
Abstract
Lignocellulosic materials (LCMs) obtained after an ozone treatment of pine wood are studied via diffuse reflectance ultraviolet (DRUV) spectroscopy, Raman spectroscopy, and fluorescence spectroscopy. The Raman spectra show that ozonation degrades lignin and hemicelluloses and lowers the content of amorphous cellulose. The Raman spectra of ozonized LCMs are deconvoluted to individual components for the first time. Based on results from analyzing the Raman and DRUV spectra, it is concluded that the aromatic structures of biomass with conjugated –С=С– and >С=О bonds simply degrade, while delignification with ozone is accompanied by a multiple increase in the fluorescence of the LCMs. It is shown that the luminescence spectral characteristics of LCMs are governed by the absorption of ozone, allowing us to identify the ranges of ozone consumption corresponding to the dominant degradation of lignin and polysaccharides in the biomaterial.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Special attention is now being given to developing innovative technologies that meet the environmental requirements. Ozone is considered an environmentally friendly reagent, since the only product of ozone decomposition is oxygen, and reactions in which it participates occur at ambient temperature and atmospheric pressure. The progress made in recent years in the field of ozone synthesis has considerably reduced the cost of ozone, naturally raising the interest in O3 and expanding its fields of application [1]. Ozone is a strong oxidizing agent that actively interacts with aromatic and unsaturated compounds, so it is used to treat pulp and paper industry wastewater of phenolic compounds (lignin (LG) derivatives). Other industries in which it is used can be found in [1, 2].
A great deal of experimental data on the interaction of plant biomass and ozone has been accumulated worldwide. The prospects for using ozone to remove LG from plant raw materials and the economic feasibility of ozonation for the subsequent production of cellulose (CL), monosaccharides, and bioethanol were demonstrated in [3, 4]. The advantages of ozone as a delignifying agent with respect to biomass include its selectivity with respect to LG, while CL and hemicelluloses (HCs) exhibit relatively high resistance to ozone [4–11].
Studies of the efficiency of delignifying biomass under different conditions of ozonation have allowed us to determine optimum conditions for using this process [5–8]. It is known that for pine wood, ozonation is most effective at a 60–65% content of moisture (MC) in the sample, at which (according to results from determining the content of LG in ozonized samples) the degree of delignification can be as high as 40% [7, 9]. High-performance liquid chromatography was used to show that the products of ozonation are aliphatic acids (e.g., formic, oxalic, and glyoxalic) that are subsequently oxidized during prolonged ozone treatment [7, 9].
Lignocellulosic materials (LCMs) from ozonized pine wood have been studied by a number of physicochemical means (infrared (IR) spectroscopy, Raman spectroscopy, diffuse reflectance ultraviolet (DRUV) spectroscopy, X-ray diffraction, thermal analysis) [6–11]. It was shown in [7] that delignification with ozone is accompanied by the degradation of HCs and the amorphous part of CL. It has been reported that the degree of polymerization of CL of LCMs falls as the period of ozonation grows.
The authors of [11–13] studied the physicochemical properties of CL-containing materials via fluorescence (FL) spectroscopy. It was first shown in [11] that the delignification of hardwood with ozone creates a material characterized by strong FL in the visible region. It was reported that FL spectroscopy can be considered one of the most sensitive methods of controlling the ozonolytic delignification of wood.
This work is a continuation of a cycle of studies of the physicochemical properties of LCMs from ozonized biomass. It focuses on studying the transformations of pine wood, which is one of the most abundant species of wood on the planet. Softwood differs from hardwood by its cellular texture and HC structure. It is characterized by strong CL polymerization and a higher content of CL than that in hardwood. The structure of softwood LG also differs considerably from the LG structure of hardwood species [14, 15]. In terms of actual practice, interest in studying the transformation of softwood is also due to the enormous amount of industrial waste that can be used as a feedstock for further chemical processing [15].
The aim of this work was to study the transformation of pine wood at different stages of ozonation via Raman, DRUV, and FL spectroscopy; and to explore the possibilities of these nondestructive methods in terms of their information content in analyzing the degradation of LG and other components of biomass.
EXPERIMENTAL
Pine sawdust (Pinus sylvestris) with particle sizes of 0.315–0.63 mm served as our test material. The MC in each sample was 60–65%, relative to the weight of oven-dried wood (o.d.w.), and MC = (\({{m}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}\)/mo.d.w.) × 100%. To prepare the samples, 0.15–0.20 mL of distilled water was added to a weighed portion (0.30–0.35 g) of air-dried wood (MC = 8%/go.d.w., determined as in [16]). The resulting mixture was kept in a closed container at t = 20°C for 5–7 days to allow the wood to swell. The final MC was controlled gravimetrically.
A set of tests was performed with different durations of the ozone treatment of pine wood samples having weights of 0.45–0.50 g. Ozonation was conducted in a flow unit equipped by temperature controller (25°C) in fixed bed reactor. The unit consisted of an ozone generator (Medozon 03/05), a photometer for determining the concentration of ozone (Medozon 254/3), and a catalytic cartridge for the decomposition of unreacted ozone described in [1]. An ozone–oxygen mixture with a 60 ± 2 mg/L concentration of ozone passed through the reactor at a flow rate of 1 × 10−3 L/s. The linear flow velocity was 1 cm/s. Due to the constancy of the sample volume, the tests were performed at a constant residence time (0.8 ± 0.05 s) in the reaction zone.
