
Abstract
Using density functional theory, the structural stabilities, electronic and magnetic properties of Fen (n = 3–6) clusters absorbed on defect armchair graphene nanoribbons are systematically investigated. We found that such systems have high stabilities and large magnetic moments. From n = 3, the binding energies are very important. This can be explained by the fact that a triangularization phenomenon between iron atoms and also more bonds with neighboring carbon atoms. Fe3-3V-AGNR exhibits a half-metal behavior with 61% spin polarization. Other doped structures (n = 4–6) exhibit semiconductor behavior.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Infinite sheets of graphene called graphene nanoribbons (GNRs) have attracted great interests in several research and applications areas [1–6]. Several methods for developing this material and controlling the quality of graphene are developed. Structural imperfections and atomic disorders can be induced by several ways. Gan et al. [7] have recently proved direct experimental evidences for the existence of these defects. They have visualized individual Au and Pt atoms incorporated into a very thin graphitic layer.
On theoretical side, several studies has been done to explore these interesting structures. Among these, using spin polarized DFT calculations, Duffy et al. [8, 9] have shown that, among the 3d transition metal atoms, only Fe, Co, and Ni are adsorbed at hollow sites [10]. Longo et al. [11] have also demonstrated that iron impurity is inducing magnetic behavior in GNRs. Experimentally, Gyamfi et al. [12] have explored the electronic properties and the adsorption site of Fe adatoms on monolayer and bilayer graphene. Eelbo et al. [13] have proved that Fe adatom is paramagnetic and exhibits an out-of-plane easy axis, with high-spin ground states. Eelbo et al. have also published other results confirming that doped graphene lattice by transition metal atoms induced magnetism [14] as well as reported and analyzed experimentally [15]. It seems that there are two ways for the introduction of transition metal (TM) atoms in GNRs and graphene: the first by embedding TM atom in vacancy defect [16–18] and the second via direct adsorption of TM atom on graphene [19, 20].
A systematic analysis of the structural and electronic properties conducted by Chan et al. [21] have targeted different metal adatoms on graphene using VASP code. Krasheninnikov et al. [22], studying all 3d transition metal (TM) atoms embedded in single and double vacancies of graphene, have found that an atom of a traditional magnet as Fe at a single vacancy of graphene is nonmagnetic, a finding that has been confirmed recently by Santos et al. [23]. Understanding the parameters involving the stability of such doped structures are explained in the interplay between the energetics and electronic properties of graphene–TM systems and their magnetic properties when deposited on pristine and defect graphene was the subject of intense recent investigations [24–29].
In this work, we have studied the electronic, structural and magnetic properties of Fen; n = 3–6 clusters with n vacancies sites in a graphene sheet by ab-initio density functional calculations. Our results show significant chemical interactions between the cluster and graphene. As a result, magnetic moments distribution appears on the Fe clusters in presence of graphene with a lower average magnetic moments compared to the free iron clusters.
COMPUTATIONAL DETAILS
The spin-polarized density functional theory calculations are carried out using SIESTA code based on numerical atomic orbitals basis set. We have employed the density functional theory (DFT) [30, 31] method to investigate the properties of our systems. Geometry relaxations are calculated within the generalized gradient approximation (GGA) as parameterized by Perdew, Burke, and Ernzerhof (PBE) [32]. The Kohn–Sham orbitals were expanded in numerical pseudo atomic localized basis sets (SIESTA package [33]) with double zeta polarization (DZP) and electron–ion interaction was included by employing norm-conserving pseudopotentials [34].
112 atoms of carbon passivated by eight hydrogen atoms compose used supercell. In order to avoid interactions between periodic images of the defective graphene layer, the size of the supercell perpendicular to the plane was set larger than 25 Å. The Monkhorst–Pack [35] used for k-sampling is equal to [1, 16]. All the atomic coordinates were always optimized until forces in all directions were smaller than 0.02 eV Å–1. The pseudopotentials for the metal atoms include nonlinear core corrections [36] for exchange and correlation. A double-ζ polarized (DZP) [37] basis set was used for the calculation of the magnetic and electronic properties. The cutoff radii of the different orbitals were obtained using an energy shift of 50 meV. For these atoms, the radii of the basis orbitals were enlarged until binding energies were converged within a few tens of meV.
