
Abstract
The thermodynamic characteristics of adsorption (TCA) of thiophene and its derivatives are determined under the conditions of equilibrium gas adsorption chromatography (GAC) on columns with graphitized thermal carbon black (GTCB) of Carbopack C HT grade. It is shown that the TCA values depend largely on the number and nature of the substituents in the main structural fragment. It is found that the surface of the GTCB is characterized by low structural selectivity for positional isomers in the series of thiophene derivatives. The effect of polar retention of an adsorbate on graphite from the gas phase due to additional intermolecular specific interactions of polar groups of the adsorbate with an easily polarizable surface of the base face of graphite is established and studied. Conclusions are reached on the applicability of the two-dimensional ideal gas model for describing the mobility of strongly polar molecules of thiophene derivatives on surfaces of graphite, and on the considerable limitations of this model when applied to the adsorption of thiophene molecules, along with its methyl and halogen derivatives.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The adsorption of thiophene and some of its derivatives on surfaces of graphite-like materials has more than once been the subject of studies by means of equilibrium gas adsorption chromatography (GAC) on columns with graphitized thermal carbon black (GTCB) [1–7]. Interest in this group of compounds is due to their relatively wide distribution among other heterocyclic compounds, their many functional derivatives, and the manifesting of different properties, including various types of biological activity. The thermodynamic characteristics of adsorption (TCA) under equilibrium GAC on columns with GTCB have been determined for thiophene [1, 2], alkyl, and halothiophenes [3, 4], arylthiophenes [5], and other compounds [6]. In addition, the parameters of the potential function of the intermolecular interaction of S atoms with C atoms of the base face of graphite were calculated, allowing us to a priori calculate equilibrium TCA values and perform chromatostructural analyses of the geometry of a number of thiophene-containing compounds adsorbed on flat graphite surfaces [5]. Among the sulfur-containing compounds of a non-thiophene series studied by GAC on GTCB, stereoisomeric perhydrothioxanthenes and perhydro-4-thia-s-indacenes are worthy of note [8, 9]. The unique capabilities of micro-packed columns with GTCB for the complete separation and identification of individual isomers of this group of compounds have been shown using GAC and molecular-statistical calculations. In addition to experimental studies, molecular-statistical calculations of equilibrium TCA have also been performed that allowed us not only to clearly identify individual isomers, but to determine the contribution from different conformations of individual isomers to the total energy of adsorption as well.
At the same time, despite the unique 2D-selective characteristics of graphite-like adsorbents and their high chemical inertness and thermal stability, the number of works on using this group of materials in gas chromatography remains small. Considerable progress along these lines was made after creating a GTCB analog for HPLC: the porous graphite-like material Hypercarb. Columns with this adsorbent are widely used to separate structural and spatial isomers of different classes of organic compounds with similar properties, including thiophene derivatives [10]. As with adsorption from the gas phase, the propensity of adsorbate molecules for planar arrangement on a flat surface of graphite plays a key role in sorption from aqueous-organic solutions, allowing us to conclude that 2D structural selectivity dominates, despite the active role of the mobile phase in sorption from medium liquid eluents.
The aim of this work was the gas chromatographic determination of the equilibrium TCA values of thiophene molecules and its various functional derivatives on a surface of GTCB. It was of interest to establish the selectivity and the possibility of separation of isomeric compounds of thiophene derivatives on columns with GTCB, and to study the propensity of strongly polar substituted thiophene molecules toward specific intermolecular interactions with an easily polarized graphite surface.