The amount of absorbed ozone Qr(t) at time t was calculated from the kinetic curves of the ozone concentration dependence according to the equation
where U is the flow rate of the gas mixture (L/s); \(C_{t}^{*}\) and \(~{{C}_{t}}\) are the current ozone concentrations at the inlet and outlet of the reactor with the test sample, respectively (mol/L); and m is the weight of o.d.w. Qr(t) (mmol/g) is calculated per gram of o.d.w. The error in determination Qr(t) was 10%.
The ozonized LCM samples were washed with distilled water to remove water-soluble products. Original air-dried sample (1) and ozonized LCMs with different durations of ozonation (О1–О6) were studied via Raman, DRUV, and FL spectroscopy.
Raman spectra were recorded on a Bruker Equinox 55/S instrument equipped with an FRA 106/S attachment. The wavelength of excitation was 1064 nm, the laser power was 850 mW, and the size of the laser spot was 0.1 mm. The spectrum was recorded with accumulation over 1024 scans at a resolution of 4 cm−1 in the region of 100–3600 cm−1. Spectra were recorded for five randomly selected points of the sample. The experimental Raman spectra were normalized to the 1096 cm−1 band according to [17], and the average intensities of certain bands in the Raman spectrum were determined. The error of determining the intensity at the maximum was 5%.
The spectrum of the original sample into individual components was deconvoluted according to Levenberg and Marquardtusing the OPUS 6.0 software (Bruker, Germany). The spectra were deconvoluted into five individual components. The choice of the number of model bands and the position of their maxima was based on literature data on the Raman spectra of LCMs [17–20]. Variations in the shape of component lines showed that the best option was 90%Gauss + 10%Lorentz for components 1–4 and 75%Gauss + 25%Lorentz for component 5. The model spectrum of the original sample was used as the first approximation of the ozonized samples. At the first stage, the intensities of the model bands were optimized while the line width and position of the maximum were fixed. At the second stage, the line width was optimized while the intensities were fixed. At the third stage, the positions of the maxima and the width of individual bands were refined. The root-mean-square error of the model spectrum’s deviation from the experimental spectrum was 10%.
The DRUV spectra of the wood samples (100 mg) were recorded on a Specord M-40 instrument (Carl Zeiss Jena, Germany) with an integrating sphere in a range of 220–820 nm. A sample of BaSO4 was used as a reference in recording the DRUV spectra. (The reflection of this sample was assumed to be 100%). The spectra were processed using the Kubelka–Munk function (F(R)), which is the ratio of the coefficient of absorption to the medium’s coefficient of scattering: F(R) = k/s = (1 – R)2/2R. This allows us to determine the absorption of an infinitely thick layer of a sample at a given wavelength. The error of F(R) determination is 10%.
Excitation and emission spectra were recorded at a temperature of 25 ± 0.2°C on a Fluorolog-3 FL spectrometer (Horiba, France) equipped with double monochromators in the channels of excitation and emission, and a 450-W high-pressure xenon lamp as the source of excitation. The signal was recorded using an FL-1073 detector based on an R928P photomultiplier operating in the mode of single photon counting at room temperature. Before recording the spectra, the air-dried samples were pressed into 150-mg pellets. Each sample was placed in the holder at an angle of 35° with respect to the beam of excitation to eliminate the effect of reflected light. The spectral width of the slits of the excitation and emission monochromators was 2 nm. The FL spectra of the wood samples were recorded for λexc = 360 and 400 nm. The spectra were integrated using the Opus 6.0 software (Bruker, Germany).
RESULTS AND DISCUSSION
Ozonation
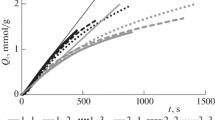
Figure 1 shows kinetic curves of ozone absorption corresponding to different durations of treatment. The curves fit into the single kinetic dependence characteristic of ozone absorption under the given conditions of treatment accepted in [7, 8] as optimal for the delignification of pine wood. Figure 1 shows the values of specific ozone absorption (Qr, mmol/g) that correspond to the end of ozonation for the labeled samples. It is evident from Fig. 1 that the rate of absorption fell as the period of ozonation grew. Of the wood components, LG reacted with ozone most actively, so it is reasonable to expect delignification to be most effective for the initial portion of the kinetic curve and samples O1 and O2 (Qr = 0.8 and 1.0 mmol/g, respectively), which were prepared under conditions of the highest rate of ozone absorption (determined from the slope of the curve). The rate of ozone absorption fell at higher values of Qr. According to [7, 9], reaction products on the surface of the wood participate in reactions with ozone at this stage of ozonation.

Kinetic curves of ozone absorption by pine wood.
