vessel, the platinum capsule with the sample was extracted from the molybdenum reactor, and the sample was used to manufacture a polished platelet for the subsequent analysis of the diffusion profile produced in the melt in the course of the experimental run. The starting samples for the glasses in the experimental runs were powders (rocks powdered in an agate mortar) of magnesian basalt from the 1975–1976 Great Tolbachik Eruption of Tolbachik volcano in Kamchatka (Bolshoe …, 1984), model basalt (Ab50Di50, mol %), and kimberlite (82 wt % silicate + 18 wt % carbonate).
The chemical compositions of the anhydrous kimberlite and basalt melts (glasses) and natural basalt used in this study to characterize principal features of their diffusion-controlled interaction are presented in Table 1. The average composition of basalts (Le Maitre, 1976) is also listed in Table 1 for comparison, as a representative composition of basalt magma. Note that the composition of kimberlite magma in the mantle and when dikes and diatremes are formed in the crust is still a matter of extensive discussion (Wyllie, 1980; Sparks et al., 2006, 2009; Kamenetsky et al., 2009; Sharygin et al., 2013). Temperature and pressure changes, dissolution of volatile components, and crystallization processes may significantly modify the compositions of kimberlite melts during their evolution.
For example, it was hypothesized that kimberlite melts may be generated by a reaction of carbonatite melts with peridotite near the mantle solidus and subsequent partial melting of the carbonatized peridotite at very low degrees of partial melting (≤1.0%) at pressures of 6–10 GPa, depths of approximately 150–300 km, and temperatures ≤1500°C (Wyllie, 1980; Dalton and Presnall, 1998; Price et al., 2000; Dasgupta and Hirschmann, 2006; Sparks et al., 2006, 2009; Kopylova et al., 2007; Michell, 2008; Kavanagh and Sparks, 2009; Kamenetsky et al., 2009; Sharygin et al., 2013; Persikov et al., 2018). Because of this, the altered kimberlite from the Udachnaya East pipe in Yakutia, Russia (Kamenetsky et al., 2009; Kopylova, et al., 2007), which was selected as a representative composition of kimberlite magma, is compared in Table 1 with the composition of our model kimberlite (82 wt % silicate + 18 wt % carbonate), which was used in the experimental study of diffusion interaction with basalt melts. Although the model kimberlite does not contain some major oxides (TiO2, Al2O3, MgO, K2O, FeO, and Fe2O3) and hence does not exactly reproduce the compositions of naturally occurring kimberlites (Table 1), we have demonstrated that this composition corresponds to natural kimberlites in terms of degree of depolymerization (100NBO/T = 313) (Persikov et al., 2018). This parameter has been proved to reliably and reasonably sensitively reflect parameters characterizing the structure and chemical composition of magmatic melts (Persikov, 1998; Persikov et al., 2018). This parameter was calculated from the chemical composition of melt (in wt %) by the method (Persikov, 1998; Persikov et al., 2018), using the equation
where T is the total number of gram-ions of network-forming cations (Si4+, Al3+, Fe3+, Ti4+, P5+, and B3+) that are tetrahedrally coordinated by oxygen and are incorporated into the anion structure of the melt; and O is the total number of oxygen gram-ions in the melt.
Our data indicate that values of 100NBO/T = 67–80 (basalt melts) correspond to the range of mafic melts (17 ≤ 100NBO/T < 200), and the value of 100NBO/T = 313 (model kimberlite) falls within the range for ultramafic melts (200 ≤ 100NBO/T ≤ 400) (Persikov, 1998; Dingwell et al., 2004; Kopylova et al., 2007) (Table 1).
The anhydrous glasses of the starting model basalt melt and natural basalt were synthesized by melting powders of stoichiometric composition (Ab50Di50, wt %) and natural basalt in platinum capsules, using the aforementioned equipment (IHPV) at a temperature of 1300°C, Ar pressure of 100 MPa, experiment duration of 3 h, and subsequent rapid isobaric quenching. The glasses of model kimberlite melt were synthesized by fusing ~1.5 g of powdered mixture of natural albite and calcite (Ab38Cal62, wt %) in the same IHPV under a pressure of dry CO2 of 100 MPa. The sample was held at a temperature of 850°C for 1 h and then at a temperature of 1300°C for 3 h. The experimental run was then rapidly quenched by switching off the internal heater of the vessel. This procedure resulted in rapid decarbonatization (T = 850°C) with mass losses (~21 wt %) for each sample. These losses were determined by calculating the mass balance of each experimental run. The mass losses (in the form of CO2) of the samples at their melting corresponded to the decarbonization reaction (Persikov et al., 2018)
The concentrations of the silicate (82 wt %) and carbonate (18 wt %) phases in the synthesized glasses of model kimberlite, and hence, the concentration of the carbonate ion \({\text{CO}}_{3}^{{2 - }}\) (10.4 wt %) in them were calculated from the obtained mass-balance data that corresponded to decarbonization reaction (2) in each run on the synthesis of the glasses. The dissolution of CO2 in the melts mostly in the form of the \(\text{CO}_{3}^{2-}\) ion was qualitatively confirmed by Raman spectroscopy (Fig. 2).