RESULTS AND DISCUSSION
Structural Study
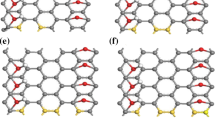
The typical relaxed geometries of the systems studied in this paper were presented on Fig. 1.

The ground-state structure of Fen/AGNR: top view and side view; n = (a) 1, (b) 2, (c) 3, (d) 4, (e) , 5 and (f) 6.
In order to gain more insights into the interaction between Fen (n = 3−6) clusters and graphene, we have studied the binding energy (Eb) for the cluster on graphene which is defined as
where n is the number of iron atoms, Esystem is the total energy of the iron–graphene nanoribbons system, \({{E}_{{{\text{F}}{{{\text{e}}}_{n}}}}}\) is the total energy of an isolated iron cluster, and Egr is the total energy of GNR with defects.
One and two iron atoms substitutions in AGNR are depicted on Figs. 1a and 1b. As it is known, many transition metal (TM) atoms can form covalent bonds with under-coordinated C atoms at a vacancy [23, 38]. It is exactly the situation of atomic configurations of Fe/SV and Fe2/DV complexes (where SV is the single vacancy and DV is the double vacancy). As the iron atomic radii are larger than those of the carbon atom, the metal atoms are displaced outward from the graphene plane. The binding energies Eb for the Fe/SV and Fe2/DV are greater than calculated by other authors in [23, 38] indicating a strong bonding and pointing to a possible use of such structures in catalysis.
The lowest energy structure of the Fe trimmer on graphene obtained from our calculations is depicted on Fig. 1c. The binding energy is very higher (14.23 eV). Two iron atoms are close to the GNR plane and the third far away, forming an isosceles triangle with 2.55 and 2.30 Å. Fe–C distances vary between 1.88 and 2.1 Å. The trimmer plane is perpendicular to the graphene plane. We note that if the number of substitution is greater than three, the clusters of atoms tend to form a three dimensional shape, with good agreement with Haldar et al. results [39, 40]. These results seem to be different from the previous studies [41, 42]. As demonstrated by Boukhvalova et al., for adatoms of transition metals, the bonding energies are larger and the charge transfer is essential and predominant. For iron, cobalt, and nickel, the distortions of the tetrahedron into carbide like configuration Me3C are very strong [43]. In the case of Fe3, the cluster plane has two different adsorption behaviors, namely, parallel or perpendicular to the AGNR. Among the optimized structures, we found the perpendicular case is energetically most stable. The parallel case is very unfavorable following results of reference [44].
For a complete study of iron substitution, four, five and six iron cases substitutions are investigated. After relaxation, all Fe atoms come out of the GNR plane. Three Fe atoms in one side and the fourth is in another side (see Fig. 1d). The ground state of the free iron tetramer is a tetrahedron with three short 2.40 Å bond lengths and three long 2.45 Å ones while this tetramer is distorted when is embedded in GNR. The calculated binding energy equals 143.3 eV. Although free Fe4 is a three-dimensional (3D) structure with D2h symmetry, it may undergo a 3D-to-2D transition after adsorption. To obtain the ground state, various possible initial structures and adsorption sites were investigated. Finally, it was found that Fe4 still keeps the tetrahedron configuration little changed after adsorption. In the ground state, the bottom three Fe atoms adsorb on one side and fourth one on the other side of AGNR and the top Fe is far away from AGNR. Similar adsorption behavior was also observed in ZGNR and graphene systems [45, 46]. In addition, the tetramer may be planar when it is on AGNR, but the energy is higher than the ground state by about 1 eV. Because Fe–Fe covalent bond is strong, Fe atoms on AGNR surface prefer to form 2D or 3D cluster structure rather than disperse to separated adsorption sites [44].