Raman Spectra
Figure 2 shows Raman spectra of the studied samples in the regions of 800–1800 cm−1 (Fig. 2a) and the stretching vibrations of C–H bonds (Fig. 2b). The spectra correspond to the Raman spectra of softwood known from the literature [17–20]. The bands are at 1374 cm−1 (δС–Н in R3CH in CL and glucomannan) [18], 1330 cm−1 (bending vibrations of Сar–OH or Сar–O–CH3), 1267 cm−1 (Сar–O–CH3 and Сar–O of the guaiacyl ring with С=О groups) [18], 1126 cm−1 (stretching vibrations of С–С and С–О in CL and glucomannan), 1096 cm−1 (νС–С and νС–О in HC and CL) [19, 20], and 899 cm–1 (C–O–C stretching vibrations of the C1 atom and four surrounding atoms in β-glycosidic structures [21]). The spectrum of original sample (1) displays intense bands of the C–C skeletal vibrations of aromatic rings (1600 cm−1) and bands at 1632 and 1656 cm−1 (C=C vibrations in coniferyl aldehyde and coniferyl alcohol, respectively) [20].

Raman spectra of LCMs from ozonized pine wood in the region of (a) 100–1800 and (b) 2600–3200 cm−1; 1 (blue line), О1 (grey dashed line), О2 (dotted line), О3 (fine black line), О4 (black dashed line), О5 (bold black line), and О6 (bold grey line). Parameters (I/I0) at (c) 1600 cm−1 and (d) 2899, 2940, and 899 cm−1; and Klason LG content [9] in the LCM as a function of specific ozone absorption.
Figure 2b shows Raman spectra in the region of C‒H stretching vibrations. The band at 3071 cm−1 was attributed to the νС–Н of the aromatic ring; the band at 2899 cm−1 corresponds to the C–H stretching vibrations of CL; and the band at 2940 cm−1 was attributed to C–H stretching vibrations in the CH3 and OCH3 groups of LG and HC [18, 20]. Compared to the Raman spectrum of original sample (1), an increase in Qr in the spectra of the ozonized samples reduced the intensity of the bands at 1600, 1632, 1660, 3071, and 1267 cm−1 (attributed to LG) and the intensity of the band at 1126 cm−1 (attributed to glucomannan). The intensity of the band at 2940 cm−1, which corresponds to the C–H stretching vibrations in the methoxy groups of LG and HC, was also reduced (Fig. 2).
Figure 2c presents the (I/I 0)1600 values (intensity I at the maximum of 1600 cm−1 of the ozonized LCM, normalized to intensity I 0 in the spectrum of the original sample) as a function of specific ozone consumption (curve 1). The figure also shows a similar dependence of the content of Klason LG (curve 2) recorded in [7]. It is evident from Fig. 2c that (I/I0)1600 fell as Qr rose, testifying to the degradation of the aromatics. The drop is most pronounced when Qr ≤ 1.0 mmol/g. (I/I 0)1600 is almost constant at higher values of Qr it is 40–42% of the initial value. In addition, curve 2 suggests that LG degrades in ozonized LCMs: the content of LG is 28.5% in the original sample and 14.8% at Qr = 1.0 mmol/g. The degradation of aromatics is most evident from the Raman spectra. This meets our expectations, since Raman spectroscopy provides information directly from the part of a sample’s surface that is delignified upon ozone treatment, in contrast to the content of LG per gram of o.d.w., which is the average content of LG in the bulk of the sample.
Figure 2d shows (I/I 0)2899 and (I/I 0)2940 values as a function of Qr. It is clear that the intensities of the bands at 2899 and 2940 cm−1 changed symbatically upon an increase in Qr, since the bands overlap. The contour of the spectrum in Fig. 1b was preserved for the ozonized samples.
The band at 1096 cm−1 (С–С and С–О stretching vibrations of CL) was attributed to crystalline CL [17, 22]. The intensity of the band at 899 cm−1, attributed to the asymmetric vibrations of the glucopyranose ring of CL, characterizes the relative content of amorphous and crystalline CL [21, 22]. It is evident from Fig. 2d that (I/I 0)899 falls at a 1.4–2.4 mmol/g rate of ozone consumption, i.e., in the range of ozone consumption where the degradation of aromatics by ozone is least effective (Fig. 2c). The drop in (I/I 0)899 testifies to the degradation of the amorphous part of the CL during ozonation. It should be noted that the same conclusion was reached in [7] from X-ray diffraction data in studying LCMs from ozonized pine wood.
Deconvolution of the Raman Spectra
The Raman spectra were deconvoluted into five individual components in the region of 1500–1700 cm−1: components 1 and 2 with maxima at 1580 and 1599 cm−1 (skeletal vibrations of the aromatic ring); component 3 at 1620 cm−1 (stretching vibrations of C=O conjugated with the aromatic ring); and components 4 and 5 at 1632–1636 and 1656 cm−1, which correspond to the C=C stretching vibrations in coniferyl aldehyde and coniferyl alcohol, respectively. The choice of bands was based on the respective structures in softwood LG [17, 20]. The position of the maxima of decomposition components 1–3 and 5 remained constant for the different samples, while it varied from 1632 to 1636 cm−1 for component 4.
As an example, Fig. 3a shows the Raman spectra of samples 1, O1, O4, and O6 in the region of 1500–1700 cm−1 and the decomposition of the spectra into components. It is evident that modelling allowed us to obtain satisfactory agreement between the experimental (blue) and model (red) spectra.