Raman spectra in the region of 1020–1120 cm–1 for the diffusion pair model kimberlite–basalt. The spectra show \(\text{CO}_{3}^{2-}\) peaks at 1073 cm–1. The continuous line is the kimberlite zone, the dashed line is the diffusion zone, Montana boundary.
The chemical composition of the quenched melts (glasses) was analyzed by a microprobe (EPMA) (Table 1). The anhydrous glasses were optically homogeneous and did not contain either any bubbles or crystallites. The small water concentrations (≤0.05 wt %) in these glasses were determined by Karl Fischer titration. The glasses were extracted from the platinum capsules, cut into 5-mm cylinders, and both flat ends of these cylinders were polished. The cylinders were used to compose diffusion pairs, which were placed into platinum capsules of same diameter as that of the capsules used to synthesize the starting glasses. The capsules were then pumped off, and flat lids were welded to their open terminations using a TIG electrode. The prepared capsules with diffusion pairs were installed in the gradient-free zone of the molybdenum reactor in the inner device of IHPV (Fig. 1).
The products of the runs were analyzed by Karl Fischer titration on a KFT AQUA 40.00 to determine the low water concentrations in the synthesized basalt and kimberlite glasses (≤0.05 wt %). Raman spectra were employed to qualitatively illustrate the concentrations of carbonates that had been dissolved in the form of the \(\text{CO}_{3}^{2-}\) carbonate ion in the model kimberlite glasses (Fig. 2).
The Raman spectra of the synthesized kimberlite glasses and glasses after the diffusion runs at the Montana boundary (Crank, 1975) were recorded on a RENIS/HAW-1000 spectrometer coupled to a Leica microscope. The gap width for obtaining spectra was 50 μm, and the spectra were processed using the standard GRAMS program.
The chemical composition of the starting samples and glasses synthesized in the experiments with diffusion pairs was analyzed on a CamScan MV2300 (VEGA TS 5130 MM) electron microscope equipped with an INCA Energy 450 and WDS Oxford INCA Wave 700 energy-dispersive spectrometric system. The analysis was conducted at an accelerating voltage of 20 kV and beam current up to 400 nA. The counting time was 50–100 s. The following standards were used: quartz for Si and O, albite for Na, microcline for K, wollastonite for Ca, corundum for Al, metallic Mn for Mn, metallic Fe for Fe, and for Mg. The EPMA data were standardized using the INCA Energy 200 and Nekrasov INCA computer programs (Table 1).
RESULTS AND DISCUSSION
We used the following two types of diffusion pairs (Table 2, 3) in the experimental runs lasting for 80–360 s. (1) Anhydrous glasses of model basalt (Ab50Di50, mol %) and model kimberlite (Sil82 Carb18, wt %) were brought in contact with each other and melted in welded platinum capsules under an Ar pressure of 100 MPa and temperature of 1300°C. The duration of the diffusion experiments was 180 and 360 s. (2) Anhydrous glasses of natural magnesian basalt and model kimberlite (Sil82Carb18, wt %) were also mounted in contact with each other and melted in welded platinum capsules under an Ar pressure of 100 MPa, 1300°C, and experimental run times of 80 and 120 s.
The distribution of major components along the diffusion profiles was measured by EPMA. Results obtained by analyzing the diffusion profiles for the pair model basalt–kimberlite are presented in Fig. 3 (runs 2131 and 2141), and results for the pair natural basalt–model kimberlite are presented in Fig. 4 (runs 2146 and 2147).

Diffusion profiles of major components and the carbonate ion \(\text{CO}_{3}^{2-}\) at interaction between model melts of basalt and kimberlite at 1300°C and total pressure of 100 MPa (dashed lines are the Montana boundary; Crank, 1975). Duration time of the diffusion experimental runs: (a) 360 s, (b) 180 s.

Diffusion profiles of major components and the carbonate ion \(\text{CO}_{3}^{2-}\) at interaction between melts of natural magnesian basalt and model kimberlite at 1300oC and total pressure of 100 MPa (dashed lines are the Montana boundary; Crank, 1975). The duration of the diffusion experimental runs was: (a) 80 s and (b) 120 s.