EXPERIMENTAL
Specific retention volumes VA,1, cm3/m2 for the adsorbates studied in this work were determined experimentally on a Crystal 4000 gas chromatograph equipped with a flame ionization detector. The carrier gas was helium (flow rate, 18–25 cm3/min). Methane was used as a non-adsorbable substance. Samples of substances were introduced at least 5 times in the form of diluted vapor–air mixtures or highly diluted solutions in ethanol and diethyl ether. Separation was performed on a glass micron-packed column with dimensions of 1.00 m × 1.5 mm. Carbopack C HT (Supelco) with a specific surface of 10 m2/g was used as our adsorbent [11]. The adsorbent mass in the column was 2.08 g. The size of the adsorbent grains was 60–80 mesh. The relative retention times of adsorbates throughout the investigated range of temperatures did not depend on the concentration of adsorbate in the gas phase, confirming the achievement of a linear portion of the adsorption isotherm (Henry region). Symmetric peaks of sorbates in the chromatograms also testified in favor of this (Fig. 1). The main TCAs were calculated out according to the standard procedure, in accordance with the basic postulate of equilibrium chromatography: VA,1 [12–15]. To process the primary chromatographic data, we used the equation
where \({\Gamma}_{{\text{i}}}^{{{\text{gas}}}}\) and \({\Gamma}_{{{\text{i}}{\text{,st}}}}^{{{\text{gas}}}}\) are the equilibrium and standard Gibbs values of adsorption (μmol/m2); \({c}_{{\text{i}}}^{{{\text{gas}}}}\) and \({c}_{{{\text{i}}{\text{,st}}}}^{{{\text{gas}}}}\) are the equilibrium and standard concentrations of the adsorbate in the gas phase, respectively (μmol/cm3); tR is the retention time of the sorbate (min); tM is the retention time of the non-absorbable substance (min); pi is the pressure of the carrier gas at the inlet to the column (atm); pa is atmospheric pressure (atm); T is the column temperature (K); Ta is room temperature (K); pw is the vapor pressure of water at a temperature of Ta (atm); \({{F}_{{{{{\text{p}}}_{{\text{a}}}}{\text{,}}{{{\text{T}}}_{{\text{a}}}}}}}\) is the volumetric velocity of the carrier gas, measured using a foam flowmeter at a pressure pa and temperature Ta (cm3/min); mA is the mass of adsorbent in the column (g); and \({{S}_{{{{{\text{N}}}_{2}}}}}\)is the specific surface area of the adsorbent, measured via static volumetry for nitrogen adsorption at a temperature of 77 K (m2/g). In determining TCA, the following values were used for the standard state of the adsorbate: \({c}_{{{\text{i}}{\text{,st}}}}^{{{\text{gas}}}}\) = 1 μmol/cm3 (in the gas phase); \({\Gamma}_{{{\text{i}}{\text{,st}}}}^{{{\text{gas}}}}\) = 1 μmol/m2 (in the adsorbed state) [14, 15].

Chromatogram of the separation of a mixture of monosubstituted thiophenes (T = 393 K, 25 cm3/min) on a column with Carbopak C HT.
To determine the values of molar differential standard heats \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) (J/mol) and the change in entropy \(\Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\) (J/(mol K)) of adsorption, we used two well-known approximations based on the dependences of lnK1,c on 1/Т [16]:
•\(\Delta \bar {C}_{{1,{\text{v}}}}^{{\text{s}}} \approx 0{\text{:}}\)
•\(\Delta \bar {C}_{{1,{\text{v}}}}^{{\text{s}}} = {\text{const}} \ne 0{\text{:}}\)
where \(\Delta \bar {C}_{{1,{\text{v}}}}^{{\text{s}}}\) = \(\bar {C}_{{{\text{ads}}}}^{{\text{o}}}\) – \(\bar {C}_{{{\text{gas}}{\text{,v}}}}^{{\text{o}}}\) is the difference between the molar differential heat capacity of the substance in the adsorbed state (\(\bar {C}_{{{\text{ads}}}}^{{\text{o}}}\), J/(mol K)) and the molar heat capacity of the substance in the equilibrium gas phase at V = const (\(\bar {C}_{{{\text{gas}}{\text{,v}}}}^{{\text{o}}}\), J/(mol K)); Tav is the middle of the investigated temperature range (K); R is the universal gas constant (8.314 J / (mol K)). The error of the experimentally determined TCA values given in Table 1, did not exceed 3.5% for K1,c, 1 kJ/mol and 6.0 J/(mol K) for values \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) and \(\Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\), respectively. Some of physicochemical parameters given in Table 2 for the studied compounds were taken from the reference literature [17–19] or calculated using known additive schemes [20, 21].