(a) Deconvolution of Raman spectra into components (samples 1, O1, O3, O6); (b) integrated intensity of the Raman spectrum (A/A0) at 1500–1700 cm−1 and relative intensities (Ac/\(A_{{\text{c}}}^{0}\)) of the decomposition components with maxima at (1) 1580, (2) 1599, (3) 1620, (4) 1632–1636, and (5) 1656 cm−1 as a function of specific ozone absorption; and (c) contribution of the decomposition components (Aс1/A, Aс2/A, Ас3/A, Ас4/A, Ас5/A, and Ас3,4,5/A) to the integrated intensity of the spectrum at the different specific ozone absorption values. Dependences of (1) Ас2/A and (2) Ас3,4,5/A on specific ozone absorption (Qr).

(Contd.)
Figure 3b shows the dependence of integrated intensity of spectrum A (defined as the area under the spectrum contour) normalized to the integrated intensity of spectrum A0 of the original sample (curve 1). The value of A/A0 fell by a factor of ~10 upon an increase in the specific ozone consumption, due to degradation of the aromatic systems. Figure 3b also shows the relative intensities (Ac/\(A_{{\text{c}}}^{0}\)) for decomposition components 1–5 as a function of specific ozone absorption. It is evident from Fig. 3b that the intensity of each decomposition component fell as Qr rose. The drop was most pronounced for component 4 with a maximum at 1632–1636 cm−1. It should be noted that the greatest drop in the intensity of the decomposition components corresponds to the ≤1 mmol/g range of ozone consumption, which is consistent with the data in Figs. 1 and 2c.
Figure 3c shows the contributions of the decomposition components to the integrated spectrum for each sample. The contribution from component 1 (Ас1/A0)(1580 cm−1) and Ас2/A was 10 and 40%, respectively, for the original sample. Around fifty percent of this contribution comes from Ас3,4,5/A. This characteristic of the spectrum is the total contribution from components with maxima at 1620, 1632–1636, and 1656 cm−1 of the aromatic structures conjugated with double bonds (–С=С–, >С=О). The dependences of Ас2/A and Ас3,4,5/A on the specific absorption of ozone (Qr) are shown by curves 1 and 2.
The contribution from the components changed along with the increase in ozone consumption. The contribution from the component with a maximum at 1599 cm−1 (Ас2/A) to the integrated spectrum grew gradually as Qr rose. It grew by 9% for the O6 sample, as is shown by curve 1, where the fraction of component 1 is virtually constant (9–10%). The total contribution from components 3–5 to the integrated intensity was reduced slightly: at Qr = 2.4 mmol/g (sample O6), the value of the Ас3,4.5/A components with maxima at 1620, 1632–1636, and 1656 cm−1 fell by 9%, relative to that of the original sample (Fig. 3c, curve 2).
These tendencies suggest that the degradation of α-carbonyl aromatic structures and aromatics participating in conjugation with C=C bonds predominate during delignification with ozone. This conclusion is based on the fraction of these structures in the Raman spectrum of the ozonized LCMs falling as the specific consumption of ozone rises. According to results from deconvolution, ozone treatment of the LCMs did not produce new aromatic structures.
Data from the Raman spectra show that HCs were partly degraded along with the aromatic structures of LG in the ozonized samples, since a drop in the fraction of amorphous CL is observed.
Diffuse Reflectance UV Spectra
Figure 4a shows DRUV spectra for the original sample and some of the ozonized samples. The inset shows the spectra in Kubelka–Munk units (F(R)). The studied LCMs absorbed in a wide range of the spectrum. They were characterized by a maximum at 280 nm, which is characteristic of aromatic compounds with oxygen-containing substituents (primarily phenols). The spectrum is a set of numerous overlapping absorption bands attributed to different structures [23–25]. Nonconjugated phenolic structures (syringyl, guaiacyl) absorb at 250 and 295–300 nm. Absorption in the region of 300–400 nm is associated with more extensive conjugated systems, primarily those that contain a guaiacyl rings, carbonyl oxygen, and a double bond in the side chain [24, 25]. Typical examples of these structures are ferulic acid (λmax = 321 nm) and coniferyl aldehyde (λmax = 342 nm) [26]. Even longer-wavelength absorption is characteristic of the o-quinone structures and stable phenoxyl radicals present in LG in trace amounts; the absorption of quinone methides formed by stilbene structures can extend to 500 nm and beyond, into the long-wavelength region of the spectrum [24, 26, 27].

(a) Diffuse reflectance UV spectra of samples 1, O1, O3, and O6; the inset shows the DRUV spectra in Kubelka–Munk units; dependences of (b) F(R) and (c) F(R)/F(R)0 on Qr at λ = 280, 360, and 400 nm: 1 (blue line), О1 (grey dashed line), О2 (dotted line), О3 (fine black line), О4 (black dashed line), О5 (bold black line), and О6 (bold grey line).
The ozone treatment of wood reduces absorption throughout the spectrum (Fig. 4a), as is clearly seen in the inset of Fig. 4a. Upon an increase in Qr, the F(R) values for wavelengths of 280, 360, and 400 nm fall and exhibit a plateau (Fig. 4b). This effect is most pronounced for F(R)360.
