
Abstract
A study is performed of IR absorption and Raman spectra of methyl 6-methoxy-2,3,4,9-tetrahydro-1H-1,4-ethanocarbazole-3-carboxylate. Optimized structures and the harmonic force fields of stable conformers are calculated using the density functional theory (the B3LYP, M062X, and BVP86 functionals combined with basis sets of different completeness). A detailed explanation of the spectra is proposed based on calculations, and the characteristic frequencies of the most stable forms of a given compound are identified. The theoretical spectra are analyzed relative to experimental data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Melatonin (N-acetyl-5-methoxytryptamine) (Fig. 1) is a neurohormone with chronobiotic, anticancer, and immunostimulating activities. It also participates in the regulation of blood pressure, body temperature, and the cardiovascular system [1]. Melatonin has been found in unicellular algae, plants [2], invertebrates, and vertebrates (including humans). The pineal gland of the brain is the main source of melatonin in vertebrate animals. Changes in melatonin production following those in daylight hours lead to daily and seasonal changes in the bodies of humans and animals. In addition, all endogenous rhythms are subordinated to melatonin production rhythms [3].

Structure of melatonin.
The use of melatonin in treating various diseases is limited because of its relatively short half-life in the body (about thirty minutes) [4]. One way of modifying the structure of melatonin is to limit the conformational mobility of its molecule through the condensation of an indole core with a rigid bicyclic framework [5]. The spatial structure of functional groups plays a key role in interactions with receptors, to which vibrational IR absorption and Raman spectra are strongly sensitive. They contain much more information and are comparatively easy to use. Finally, Raman spectroscopy is of special importance when studying melatonin analogs in aqueous solutions. Reliable spectral means for the identification of new compounds are needed to study the mechanisms of their action in real conditions. As a result, it is important to obtain as much information as possible on the structure of molecules of conformationally constrained melatonin analogs.
In this work, we studied and interpret IR absorption and Raman spectra of methyl 6-methoxy-2,3,4,9-tetrahydro-1H-1,4-ethanocarbazole-3-carboxylate (A), allowing for possible rotational isomerism (Fig. 2).

Structures of exo-isomers and endo-isomers of A.
Our test substance was synthesized at the Department of Chemistry, Moscow State University as a part of the program to find substances that act on melatonin receptors. Compound A is an intermediate in the synthesis of a conformationally constrained analog of melatonin N-(exo-6-methoxy-2,3,4,9-tetrahydro-1H-1,4-ethanocarbazol-3-yl)acetamide [6]. The BVP86/TZVP level of theory [7–12] was used as the basis for explaining the experimental spectra [7–12]. It proved to be highly efficient, due to its low computational costs in performing calculations for polycyclic organic compounds [13, 14]. The theoretical spectra were analyzed relative to experimental ones obtained in the 400–3600 (IR) and 50–3600 cm−1 (Raman) wavenumber ranges.
EXPERIMENTAL
Methyl 6-methoxy-2,3,4,9-tetrahydro-1H-1,4-ethanocarbazole-3-carboxylate A is a mixture of endo- and exo-isomers in an approximately 1 : 1 ratio. The sample of the studied compound is a brown resin at room temperature. The purity of the substance was ~95%. The IR absorption spectrum of the substance pressed into a pellet with potassium bromide was recorded on a Bruker Tensor-27 Fourier spectrometer (Germany) with a resolution of 1 cm−1. The Fourier transform Raman spectrum of the solid compound was obtained on a Bruker EQINOX 55 spectrometer equipped with an FRA-106 attachment (Germany). The excitation source was a Nd:YAG laser (1064 nm) with a power of 500 mW. The resolution was 2 cm−1. The signal was averaged over a thousand scans. Figures 3a and 3b show the resulting spectra.

Comparison of the experimental (a) IR absorption and (b) Raman spectra of a mixture of isomers A to ones calculated (BVP86/TZVP) for conformers (c, d) exo-1, (e, f) endo-1, (g, h) endo-2, and (i, j) exo-2.
CALCULATIONS
Quantum-mechanical calculations were performed using the density functional theory and the Gaussian 09 software (version D.01) [15]. Quantum-chemical calculations of the stable conformations of molecules were performed with B3LYP [16], M062X [17], and BVP86 hybrid functionals [11–14], coupled with the 6-31G**, 6-31+G** [18–20], and TZVP basis sets [11, 12], respectively. Optimized geometries were obtained for all possible conformers (with no symmetry restrictions), and their harmonic force fields, the vibration frequencies, the intensities of IR absorption bands in the gas phase, and the activities in Raman spectra were calculated. In addition, the vibration frequencies in an anharmonic approximation were calculated for all conformers. All energy differences given below were calculated with allowance for corrections for the zero vibrational level. The Chemcraft program was used to visualize the results from quantum-mechanical calculations [21]. The quantum-chemical matrices of the force constants were converted from Cartesian coordinates to a dependent system of internal coordinates, the normal vibrations were analyzed, and the distribution of potential energy over vibrations of the conformers was calculated with the SPEKTR program [22, 23]. Table 1 presents the internal coordinates.
RESULTS AND DISCUSSION
A number of conformations were possible for the test compound that had different orientations of the methoxy and methoxycarbonyl groups. Eight conformations of the test compound that arose for the syn- and anti-positions of a methyl group with fixed syn- or anti-orientations relative to the C=O bond of a five-membered cycle and a methoxy group bound directly to an indole core were considered for the exo- and endo-isomers of A.
The structures were optimized and the harmonic force fields were calculated along with the vibration frequencies of sixteen stable structures at the B3LYP/6-31G**, B3LYP/6-31+G**, М062Х/6-31+G**, and RBVP86/TZVP levels of theory.
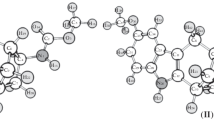
The results from calculations show that the anti-syn-syn-conformations of (a) exo-1 and (b) endo-1 isomers were the ones most stable (Fig. 4). The energy difference between them was only ~0.04 kcal/mol (RBVP86/TZVP). The energy difference between the ones most stable and the syn-syn-syn conformers of exo-2 and endo-2 was ~0.6 kcal/mol for the exo-isomer and ~0.5 kcal/mol for the endo-isomer, which corresponds to the relative amount in mixtures of 3 : 1 and 7 : 3, respectively. A detailed explanation of the vibrational spectrum is proposed for the most stable conformations, based on an analysis of normal vibrations (Table 2).