RESULTS AND DISCUSSION
Table 1 shows the TCA values of thiophene, benzene, and their functional derivatives. From the presented data, it is seen that the values of lnK1,с and \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) for thiophene derivatives are notably lower than the corresponding values for benzene derivatives. The difference between values \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) for benzene and thiophene is 3.5 kJ/mol, which exceeds value \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) of the experimental measurement error. In the series of methyl-, chloro-, bromo-, and iodo- derivatives of benzene and thiophene, the difference in values \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) decreases from 3.2 to 1.9 kJ/mol, respectively. The reason for the observed features is probably due to the value of the Van der Waals radius of the S atom (0.185 nm [22]) being greater than the half-thickness of the benzene ring (0.177 nm) [23]. The CH groups in the thiophene molecule in close proximity to the S atom (ring positions 2 and 5) are thus elevated above the flat surface of graphite, weakening their interaction with the adsorbent and reducing ln K1,c and \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\). The conclusion that the sulfur atom raises the plane of the thiophene ring above the graphite surface confirms the presence of two separate dependences \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) = f(αМ) for mono derivatives of benzene and thiophene (Fig. 2): the dependence for derivatives of thiophene in the graph coordinates \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) = f(αМ) lies below the corresponding dependence for benzene derivatives. Similar effects were observed in [24, 25] while studying the adsorption of other aromatic compounds with bulky substituents on a surface of graphite. Differences in the adsorption of the main structural fragments (benzene and thiophene rings) also influence the contributions from functional groups to \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\). Table 3 shows the values equal to the contribution from different substituents to the heat of adsorption in the series of thiophene derivatives are higher than for the corresponding benzene derivatives.

Dependences of heat of adsorption \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\), kJ/mol, on molecular polarizability αM, Å3 in the series of mono derivatives of thiophene (s) and benzene (d).
Unlike monosubstituted benzene, positional isomerism is possible in the series of monosubstituted thiophene, due to the difference between the relative positions of the S atom and substituent in the ring (2- and 3-isomers, respectively). From the data of Table 1, it follows that the 2- and 3-isomers are characterized by close TCA values. This prevents their satisfactory separation on the microwell columns with GTCB used in this work. Despite this, the lnK1,с values for the 2-isomer are slightly higher than those for 3-isomer (e.g., 2‑ and 3-methylthiophenes). However, an increase in the Van der Waals radius of the substituent results in the difference between the values of lnK1,с for 2-/3-isomers (e.g., 2- and 3-bromothiophenes) virtually disappearing. With disubstituted derivatives of thiophene, the TCA values are almost indistinguishable, as can be easily seen by comparing the TCA values for the molecules of 2,4- and 2,5-dinitrothiophenes.
Analysis of lnK1,с values shows (Table 1) that GTCB can be used to separate effectively the considered thiophene derivatives with various functional groups. The separation chromatogram of the model mixture shown in Fig. 1 confirms this conclusion. At the same time, compounds with similar molecular polarizabilities (methyl and chlorothiophenes, along with isomers of the 2-, 5-, 13-4 positions) are characterized by almost identical TCA values, so mixtures of these compounds cannot be divided into the micronodules used in the work columns with Carbopack C HT. The GTCB surface is thus characterized by low structural selectivity for position isomers in the series of mono- and disubstituted derivatives of thiophene, which is in good agreement with low ortho-/meta-/para-selectivity in the series of corresponding benzene derivatives [24].
Upon moving to derivatives containing several different substituents (particularly 2-iodo-5-nitrothiophene, 2-nitro-3-bromothiophene, and 2-aceto-5-nitrothiophene molecules), additivity of the substituent contributions to value \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) is observed (the slight difference between the experimental values and \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) calculated from contributions fit into the error range of its gas chromatographic determination). An exception to this pattern is the 2,4-dinitro-5-iodothiophene molecule, for which \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) calculated from contributions (∼76 kJ/mol) is considerably higher than the experimental value of 57.6 kJ/mol. One explanation for this is apparently the large Van der Waals size of atom I (0.215 nm [22]), which lifts a flat thiophene ring with two conjugated NO2 groups above the graphite surface, as a result of which the intermolecular adsorbate–graphite energy falls sharply.
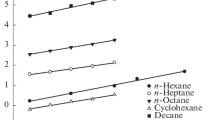
Of particular interest in light of the problem posed in this work is studying the nature of intermolecular interactions between thiophene derivatives with different polarities and a surface of graphite. It is known that according to the classification of Kiselev, GTCB is a type I adsorbent [24] on whose surface only dispersive intermolecular interactions can proceed. A reliable criterion for the occurrence of predominantly dispersive intermolecular interactions in adsorption is a correlation between values \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) of the adsorbates and their molecular polarizability αM. With molecules of thiophene, methylthiophenes, halogeniophenes (a group of weakly polar compounds for which μ ≤ 1.5 D), and n-alkanes, the correlation between experimental values \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) (Fig. 3) and αM values is characterized by a high determination coefficient (r2 = 0.98). In addition, the deviations of values \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) of these compounds from dependence \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) = f(αM) for n-alkanes are small. The above testifies to the predominant contribution from dispersion intermolecular interactions between thiophene, methylthiophene, and halogeniophene molecules and the surface of the base face of graphite.