Comparison of the F(R) values normalized to that of F(R)0 for the original sample allows us to compare the efficiency of ozone treatment with respect to aromatic structures that absorb in the different spectral regions. It is clear from Fig. 4c that F(R)360/F(R)\(_{{360}}^{0}\)~ 45% of the initial value at, e.g., Qr = 2.0 mmol/g. The curves of the dependence of F(R)/F(R)0 on Qr are similar for the absorption bands at 280 and 400 nm. At Qr = 2.0–2.4 mmol/g, F(R)280/F(R)\(_{{280}}^{0}\) and F(R)400/F(R)\(_{{400}}^{0}\) are 65–70 and 50–55%, respectively, relative to the initial value. This means chromophores that absorb in these regions of the UV spectrum are less effectively degraded by ozone. Using DRUV spectra, it was shown that chromophores which absorb near 360 nm (structures with an α,β-double bond and a conjugated carbonyl group, stilbene structures) are predominantly degraded by ozone. This is consistent with the familiar selectivity of the action of ozone on double bonds and data on the deconvolution of Raman spectra, which show that ozonation results mainly in the degradation of aromatic structures that participate in conjugation with C=C and C=O bonds.
In studying DRUV spectra of ozonized aspen wood, the authors of [11] also observed the predominant degradation of chromophores that absorb at λabs = 360 nm, while structures that absorb at 280 and 400 nm account for 30–35 and 40% of the initial value, respectively. The conventional orders of the drop in the effect of ozone treatment are 360 → 280 → 400 nm for aspen wood and 360 → 400 → 280 nm for pine wood. It is therefore clear that nonconjugated phenolic structures with λabs = 280 nm in pine wood display more resistance to ozone than the nonconjugated phenolic structures in aspen wood. This difference can be attributed to the different structures of softwood (guaiacyl) and hardwood LG (syringyl–guaiacyl).
Fluorescence Spectra
The FL excitation spectra of a set of pine wood samples (Fig. 5a) are characterized by a maximum at 400 nm. They are identical for all samples except for their intensity. Shoulders at 430, 460, and 520 nm that indicate different types of fluorophores are observed for all samples. The obvious difference between the spectra in Fig. 5a and the absorption spectra in Fig. 4a with a maximum near 280 nm can be attributed to the inhomogeneity of the chemical structure of LG and the relatively low FL quantum yield of nonconjugated phenolic structures that absorb in the given spectral region. Another reason for the mismatch between the positions of the maxima in the absorption and FL excitation spectra could be the so-called “inner filter effect” associated with the suppression of FL due to the high absorbance of the sample, which is strongest in the range of 260–300 nm. The most intense FL is characteristic of biphenyl groups, certain phenylcoumarane and stilbene structures [28], and quinone methide groups. The last groups absorb near 500 nm and are apparently responsible for the small shoulders in the region of 500–550 nm seen in the excitation spectra shown in Fig. 4 [27].

(a) Fluorescence excitation spectra of LCM samples, (b) FL spectra of LCM samples from pine wood (λexc = 360 and 400 nm), and dependences of (c) FL intensity Inorm and (d) relative FL intensity (Inorm/\(I_{{{\text{norm}}}}^{0}\)) on Qr; λexc = (1) 360 and (2) 400 nm; data of [11]: λexc = (3) 360 and (4) 400 nm; 1 (blue line), О1 (grey dashed line), О2 (dotted line), О3 (fine black line), О4 (black dashed line), О5 (bold black line), and О6 (bold grey line).
The FL spectra of the LCM samples shown in Fig. 5b are characterized by an emission maximum at 452–453 (λexc = 360 nm) and 470–475 nm (λexc = 400 nm). The dependence of the position of the FL band of wood on the quantum energy of radiation incident on the surface of the wood is characteristic of plant substrates. This effect was observed in studying the structure of softwood and hardwood in [29] and the FL spectra of LCMs from ozonized aspen wood in [11].
The FL properties of LCMs thus differ from those of LG preparations, which behave as a single fluorophore where the electronic energy of excitation is transferred according to the Förster mechanism to energy acceptor structures with lower singlet levels. The radiative transition from these levels competes with the intersystem crossing to triplet energy levels [25–31]. This mechanism of energy transfer between fluorophores is partly inherent in the studied LCM as well, which is supported by a fairly large difference between the energies of FL excitation and emission. We may also assume the FL maximum at ~450 nm (λexc = 360 nm) can be attributed to structures activated by the secondary absorption of radiation emitted by fluorophores with λexc = 360 nm.
It is evident from Fig. 5b that the FL spectra of the ozonized samples are characterized by greater intensity that is not accompanied by a change in the position of the maximum or the contour of the spectrum. This finding is consistent with the data from the Raman spectra, showing that new aromatic structures do not form upon ozonation of the LCM.
The intensity of FL depends on that of the absorption of exciting radiation, so the normalized values (Inorm) as a function of specific ozone absorption are discussed below (Inorm is the intensity of FL normalized to F(R) at λexc = 360 nm (Inorm = I/(F(R)360) and λexc = 400 nm (Inorm = I/(F(R)400)). The dependences of Inorm on Qr are shown in Fig. 5c. A comparison of curves 1 and 2 shows that the Inorm values for λexc = 400 nm are higher throughout the region of Qr. This is consistent with the spectra of excitation, which display a maximum at λabs = 400 nm.