By adding Fe atoms, the coordination number of Fe atoms increases, leading to higher fraction of triangular lattices. We can easily see on Fig. 1e, for five iron atoms that three atoms form a triangle on one side of the graphene and the other two atoms go on the other side. Bond lengths vary from 2.24 to 2.89 Å with a broken symmetry. The free Fe5 has symmetry of distorted trigonal bipyramid. The calculated binding energy is less than previous cases. It equals to 139.9 eV. This effect is expected. In principle, one expects the binding energies decrease as the triangular lattices enlarge. However, the formation energies per Fe atom for high-symmetry Fe increases as the number of newly added Fe atoms increases, while the formation energies per Fe atom for low-symmetry are nearly unchanged. Earliest, Srivastava et al. [42] proposed that a square pyramidal structure of the Fe pentamer on graphene is the ground-state structure.
A substitution with six iron atoms still gives support to our hypothesis or reasoning. After relaxation, the six iron atoms that are initially in GNR plane, go forward and form two bonded iron trimmer via GNR plane. This structure seems to be the most stable among other with binding energy of 148.1 eV. To the best of our knowledge, only one study for Fe hexamer on graphene have been published. The structure has two layers of Fe atoms with three Fe atoms in the first layer sitting above the B sites and the other layer in opposite face of graphene, in good agreement with [40].
Magnetic and Electronic Properties
Fen/GNR systems preserve in almost cases, the semiconducting nature of the pristine nanoribbon. The Fe clusters get strong positive atomic spin magnetic moments [45] because most Fe states near the Fermi level are spin-down while the corresponding spin-up states are well below the Fermi level. From Fig. 2, we see that all our structures are semiconductor except the Fe3-AGNR one which exhibits a metallic behavior.

Bands structure of Fen/AGNR: n = (a) 1, (b) 2, (c) 3, (d) 4, (e) 5, and (f) 6.
The Fe3-AGNR has a total magnetic moment averaging 3.21 μB. An interesting result must be noted. We have found that the three iron atoms exhibit antiparallel magnetic moments. The iron atom which is the most distant from the GNR plane has a negative magnetic moment equal to –2.81 μB. This structure is typically half-metal. In such system, the spin-up electron will be blocked but the spin-down electron can easily pass through it.
Fe4-AGNR structure exhibits a total magnetic moment of about 10 μB. In this case, all magnetic moment is carried by iron atom are parallel and the set of carbon atoms surrounding the hole carries an anti-parallel moment. From band structures (Fig. 2d), we see that this structure is semiconductor.
The same observation can be made for Fe5-AGNR structure. This structure exhibits 14 μB for the magnetic moment. The surrounding carbon atoms and iron atoms exhibit anti-parallel spins. This structure exhibits a semiconductor behavior with a little band gap (0.15 eV).The last structure (Fe6-AGNR) is also semiconductor, with a total magnetic moment of about 18 μB. We note that the magnetic moments of the last three structures are less than those obtained by reference [39].
In Table 1 we compare the magnetic moments of the Fe clusters in their free-standing form (in parenthesis) and on graphene. We can see that the magnetic moments of all clusters on graphene are reduced from its value in the freestanding. A drastic diminution on magnetic moment is noted for iron trimmer on AGNR. Le magnetic moment of the cluster equals 2.97 μB while it equals 11.9 μB for the free one. Fe4 and Fe5 cluster on AGNR know also an important reduction of the magnetic moments. The magnetic moment reduction is very significant. The magnetic moments are reduced by ∼5 and 4 μB, respectively.