The interdiffusion coefficients (D) of all major components and the carbonate ion \(\text{CO}_{3}^{2-}\) in the melts (Tables 2, 3) were determined in a linear approximation, by minimizing the functional with the application of the least-squares method
where Ci is the experimentally determined concentration of chemical element i, and C(xi, t) is the value of the model function describing the diffusion profile at points with coordinates xi at time t.
The used model function C(x, t) was the transcendental equation
where C0 = (Cmax + Cmin)/2, ΔC = (Cmax – Cmin)/2, Cmin and Cmax are the minimum and maximum values of the concentration of component i, x0 is the Montana boundary, t is the time of the experimental run, and D(C) is the function to describe the dependence of the diffusion coefficient of component i on its concentration.
Herein we mostly used an exponential dependence of the diffusion coefficient on the concentration of the component, i.e.,
where D0 is the starting value of the diffusion coefficient at C = 0, and α is the exponent. For comparison also considered the situation when the diffusion coefficient is independent of the concentration of the component, i.e., D(C) = constant.
Our results on the diffusion of various melt components were compared and estimated using the average values of the diffusion coefficient for the studied composition range: from Cmin to Cmax
Analysis of the results led us to the following conclusions. The interdiffusion rates of all of the major components (SiO2, Na2О, CaО, and MgО) are practically exactly identical (within the errors of ±20 rel. %) at interaction between model anhydrous basalt and kimberlite carbonate-bearing melts (Table 2) and are approximately one order of magnitude greater than the diffusion rates of components in the model system Ab75Di25–Ab50Di50 (Persikov et al, 2011). This significant increase in the interdiffusion rates of major components is explained, first of all, by the much lower viscosity of the boundary melt that is formed at interaction between model basalt and kimberlite melts. Estimates of the viscosity of this melt by our model for predicting the viscosity of silicate melts (Persikov et al., 2020), with regard to newly acquired experimental data on the viscosities of basalt and kimberlite melts (Persikov et al., 2018), confirm this hypothesis. According to these evaluations, the viscosity of the boundary melt (Montana boundary; Crank, 1975) of the basalt−kimberlite model system is one order of magnitude lower than the viscosity of the boundary melt of the system Ab75Di25–Ab50Di50 at the parameters of these experiments. The equal rates of the chemical interdiffusion of all of the major components (SiO2, Na2O, CaO, and MgO) indicate that interdiffusion proceeds by means of diffusion of end members, i.e., interdiffusion at equal rates of the diopside and calcite molecules (in the opposite directions) at interaction between model basalt and carbonate-bearing kimberlite melts. The diffusion rate of a network-forming anion (SiO2) is approximately two orders of magnitude higher than the rate of the tracer diffusion (Watson and Baker, 1991). Conversely, the diffusion rates of network-modifying cations (Na2O, CaO, and MgO) are roughly one order of magnitude lower than the rate of tracer diffusion (Watson and Baker, 1991). Correspondingly, the end-member character of the diffusion of albite molecules that has been detected at interaction of model melts in the andesite−basalt system (Persikov et al, 2011) does not operate in the model system basalt−kimberlite.
Note that no equal interdiffusion rates of all of the major components (SiO2, Na2O, CaO, MgO, Al2O3, and FeO) were detected in the diffusion pair of natural basalt−model kimberlite melts, in contrast to what was determined in the pure model system. The diffusion mobility of most components in this pair is approximately four times higher than in the pure model diffusion pair (Tables 3, 4) because of the lower viscosity of the boundary melt (Montana boundary; Crank, 1975) of this pair. The viscosity of this melt estimated by our model for predicting the viscosities of magmatic melts (Persikov et al., 2020), with regard to newly obtained experimental data on the viscosity of basalt and kimberlite melts (Persikov et al., 2018), confirms this hypothesis (Table 2).
A concentration dependence of the diffusion-controlled mobility of components has been determined based on numerical computations using Eqs. (3), (4), and (5). According to these results, the diffusion mobility of all components practically does not depend (within the error of ±20 rel. %) on the concentrations of these components in the melts of the diffusion pairs (Figs. 5, 6). For example, Fig. 6 shows the concentration dependence of the diffusion coefficients of CaO and \({\text{CO}}_{3}^{{2 - }}\) at interaction between kimberlite and basalt melts, with a calculated error of 1 rel. %, which is much lower than the real experimental errors (~20 rel. %). Nevertheless, even in this situation, the two types of the concentration dependences [D(i) is a constant and D(i) is an exponential function] are practically indiscernible from each other.