The Inorm/\(I_{{{\text{norm}}}}^{0}\) values (where \(I_{{{\text{norm}}}}^{0}\) is the intensity of the original sample’s FL) allows us to assess the effect ozone treatment has on the FL of the LCM. Figure 5d shows that compared to the original sample, Inorm/\(I_{{{\text{norm}}}}^{0}\) grows by 3–4 and 6–7 times at λexc = 400 and 360 nm, respectively, while approaching saturation upon an increase in Qr. The stronger increase in (Inorm/\(I_{{{\text{norm}}}}^{0}\))360 meets expectations and is attributed to the data of UV spectra, which show the LCM structures that absorb at 360 nm suffered the greatest degradation by ozone.
It is evident from Figs. 5c and 5d that the FL grows substantially upon ozone consumption of ≤1 mmol/g. This range of ozone consumption corresponds to the linear section of the curve in Fig. 1. The rate of ozone absorption is greatest in this part of the curve because ozone reacts mostly with LG, and this region of Qr corresponds to the greatest drop in the content of LG in the LCM (Fig. 2c). The FL is virtually constant in the range of Qr = 1.4–2.4 mmol/g (Figs. 5c, 5d), where the content of aromatic structures does not change (Fig. 2c) and polysaccharides are partially degraded (Fig. 2d). The rate of ozone absorption also falls in this part of the curve in Fig. 1. The above correlations between the dependence of FL intensity and the kinetic curve of ozone absorption suggest that the luminescence properties of LCMs can be controlled by varying the conditions of ozone treatment for the LCMs.
A multiple increase in the FL intensity of the LCM sample after ozone treatment was observed when studying ozonized aspen wood in [11], as is shown in Fig. 5d (curves 3, 4). Since FL is attributed to the presence of LG, which is the component most effectively degraded by ozone, and the formation of new aromatic structures resistant to the action of O3 is not observed, the change in the properties of the LG-containing material formed after ozone treatment was proposed as a factor responsible for the enhancement of FL.
When discussing data on the ozonation of pine wood in this context, it should be noted that along with intra- and intermolecular hydrogen bonds, the ozonation-induced degradation of LG and LG–carbohydrate covalent bonds, which was observed in [7, 9, 10], hinders the nonradiative mechanisms of energy loss in the form of vibrations and contributes to enhancement of the emission of electromagnetic radiation. The FL of a particular fluorophore is determined by the ratio of the probabilities of the radiative and nonradiative dissipation of the energy of electronic excitation [29, 30], so the environment of the fluorophore is an important factor. IR spectroscopy in [7, 9] showed that the delignification of wood with ozone is accompanied by the formation of numerous aliphatic carbonyl and carboxyl compounds. The resulting drop in the pH of the medium can play an important role in changing the intensity of emissions by shifting the protolytic equilibria toward the formation of nonionized species of the phenolic structures of LG.
In addition, the increase in the intensity of the FL of ozonized LCMs can be associated with a drop in the abovementioned inner filter effect upon the removal of LG capable of actively absorbing emitted radiation and quenching FL, in analogy with concentrated solutions of fluorophores. This version is supported by the data in [12, 13], where an increase in the FL intensity was observed upon a reduction in the content of LG in the LCM.
The drop in the concentration of structures with a low yield of FL after their degradation by ozone is apparently an important factor as well, which can contribute to an increase in the intensity of FL. These structures can be C=O groups conjugated with an aromatic ring, since the authors of [13, 30] reported that a reduced content of aryl–carbonyl groups in LG preparations enhances their FL. This argument is supported by data on the deconvolution of Raman spectra that revealed their degradation by ozone. It is obvious that reliable determination of the role of the above mechanisms of changes in FL in the effect observed upon ozone treatment requires more study with modern physical and physicochemical means to explore the structure of LGs in LCMs and is beyond the scope of this study.
Figure 6 shows the dependence of FL (Inorm/\(I_{{{\text{norm}}}}^{0}\)) on intensity (I/I0)1600 of the 1600 cm–1 band in the Raman spectrum of the LCM. Since (I/I0)1600 characterizes the content of LG, the data in Fig. 6 characterize the dependence of FL on the content of LG in the LCM. (I/I0)1600 = 1 and (Inorm/\(I_{{{\text{norm}}}}^{0}\)) = 1 for the original sample. The intensity of FL grows by several times for the ozonized LCMs. The increase at λexc = 360 nm upon a drop in (I/I0)1600 is double the one at λexc = 400 nm (curves 1, 2).

Dependences of the relative FL intensity (Inorm/\(I_{{{\text{norm}}}}^{0}\)) of the LCM on (I/I0)1600 of the band at 1600 cm−1 in the Raman spectra; λexc = (1) 360 and (2) 400 nm; curves 3 and 4 are data of [11], λexc = (3) 360 and (4) 400 nm.
Figure 6 also suggests that in the same range of (I/I0)1600 values, the increase in the FL of the LCM from ozonized pine wood half that of LCMs from ozonized aspen wood (curves 3, 4 from [11]). From a formal viewpoint, these differences can be simply attributed to the abovementioned changes in the UV spectra of LCMs from ozonized aspen and pine wood.
According to [9], the content of LG in pine wood is ~5% higher than in aspen wood. Approximately the same difference in LG content is preserved for the ozonized samples. The higher LG content in the ozonized pine wood samples contributes to the occurrence of the abovementioned inner filter effect and could be responsible for the lower FL of the LCMs from pine wood in Fig. 6.