Dependence of heat of adsorption \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\), kJ/mol, on molecular polarizability αM, Å3 of the studied thiophene derivatives.
A completely different picture is observed for the adsorption of molecules containing highly polar functional groups (e.g., nitro-, acetyl-, and acetamido-molecules—a group of strongly polar compounds for which μ ≥ 2.5 D). As can be seen from Fig. 3, values \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) of these compounds do not correlate well with αM. The corresponding points in the coordinate plane \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) = f(αМ) lie much higher than the straight line corresponding to n-alkanes and malopolar derivatives of thiophene. Such unexpected adsorption behavior of nitro-, acetyl-, and other strongly polar derivatives of thiophene indicates the manifestation of specific intermolecular interactions additional to dispersion. The most likely explanation for this feature could be that polar (e.g., NO2– and CH3CO–) groups, along with dispersion interactions, can join in a specific induction interaction with an easily polarizable system of π-electrons on a graphite surface. Similar interaction was recently observed in studying the adsorption of strongly polar adsorbates on a graphite-like surface under the conditions of HPLC [26–28]. This interaction was labeled the polar retention effect on graphite (detected on graphite-like adsorbent Hypercarb for HPLC) [28]. Figure 4 schematically shows the deformation of a delocalized system of p-electrons under the action of a strongly polar group.

(Color online) Schematic representation of an undeformed graphite surface for (a) nonspecific adsorption and (b) deformation of a delocalized system of p-electrons under the action of a strongly polar group.
In this work, we attempt to quantify the effect of polar retention. Using the dependence \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) = f(αM) obtained for n-alkanes,
and based on the known values of αM, we calculated theoretical values \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) corresponding to the contribution from only dispersion interactions to the heat of adsorption. The obtained values are given in Table 2 (column I). Table 2 shows that the values of \(\delta {{\bar {q}}_{{{\text{dif}}{\text{,1}}}}} = {{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) (theor.) − \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) (exp.) for thiophene, methylthiophene and halogeniophene molecules are comparable to experimental error values \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\). In contrast, this difference varies from 3.9 to 14.0 kJ/mol for compounds 8 to 17. With adsorbates that have strongly polar groups (particularly nitrothiophenes), there is specific interaction during adsorption on GTCB, the contribution from which to value \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) grows along with the number of such groups. Our values of \(\delta {{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) can serve as a quantitative characteristic of the polar retention on graphite effect of the considered strongly polar derivatives of thiophene on graphite.
Along with the value of αM in property–retention correlations, parameter \({{{{T}_{{\text{c}}}}} \mathord{\left/ {\vphantom {{{{T}_{{\text{c}}}}} {\sqrt {{{P}_{{\text{c}}}}} }}} \right. \kern-0em} {\sqrt {{{P}_{{\text{c}}}}} }}\), where Tc and Pc are the critical constants of substances, is widely used. The equation linking \({{{{T}_{{\text{c}}}}} \mathord{\left/ {\vphantom {{{{T}_{{\text{c}}}}} {\sqrt {{{P}_{{\text{c}}}}} }}} \right. \kern-0em} {\sqrt {{{P}_{{\text{c}}}}} }}\) and \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) obtained in [29] is
where D = 0.446 kJ bar0.5/K for graphite. The values calculated using Eq. (5)\({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) (theor.) are given in Table 2 (column II). Figure 5 shows the resulting dependence: \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}} = f({{{{T}_{{\text{c}}}}} \mathord{\left/ {\vphantom {{{{T}_{{\text{c}}}}} {\sqrt {{{P}_{{\text{c}}}}} }}} \right. \kern-0em} {\sqrt {{{P}_{{\text{c}}}}} }})\). We can see that the location of the points for strongly polar compounds relative to those for low-polar derivatives of thiophene differs from the similar graph in coordinates \({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) = f(αМ) (Fig. 3). Values \(\delta {{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) were also calculated (Table 2, column II). It can be seen from the presented data that values \(\delta {{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\) obtained on the basis of parameter \({{{{T}_{{\text{c}}}}} \mathord{\left/ {\vphantom {{{{T}_{{\text{c}}}}} {\sqrt {{{P}_{{\text{c}}}}} }}} \right. \kern-0em} {\sqrt {{{P}_{{\text{c}}}}} }}\) distinguish molecules of the considered adsorbates from their capability of specific interactions with the graphite surface much worse, since the values of Tc and Pc not only contain information about the ability of substances to disperse interactions, but also reflect the entire spectrum of possible intermolecular interactions. When determining the contribution from specific interactions to the total heat of adsorption, molecular polarizability αM is therefore the best correlated parameter.