An example of approximating the diffusion profiles of all components at interaction between kimberlite and basalt melts (the dashed line is practically indiscernible at D(i) = constant, and the continuous lines at D(i) are a weak exponential dependence).

Example of comparison of two concentration dependences of CaO and \(\text{CO}_{3}^{2-}\) diffusion at interaction between kimberlite and basalt melts (run 2146, dashed lines are parallel to the abscissa axis D(i) = constant, the dashed line is the exponential dependence).
Results of particular interest were recently acquired on the diffusion mobility of the carbonate ion \({\text{CO}}_{3}^{{2 - }}\) at interaction of carbonate-bearing melts with natural and model basalt melts. Raman spectroscopic data indicate that absolutely no molecular CO2 is formed when the diffusion pair interacts (Fig. 2). It has been established that the diffusion mobility of the carbonate anion \({\text{CO}}_{3}^{{2 - }}\) under these conditions is equal (within the error of ~20 rel. %) to the diffusion rate of CaO (Tables 2, 3). This proves that calcite molecules diffuse from the carbonate-bearing melt (model kimberlite) into basalt melt (both model and natural) by the mechanism of end-member diffusion. This diffusion mechanism of Na carbonate spiked with a radioactive tracer (\({\text{Na}}_{2}^{{14}}\)CO3) in model basalt melt has been earlier obtained in (Watson et al., 1982) under a pressure of 500 MPa. These authors have derived a value for the sodium carbonate diffusion coefficient approximately one order of magnitude greater than that according to our data on the diffusion of calcium carbonate (CaCO3) into basalt melts (Tables 2, 3).
CONCLUSIONS
We have obtained new experimental data on the chemical interdiffusion of major components (SiO2, Na2O, CaO, MgO, Al2O3, and FeO) and the carbonate ion \({\text{CO}}_{3}^{{2 - }}\) at immediate interaction between melts of carbonate-bearing model kimberlite and basalt at a pressure of 100 MPa and temperature of 1300oC. The diffusion of calcium carbonate from model kimberlite melt into model and natural basalt melts was proved to proceed according to an end-member mechanism. The weak exponential concentration dependence of the chemical diffusion of all other of the studied components of the interacting melts is close to D(i) = constant, which is typical of both the model system and the system of model kimberlite and natural basalt.
REFERENCES
Bol’shoe treshchinnoe Tolbachinskoe izverzhenie 1975–1976 gg., Kamchatka (Great Tolbachik Fissure Eruptions. Kamchatka), Moscow: Nauka, 1984.
Bowen, N.L., Diffusion in silicate melts, J. Geol., 1921, vol. 29, pp. 295–317.
Crank, J., The Mathematics of Diffusion, London: Oxford University Press, 1975.
Dalton, J.A. and Presnall, D.C., The continuum of primary carbonatitic–kimberlite melt compositions in equilibrium with lherzolite: data from the system CaO–MgO–Al2O3–SiO2–CO2 at 6 GPa, J. Petrol., 1998, vol. 39, pp. 1953–1964.
Dasgupta, R. and Hirschmann, M.M., Melting in the earth’s deep upper mantle caused by carbon dioxide, Nature, 2006, vol. 440, pp. 659–662.
Dingwell, D.B., Copurtial, P., Giordano, D., and Nichols, A.R.L., Viscosity of peridotite liquid, Earth Planet. Sci. Lett., 2004, vol. 226, pp. 127–138.
Kamenetsky, V.S., Kamenetsky, M.B., Weiss, Y., et al., How unique is the Udachnaya-East kimberlite? Comparison with kimberlites from the Slave Craton (Canada) and SW Greenland, Lithos, 2009, vol. 112S, pp. 334–346.
Kavanagh, J.L. and Sparks, R.S.J., Temperature changes in ascending kimberlite magma, Earth Planet. Sci. Lett., 2009, vol. 286, pp. 404–413.
Kopylova, M.G., Matveev, S., and Raudseep, M., Searching for parental kimberlite melt, Geochim. Cosmochim. Acta, 2007, vol. 71, pp. 3616–3629.
Kress, V.C. and Ghiorso, M.S., Multicomponent diffusion in basaltic melts, Geochim. Cosmochim. Acta, 1995, vol. 59, pp. 313–324.
Liang, Y., Multicomponent diffusion in molten silicates: theory, experiments, and geological applications, Rev. Mineral. Geochem., 2010, vol. 72, pp. 409–446.
Le Maitre, R.W., The chemical variability of some common igneous rocks, J. Petrol., 1976, vol. 117, no. 4, pp. 589–637.