Structures of the stable conformations of isomers A: (a) exo-1, (b) endo-1, (c) endo-2, and (d) exo-2.
CONCLUSIONS
A comparison of our theoretical and experimental vibration frequencies (Fig. 3 and Table 2) shows that the BVP86/TZVP level satisfactorily reproduces both the structural and spectral data for the considered compound, which confirms the possibility of using this approximation.
A comparison of theoretical spectra of the isomers shows there are slight differences between the vibrational frequencies in the 1700–3000 and 500–630 cm−1 ranges. Minor differences in the fingerprint region are apparent mainly in the relative intensity of the bands.
REFERENCES
B. Stauch, L. C. Johansson, J. D. McCorvy, et al., Nature (London, U.K.) 569, 284 (2019).
D. X. Tan, R. Hardeland, L. C. Manchester, et al., J. Exp. Bot. 63, 577 (2012).
A. Yu. Bespyatykh, V. Ya. Brodskii, O. V. Burlakova, et al., Melatonin: Theory and Practice (Medpraktika-M, Moscow, 2009) [in Russian].
J. B. Fourtillan, A. M. Brisson, P. Gobin, et al., Biopharm. Drug Dispos. 21, 15 (2000).
D. P. Zlotos, Arch. Pharm. Chem. Life Sci. 338, 229 (2005).
O. N. Zefirova, T. Yu. Baranova, A. A. Ivanova, et al., Bioorg. Chem. 39, 67 (2011).
A. D. Becke, Phys. Rev. A 38, 3098 (1988).
A. D. Becke, J. Chem. Phys. 98, 5648 (1993).
M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian 03 (Gaussian, Inc., Pittsburgh, PA, 2003).
M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian 09, Revision A.1 (Gaussian, Inc., Wallingford, CT, 2009).
A. Schaefer, H. Horn, and R. Ahlrichs, J. Chem. Phys. 97, 2571 (1992).
A. Schaefer, C. Huber, and R. Ahlrichs, J. Chem. Phys. 100, 5829 (1994).
V. Pomogaev, A. Pomogaeva, P. Avramov, et al., Theor. Chem. Acc. 130, 609 (2011).
D. Kosenkov, Y. Kholod, L. Gorb, et al., J. Phys. Chem. A 113, 9386 (2009).
M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian 09, Revision D.01 (Gaussian Inc., Wallingford, CT, 2013).
A. D. Bekke, J. Chem. Phys. 96, 2155 (1992).
Y. Zhao and D. G. Truhlar, Theor. Chem. Acc, 120, 215 (2006).
G. A. Petersson, A. Bennett, T. G. Tensfeldt, et al., J. Chem. Phys. 89, 2193 (1988).
G. A. Petersson and M. A. Al-Laham, J. Chem. Phys. 94, 6081 (1991).
V. A. Rassolov, M. A. Ratner, J. A. Pople, et al., J. Comput. Chem. 22, 976 (2001).
ChemCraft, Version 1.5. http://www.chemcraftprog.com.
G. M. Kuramshina, F. A. Weinhold, I. V. Kochikov, et al., J. Chem. Phys. 100, 1414 (1994).
I. V. Kochikov, A. G. Yagola, G. M. Kuramshina, et al., Spectrochim. Acta, A 41, 185 (1985).
Funding
This work was financially supported by the Russian Foundation for Basic Research, project no. 18-33-00826.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by A. Tulyabaev
Rights and permissions
About this article

Cite this article
Davydova, I.B., Senyavin, V.M., Zefirova, O.N. et al. Vibrational Spectra and Stable Conformations of Methyl 6-Methoxy-2,3,4,9-Tetrahydro-1H-1,4-Ethanocarbazole-3-Carboxylate. Russ. J. Phys. Chem. 94, 2255–2264 (2020). https://doi.org/10.1134/S0036024420110047
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036024420110047