Dependence of the heat of adsorption (\({{\bar {q}}_{{{\text{dif}}{\text{,1}}}}}\), kJ/mol) on critical parameters \({{T}_{{\text{c}}}}{\text{/}}\sqrt {{{P}_{{\text{c}}}}} \), K/bar0.5 of the studied derivatives of thiophene. White dots represent experimental values; black dots, values calculated with Eq. (5).
The mobility of the molecules of the studied thiophene derivatives on the surface of Carbopack C HT was estimated using the two-dimensional ideal gas model with values \(\Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\)(theor.) calculated according to the formula [30]
In this model, it is assumed that when molecules are adsorbed on a flat surface, they lose only one degree of freedom of translational motion directed perpendicular to the surface. Values \(\Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\) calculated in this way (theor.) are given in Table 4. It can be seen that \(\Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\) (exp.) is notably lower \(\Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\) (theor.), indicating part of the vibrational degrees of freedom are preserved when the adsorbate transitions from the equilibrium gas phase to the state adsorbed on the GTCB. It should be noted that the greatest differences in the values of \(\Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\) (exp.) and \(\Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\) (theor.) are observed for compounds where mainly dispersion interactions with the graphite surface are observed. At the same time, for compounds with several functional groups (8, 12–17) and bithiophene (9), value \(\delta \Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\) is comparable to the experimental error in determining \(\Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\) (the boundaries of the experimental error are indicated by the dashed line in Fig. 6), which makes the two-dimensional ideal gas model applicable to describing the adsorption of these compounds on graphite. We may assume the specific interaction between the polar groups and the graphite surface realized above prevents oscillations from being perpendicular to the flat surface of the adsorbent. In the adsorbed state, the molecules of thiophenes with polar groups are thus pressed more to the flat surface of Carbopack C HT, compared to low-polar compounds. We may conclude that for low-polar molecules of thiophene derivatives, the two-dimensional ideal gas model is a somewhat rough approximation. The latter was in good agreement with the results in [7] from analyzing quantities \(\Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\) for unsubstituted thiophene and 2,2′-bithiophene molecules.

Dependence of values \(\Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\) on the molecular weight of the studied derivatives of thiophene. White dots represent experimental values; black dots, values calculated with Eq. (6).
CONCLUSIONS
Under the conditions of equilibrium GAC on a Carbopack C HT grade hydraulic system in a wide temperature range, the TCA values of thiophene molecules and its various derivatives were determined for the first time. It was shown that the TCA values depend largely on the number and nature of the substituents in the main structural fragment. It was found that the surface of GTCB is characterized by low structural selectivity for positional isomers in the series of thiophene derivatives.
Substituted benzenes that are isostructural to the thiophene derivatives and have similar physicochemical characteristics are characterized by higher TCA values, due to the smaller contact area of the thiophene derivatives with a graphite-like surface as a result of the large Van der Waals dimensions of the S atom. Based on chromatographically determined heats of adsorption of nitro-, aceto- and acetamidothiophenes, the effect of polar retention on graphite from the gas phase was determined and studied for the first time by implementing additional polar intermolecular interactions of the polar groups in the adsorbate with the easily polarized surface of the base face of graphite, and the quantitative contribution from this effect to the heats of adsorption was established.
It was shown that the two-dimensional ideal gas model satisfactorily describes the experimental values \(\Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\) in the case of adsorption of polar molecules of thiophene derivatives on the surface of Carbopack C HT, however, this model has serious limitations to describe the mobility of thiophene molecules and its methyl and halogen derivatives and cannot be used for a priori calculation of quantities \(\Delta {{(\bar {S}_{{{\text{1}}{\text{,c}}}}^{{\text{o}}})}^{{\text{s}}}}\).