Ozonolytic Delignification of Wood
Studies of the laws governing ozone absorption by different types of biomass [6–10] have shown that ozone reactions occur on the surface of the porous structure of a biomaterial that is accessible to ozone molecules. In this work, ozone treatment was carried out under the optimum conditions of biomass delignification, where LG mostly reacts with molecular ozone dissolved in water in the porous structure of the biomass [5, 6, 8]. Molecular ozone undergoes electrophilic cycloaddition reactions with subsequent opening of the aromatic ring and the formation of aliphatic acids [9–11]. Molecular ozone reacts mostly with functional groups that have high electron density [32]. A comparison of the activity of organic compounds in reactions with ozone showed that the highest activity is exhibited by unsaturated structures and stilbenes. Next in the order of activity are syringyl and guaiacyl structures and carbonyl-containing and polyaromatic compounds [33, 34]. Aliphatic bonds in hydrocarbons and carbohydrates undergo relatively slow oxidation by ozone [32, 33].
Our ozonized samples differ in the values of Qr obtained at different durations of ozonation, allowing us to study the degradation of the biomaterial in stages. The interaction between ozone and wood occurs in the amorphous regions of a lignocarbohydrate complex consisting mostly of polysaccharides and LG [7]. Aromatic structures accessible to the reagent constitute the most reactive part of the structural organization, so they are the first to degrade, as we can see from the data of the Raman, UV, and FL spectra. It is shown that conjugated aromatic structures are predominantly degraded, testifying to the selective effect ozone has on the functional groups of LG in wood.
A comparison of these results and data in [11] from studying properties of the luminescence spectra of LCMs from ozonized aspen wood also suggests that ozone has a selective effect on different aromatic structures of LG. Hardwood LG contains guaiacyl and syringyl structural subunits (ratio of 1 : 1), while ~90% of softwood LG consists of guaiacyl structures [35]. It is known that the rate constants of the reaction with ozone differ by several orders of magnitude (105 and >107 L/(mol s) for guaiacol and syringol, respectively) for guaiacol and 2,6-dimethoxyphenol, which mimic guaiacyl and syringyl residues, respectively [34]. The stronger impact of ozone on hardwood and the modification of the physicochemical properties of the LCMs are determined by the structure of the native LG.
The predominance of guaiacyl structural units, and thus the lower content of methoxy groups and the stronger tendency of condensation due to free position 5 in the aromatic ring of the guaiacylpropane unit, play an important role in radical processes involving phenoxyl radicals [24, 25]. Phenoxyl radicals also form in reactions between ozone and phenolic compounds [32]. The role of radical processes is minimized in the ozonation of softwood under conditions favorable for delignification with the participation of molecular ozone [7–9].
In this work, UV, Raman, and FL spectroscopy were used to obtain information on the properties of an LCM surface, a portion of which was modified with ozone. The use of these techniques allowed us to observe the degradation of the aromatic structures of LG and reveal the selective pattern of this process under the action of a strong oxidizing agent (ozone). Delignification was accompanied by a substantial increase in the FL of the material. The degradation of LG by ozone also results in the removal of glucomannan—one of the structures of softwood HCs covalently bound to LG—from the LCM.
At higher Qr values, ozone is consumed for both the degradation of accessible aromatics and reactions with products of delignification and the degradation of polysaccharides. Oxidation of amorphous CL was also observed. The revealed laws governing the change in the physicochemical properties of LCMs correspond to the activity of ozone with respect to a number of organic compounds.
CONCLUSIONS
Regularities governing the decomposition of the biomass structure during ozonolytic delignification were studied using nondestructive means of research. It was shown that the luminescence spectral properties of LCMs are governed by the process of ozone absorption, which determines the ranges of ozone consumption corresponding to the predominant degradation of LG and polysaccharides of the biomaterial. It was shown that specific features of modifying the physicochemical properties of LCMs during ozonation are mainly determined by the structure of the native LG.
We may assume our combined ways of studying the transformation of wood under the action of ozone by means of Raman and fluorescence methods can also be used to study the delignification of plant biomass by other means.
REFERENCES
V. G. Samoilovich, S. N. Tkachenko, I. S. Tkachenko, and V. V. Lunin, Theory and Practice of Obtaining and Using Ozone, Ed. by V. V. Lunin (Mosk. Gos. Univ., Moscow, 2016) [in Russian].
N. A. Mamleeva, E. M. Ben’ko, and V. V. Lunin, Methods for the Neutralization of Wastewater, Gas Emissions and Waste from Production and Consumption, Ed. by V. V. Lunin (Mosk. Gos. Univ., Moscow, 2019) [in Russian].
O. M. Perrone, F. Colombari, J. Rossi, et al., Bioresour. Technol. 218, 69 (2016).
M. T. Garcia-Cubero, L. G. Palacin, G. Gonzalez-Benito, et al., Bioresour. Technol. 107, 229 (2012).
C. Li, L. Wang, Z. Chen, Y. Li, et al., Bioresour. Technol. 183, 240 (2015).
. E. V. Benko, D. G. Chukhchin, and V. V. Lunin, Holzforschung 74, 1157 (2020).