In order to understand how the Fe clusters interact with graphene and how the graphene affects the magnetic moments of the Fe clusters, we performed a chemical bonding analysis to study the electron transfer and bonding characteristics among the different orbitals of Fe atoms and graphene within Mulliken charge analysis method. We can see that the s orbitals of Fe atoms transfer their electrons to the p and d orbitals of Fe (in both freestanding and adsorbed clusters) and to the pz orbitals of graphene in the adsorption case (not mentioned in Table 1). The origin of the magnetic moment reduction can be attributed to the charge transfer and redistribution among different orbitals within the Fe clusters and between the clusters and graphene upon adsorption. We can also see that magnetization on each carbon atom in the graphene is also strongly correlated with the lattice distortion of the carbon atoms in good agreement with results of [40]. Iron cluster adsorbed on AGNR exhibits strong covalent bonding with graphene. The majority of the contributions to the covalent bonds are from strong hybridization between the \({{d}_{{x2 - y2}}}\) and dyz orbitals of the 3d-transition-metal adatoms and pz orbitals of the carbon atoms. The strong covalent bonds cause large in-plane lattice distortions in the graphene layer. Charge redistributions upon adsorptions also induce significant electric dipole moments and affect the magnetic moment [47]. Carbon pz orbitals (π bonded states) were found to hybridize strongly with the d orbitals of the adsorbed iron atoms. Generally, the magnetic moment of any iron atom on the AGNR surface depended strongly on the adsorption site with good agreement with references [10, 48, 49]. For the spin polarization, all structures (with one and two Fe atoms) have a zero spin polarization since they are semiconductors except Fe3-GNR, which have a half-metal behavior with a spin polarization equal to 61% (Fig. 3).

Total density of states of Fen/AGNR: n = (a) 1, (b) 2, (c) 3, (d) 4, (e) 5, and (f) 6.
CONCLUSIONS
In present paper, we studied the structural, electronic and magnetic properties of Fen embedded in defect vacancies of AGNR (n = 1–6) systems by using density functional theory. It is found that the Fen/AGNR systems have high stabilities and large magnetic moments. We have found that depending on the number of iron atoms Fen/AGNR may be half metal with 61% spin polarization (n = 3) or semiconductor (n = 1 and 2). These fascinating electronic and magnetic properties show that these systems are promising materials for designing new nanodevices.