REFERENCES
A. V. Kiselev, I. A. Migunova, and Ya. I. Yashin, Zh. Fiz. Khim. 42, 1235 (1968).
P. A. Elkington and G. Curthoys, J. Phys. Chem. 78, 2321 (1969).
A. K. Buryak, P. B. Dallakyan, and A. V. Kiselev, Dokl. Akad. Nauk SSSR 282, 350 (1985).
P. B. Dallakyan, Extended Abstract of Cand. Sci. (Chem.) Dissertation (Mosc. State Univ., Moscow, 1986).
P. B. Dallakyan and A. V. Kiselev, Zh. Fiz. Khim. 59, 1277 (1985).
P. B. Dallakyan and A. V. Kiselev, Zh. Fiz. Khim. 59, 1278 (1985).
A. A. Lopatkin and P. B. Dallakyan, Russ. J. Phys. Chem. A 71, 1195 (1997).
V. G. Adeeva, M. S. Bobyleva, N. S. Kulikov, and V. G. Kharchenko, J. Chem. Soc., Perkin Trans. 2, No. 6, 965 (1992).
N. S. Kulikov and M. S. Bobyleva, J. Chem. Soc., Perkin Trans. 2, No. 3, 571 (2000).
N. S. Emel’yanova, Extended Abstract of Cand. Sci. (Chem.) Dissertation (Samar. State Univ., Saratov, 2013).
S. Bernes, M. M. Davila-Jimenez, M. P. Elizalde-Gonzalez, et al., Mater. Chem. Phys. 85, 347 (2004).
Experimental Methods in Adsorption and Molecular Chromatography, Ed. by Yu. S. Nikitin and R. S. Petrova (Mosk. Gos. Univ., Moscow, 1990) [in Russian].
S. N. Yashkin, D. A. Svetlov, O. V. Novoselova, and E. A. Yashkina, Russ. Chem. Bull. 57, 2472 (2008).
A. A. Lopatkin, Ross. Khim. Zh. 41 (3), 85 (1997).
S. N. Yashkin, Russ. Chem. Bull. 63, 582 (2014).
S. N. Yashkin, D. A. Svetlov, and B. A. Murashov, Russ. J. Appl. Chem. 86, 432 (2013).
The Chemistry of Heterocyclic Compounds, Vol. 44: Thiophene and Its Derivatives, Ed. by S. Gronowitz (Wiley, New York, 1991), p. 1.
Thermophysical Properties of Chemicals and Hydrocarbons, Ed. by C. L. Yaws (Elsevier, Amsterdam, 2014).
B. V. Aivazov, S. M. Petrov, V. R. Khairullina, and V. G. Yapryntseva, Physicochemical Constants of Organosulfur Compounds (Khimiya, Moscow, 1964) [in Russian].
R. Todeschini and V. Consonni, Handbook of Molecular Descriptors (Wiley-VCH, Weinheim, 2000).
www.chemspider.com.
Yu. V. Zefirov, Crystallogr. Rep. 42, 111 (1997).
O. A. Reutov, A. L. Kurts, and K. P. Butin, Organic Chemistry (Mosk. Gos. Univ., Moscow, 1999), Part 1, p. 78 [in Russian].
A. V. Kiselev and Ya. I. Yashin, Adsorption Gas and Liquid Chromatography (Khimiya, Moscow, 1979) [in Russian].
S. N. Yashkin, B. A. Murashov, and Yu. N. Klimochkin, Russ. J. Phys. Chem. A 85, 677 (2011).
C. West, C. Elfakir, and M. Lafosse, J. Chromatogr. A 1217, 3201 (2010).
L. Pereira, J. Liq. Chromatogr. Rel. Technol. 31, 1687 (2008).
J. H. Knox and P. Ross, Adv. Chromatogr. 37, 73 (1997).
G. I. Berezin, Dokl. Akad. Nauk 217, 843 (1974).
A. A. Lopatkin, Ross. Khim. Zh. 40 (2), 5 (1996).
ACKNOWLEDGMENTS
The authors are grateful to A.V. Yudashkin and V.V. Meshkova for providing samples of thiophene derivatives used in this work.
Funding
This work was supported by the Russian Foundation for Basic Research, project no. 16-43-630634 p_a; and by the RF Ministry of Science and Higher Education as part of State Task no. 4.6328.2017/8.9 for Samara State University, “Thermodynamics of Intermolecular Interactions in Systems with 2D and 3D Types of Structural Selectivity.”
Author information
Authors and Affiliations
Corresponding authors
Additional information
Translated by M. Aladina
Rights and permissions
About this article

Cite this article
Yashkin, S.N., Yashkina, E.A., Svetlov, D.A. et al. Adsorption and Chromatographic Separation of Thiophene Derivatives on Graphitized Thermal Carbon Black. Russ. J. Phys. Chem. 93, 2482–2489 (2019). https://doi.org/10.1134/S0036024419120355
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036024419120355