Michell, R.H., Petrology of hypabyssal kimberlites: relevance to primary magma compositions, J. Volcanol. Geotherm. Res., 2008, vol. 174, pp. 1–8.
Persikov, E.S., Viscosity of model and magmatic melts under P-T parameters, of the Earth’s crust and upper mantle, Russ. Geol. Geophys., 1998, vol. 39, no. 12, pp. 1780–1792.
Persikov, E.S. and Bukhtiyarov, P.G., Viscosity of magmatic melts: improved structural–chemical model, Chem. Geol., 2020, vol. 556. https://doi.org/10.1016/j.chemgeo.2020.119820
Persikov, E.S., Bukhtiyarov, P.G., and Nekrasov, A.N., Osobennosti vstrechnoi khimicheskoi diffuzii petrogennykh komponentov (SiO2, Al2O3, Na2O, CaO, MgO) in model and naturalmelts of the andesite–basalti system at high pressures in relation with their viscosity, XII Mezhdunarodnaya konferentsiya: Fiziko-khimicheskie i petrofizicheskie issledovaniya v naukakh o Zemle (12th International Conference on Physicochemical and Petrophysical Studies in the Earth’s Sciences), Moscow: IGEM RAN, 2011, pp. 236–239.
Persikov, E.S., Bukhtiyarov, P.G., and Sokol, A.G., Viscosity of haplokimberlite and basaltic melts at high pressures, Chem. Geol., 2018, vol. 497, pp. 54–63.
Price, S.E., Russell, J.K., and Kopylova, M.G., Primitive magma from the Jericho Pipe, N.W.T., Canada: constraints on primary kimberlite melt chemistry, J. Petrol., 2000, vol. 47, pp. 789–808.
Sharygin, I.S., Litasov, K.D., Shatsky, A.F., et al., Melting of kimberlite of the Udachnaya-East Pipe: experimental study at 3–6.5 GPa and 900–1500C, Dokl. Earth Sci., 2013, vol. 448, no. 2, pp. 200–205.
Sparks, R.S.J., Baker, L., Brown, R.J., et al., Dynamical constraints of kimberlite volcanism, J. Volcanol. Geotherm. Res., 2006, vol. 155, pp. 18–48.
Sparks, R.S.J., Brooker, R.A., Field, M., et al., The nature of erupting kimberlite melts, Lithos, 2009, vol. 112, pp. 429–438.
Watson, E.B., Diffusion in volatile-bearing magmas, Volatiles in Magmas, Carrol, M.R. and Holloway, J.R. Eds., Rev. Mineral. Geochem., 1994, vol. 30, pp. 371–411.
Watson, E.B. and Baker, D.R., Chemical diffusion in magma: an overview of experimental results and geochemical applications, Physical Chemistry in Magma, Perchuk, L.L. and Kushiro, I., Eds., New York: Springer, 1991.
Watson, E.B., Sneeringer, M.A., and Ross, A., Diffusion of dissolved carbonate in magmas: experimental results and applications, Earth Planet. Sci. Lett., 1982, vol. 61, pp. 346–358.
Wyllie, P.J., The origin of kimberlite, J. Geophys. Res., 1980, vol. 85, pp. 6902–6910.
Yoder, H.S., Contemporaneous basaltic and rhyolitic magmas, Am. Mineral., 1973, vol. 5, pp. 153–171.
ACKNOWLEDGMENTS
The authors thank G.V. Bondarenko (Korzhinskii Institute of Experimental Mineralogy, Russian Academy of Sciences) for assistance with using Raman spectroscopy in analyzing the dissolution forms of CO2 in melts. L.Ya. Aranovich (Institute of Geology of Ore Deposits, Petrography, Mineralogy, and Geochemistry, Russian Academy of Sciences) is thanked for valuable comments that undoubtedly led us to improve the content of the manuscript.
Funding
This study was carried out under government-financed research project FMUF-2022-0004 for the Korzhinskii Institute of Experimental Mineralogy, Russian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by E. Kurdyukov
Rights and permissions
About this article

Cite this article
Persikov, E.S., Bukhtiyarov, P.G. & Nekrasov, A.N. Experimental Study of the Multicomponent Chemical Diffusion of Major Components (SiO2, Al2O3, Na2O, CaO, MgO, and FeO) and the \(\text{CO}_{3}^{2-}\) Anion at Interaction between Basalt and Kimberlite Melts under a Moderate Pressure. Petrology 30, 325–335 (2022). https://doi.org/10.1134/S0869591122020060
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0869591122020060