REFERENCES
P. Gambardella, A. Dallmeyer, K. Maiti, et al., Nature (London, U. K.) 416, 301 (2002).
J. Dorantes-Dávila and G. M. Pastor, Phys. Rev. Lett. 81, 208 (1998).
A. Vindigni, A. Rettori, M. G. Pini, et al., Appl. Phys. A 82, 385 (2006).
A. Delin, E. Tosatti, and R. Weht, Phys. Rev. Lett. 92, 057201 (2004).
H. Lee, Y. W. Son, N. Park, et al., Phys. Rev. B 72, 174431 (2005).
M. Fujita, K. Wakabayashi, K. Nakada, and K. Kusakabe, J. Phys. Soc. Jpn. 65, 1920 (1996).
Y. Gan, L. Sun, and F. Banhart, Small 4, 587 (2008).
D. M. Duffy, J. A. Blackman, P. A. Mulheran, and S. A. Williams, J. Magn. Magn. Mater. 177–181, 953 (1998).
D. M. Duffy and J. A. Blackman, Phys. Rev. B 58, 7443 (1998).
H. Valencia, A. Gil, and G. Frapper, J. Phys. Chem. 114, 14141 (2010).
R. C. Longo, J. Carrete, and L. J. Gallego, Phys. Rev. B 83, 235415 (2011).
M. Gyamfi, T. Eelbo, M. Wasniowska, and R. Wiesendanger, Phys. Rev. B 84, 113403 (2011).
T. Eelbo, M. Wasniowska, and P. Thakur, Phys. Rev. Lett. 110, 136804 (2013).
T. Eelbo, M. Waśniowska, M. Gyamfi, et al., Phys. Rev. B 87, 205443 (2013).
A. W. Robertson, B. Montanari, and K. Heetal, Nano Lett. 13, 1468 (2013).
V. Krasheninnikov, P. O. Lehtinen, A. S. Foster, et al., Phys. Rev. Lett. 102, 126807 (2009).
E. J. G. Santos, D. Sánchez-Portal, and A. Ayuela, Phys. Rev. B 81, 125433 (2010).
S. Lisenkov, A. N. Andriotis, and M. Menon, Phys. Rev. Lett. 108, 187208 (2012).
H. Sevinçli, M. Topsakal, E. Durgun, and S. Ciraci, Phys. Rev. B 77, 195434 (2008).
V. A. Rigo, T. B. Martins, A. J. R. da Silva, et al., Phys. Rev. B 79, 075435 (2009).
K. T. Chan, J. B. Neaton, and M. L. Cohen, Phys. Rev. B 77, 235430 (2008).
A. V. Krasheninnikov, P. O. Lehtinen, A. S. Foster, et al., Phys. Rev. Lett. 102, 126807 (2009).
E. J. G. Santos, A. Ayuela, and D. Sánchez-Portal, New J. Phys. 12, 053012 (2010).
S. Sahoo, M. E. Gruner, S. N. Khanna, and P. Entel, J. Chem. Phys. 141, 074707 (2014).
I. Cabria, M. López, and J. Alonso, Phys. Rev. B 81, 035403 (2010).
P. Błoński and J. Hafner, J. Chem. Phys. 134, 154705 (2011).
F. Banhart, J. Kotakoski, and A. V. Krasheninnikov, ACS Nano 5, 26 (2011).
M. J. López, I. Cabria, and J. A. Alonso, J. Phys. Chem. C 118, 5081 (2014).
Xielong Hu and Fanyan Meng, J. Nano Mater. 2016, 2672816 (2016).
P. Hohenberg and W. Kohn, Phys. Rev. B 136, 864 (1964).
W. Kohn and L. J. Sham, Phys. Rev. A 140, 1133 (1965).
J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996).
J. M. Soler, E. Artacho, J. D. Gale, et al., J. Phys.: Condens. Matter 14, 2745 (2002).
N. Troullier and J. L. Martins, Phys. Rev. B 43, 1993 (1991).
H. J. Monkhosrt and J. D. Pack, Phys. Rev. B 13, 5188 (1976).
S. G. Louie, S. Froyen, and M. L. Cohen, Phys. Rev. B 26, 1738 (1982).
J. Junquera, O. Paz, D. Sanchez-Portal, and E. Artacho, Phys. Rev. B 64, 235111 (2001).
A. V. Krasheninnikov, P. O. Lehtinen, A. S. Foster, et al., Phys. Rev. Lett. 102, 126807 (2009).
S. Haldar, B. S. Pujari, S. Bhandary, et al., Phys. Rev. B 89, 205411 (2014).
X. Liu, C.-Z. Wang, H.-Q. Lin, et al., Phys. Rev. B 90, 155444 (2014).
H. Johll, H. C. Kang, and E. S. Tok, Phys. Rev. B 79, 245416 (2009).
M. K. Srivastava, Y. Wang, A. F. Kemper, and H.‑P. Cheng, Phys. Rev. B 85, 165444 (2012).
D. W. Boukhvalova and M. I. Katsnelson, Appl. Phys. Lett. 95, 023109 (2009).
D. Wang, Zh. Yang, L.-Ch. Xu, et al., Comput. Theor. Chem. 1062, 84 (2015).
R. C. Longo, J. Carrete, J. Ferrer, and L. J. Gallego, Phys. Rev. B 81, 115418 (2010).
H. Johll, J. Wu, S. W. Ong, and H. C. Kang, Phys. Rev. B 83, 205408 (2011).
X. Liu, C. Z. Wang, Y. X. Yao, et al., Phys. Rev. B 83, 235411 (2011).
D. M. Duffy, J. A. Blackman, P. A. Mulheran, and S. A. Williams, J. Magn. Magn. Mater. 177–181, 953 (1998).
D. M. Duffy and J. A. Blackman, Phys. Rev. B 58, 7443 (1998).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Lamiri, L., Benchallal, L., Boubenider, F. et al. Electronic and Magnetic Properties of Iron Doped Graphene Nanoribbons. Russ. J. Phys. Chem. 96 (Suppl 1), S132–S138 (2022). https://doi.org/10.1134/S0036024422140151
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036024422140151