N. A. Mamleeva, A. N. Kharlanov, D. G. Chukhchin, et al., Khim. Rastit. Syr’ya, No. 1, 85 (2019).
N. A. Mamleeva, A. N. Kharlanov, S. Yu. Kupreenko, and D. G. Chukhchin, Russ. J. Phys. Chem. A 95, 2214 (2021).
N. A. Mamleeva, E. M. Ben’ko, A. N. Kharlanov, A. V. Shumyantsev, and D. G. Chukhchin, Russ. J. Phys. Chem. A 95, 577 (2021).
N. A. Mamleeva, A. N. Kharlanov, and V. V. Lunin, Russ. J. Phys. Chem. A 93, 2550 (2019).
N. A. Mamleeva, A. N. Kharlanov, M. V. Kuznetsova, and D. S. Kosyakov, Russ. J. Phys. Chem. A 96, 2043 (2022).
E. Billa, E. Koutsoula, and E. G. Koukios, Biores. Technol. 67, 25 (1999).
A. N. Zakazov and E. I. Chupka, Khim. Drev., No. 2, 52 (1983).
A. N. Papadopoulos, C. A. S. Hill, and A. Gkaraveli, Holz als Roh- Werlag 61, 453 (2003).
V. I. Azarov, Chemistry of Wood and Synthetic Polymers (St. Petersburg, 1999) [in Russian].
A. V. Obolenskaya, Z. P. El’nitskaya, and A. A. Leonovich, Laboratory Work on the Chemistry of Wood and Cellulose (Ekologiya, Moscow, 1991) [in Russian].
U. P. Agarwal, Front. Plant Sci. 5, 490 (2014). https://doi.org/10.3389/fpls.2014.00490
Zhe Ji, Jianfeng Ma, and Feng Xu, Microsc. Microanal. 20, 566 (2014).
M. Kihara, M. Takayama, H. Wariishi, and H. Tanaka, Spectrochim. Acta, Part A 58, 2211 (2002).
J. S. Lupoi, S. Singh, R. Parthasarathi, et al., Renewable Sustainable Energy Rev. 49, 871 (2015).
D. Ciolacu, F. Ciolacu, and V. Popa, Cellulose Chem. Technol. 45, 13 (2011).
C. E. Molina-Guerrero, G. de la Rosa, H. Castillo-Michel, et al., Chem. Eng. Technol. 41, 1350 (2018).
Physical Chemistry of Lignin, Ed. by K. G. Bogolitsyn and V. V. Lunin (Arkhang. Gos. Tekh. Univ., Arkhangel’sk, 2009) [in Russian].
H. Sadeghifar and A. Ragauskas, Polymers 12, 1134 (2020). https://doi.org/10.3390/polym12051134
M. Paulsson and J. Parkas, BioResources 7, 5995 (2012).
D. S. Kosyakov, N. S. Gorbova, K. G. Bogolitsyn, and L. V. Gusakov, Russ. J. Phys. Chem. A 81, 1076 (2007).
E. I. Chupka and V. M. Burlakov, Khim. Drev., No. 2, 31 (1984).
B. Albinsson, S. Li, K. Lundquist, and R. Stomberg, J. Mol. Struct. 508, 19 (1999).
L. Donaldson, IAWA J. (2013). https://doi.org/10.1163/2294193200000002
M. V. Panfilova, D. S. Kosyakov, and K. G. Bogoltsin, in Proceedings of the Europe Workshop on Lignocelulosics and Pulp. EWLP, Seville, Spain, June 24–27, 2014, p. 627.
M. V. Kuznetsova, D. S. Kosyakov, N. S. Gorbova, and K. G. Bogolitsyn, Russ. J. Phys. Chem. A 94, 1587 (2020).
S. D. Razumovskii and G. E. Zaikov, Ozone and Its Reactions with Organic Compounds (Nauka, Moscow, 1974), p. 219.
C. Olkkonen, Y. Tylli, I. Forsskåhl, et al., Holzforschung 54, 397 (2000).
H. Kaneko, S. Hosoya, K. Iiyama, and J. Nakano, J. Wood Chem. Technol. 3, R399 (1983).
J. E. Holladay, J. J. Bozell, and J. F. White, Top Value-Added Chemicals from Biomass (USA, 2007), Vol. 2. http://www.ntis.gov/ordering.htm.
ACKNOWLEDGMENTS
The authors thank Prof. N.G. Bazarnova (Altai State University) for providing our samples of wood.
Funding
This work was performed on equipment at the shared resource center “Nanochemistry and Nanomaterials” of Moscow State University’s Faculty of Chemistry. It was supported by the RF Ministry of Education and Science as part of State Task no. AAA-A21-121011990019-4 “Physical Chemistry of Surfaces, Adsorption, and Catalysis” (ozonolytic delignification of the LCMs, UV and Raman spectro-scopy); and by the shared resource center “Arktika” of Northern (Arctic) Federal University as part of State Task no. FSRU-2021-0009 “Studying the Luminescence Characteristics of Wood Components.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare they have no conflict of interest.
Additional information
Translated by M. Timoshinina
Rights and permissions
About this article

Cite this article
Mamleeva, N.A., Kharlanov, A.N., Kuznetsova, M.V. et al. Physicochemical Properties of Lignocellulosic Materials from Ozonized Wood. Russ. J. Phys. Chem. 97, 790–800 (2023). https://doi.org/10.1134/S0036024423040210
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036024423040210