
Abstract
A comparison of the experimental IR absorption spectrum of the synthesized graphite oxide specimen with the calculated (using ab initio approaches) spectra of CnOmHk model systems containing the basic C54 fragment of the graphite layer and various modifying functional groups (hydroxyl, epoxy, carboxyl, and keto groups) in various numbers and combinations in the presence of a small number of solvating water molecules made it possible to clarify the nature of some signals in the medium and characteristic ranges of the IR spectrum of graphite oxide and reveal the role of hydrogen bonding between the functional groups and the water molecules.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
EXPERIMENTAL DATA AND STRUCTURAL MODELS OF GRAPHITE OXIDE
The development of both the experimental methods of structural studies and the computational schemes for ab initio modeling of the stationary fragments of complex macrosystems of an irregular structure in addition to modeling large individual molecules or molecular clusters and agglomerates has stimulated more detailed research into chemical systems of practical importance. The combination of experiment and theory is very effective in those cases when the independent use of these approaches does not lead to the construction of a sound correct model of a system or process because of some internal limitations. The problem of the structure of graphite oxide is a vivid example illustrating this.
More than a century that passed since 1855 when graphite oxide was first synthesized [1] has seen many times the growth, decline, and revival of interest in its structure and potential applications. In recent decades, the appearance of new methods of investigation or significant improvements in the accuracy of the existing methods have stimulated studies of graphite oxide, which revealed new structural features of this compound. The results of these studies served as a start for formulating new ideas regarding the mechanism of formation of graphite oxide, the role of certain reagents, the nature of intermediates (isolated under certain conditions), the structure of graphite oxide layers, and the nature of those functional groups that determine the properties of this substance.
Graphite oxide can hardly be called a compound because its composition significantly depends on the synthesis conditions: the size of the initial graphite particles, reagent mixtures used for their oxidation, their concentration, the ratio of the amounts of graphite and oxidant, duration of oxidation, and specimen washing procedure. There are three main methods for the synthesis of graphite oxide, which have many variations and modifications. Historically, Brodie’s method was the first procedure [1], in which potassium chlorate in fuming nitric acid (KClO3 + conc. HNO3) was used as the oxidant. In Staudenmaier’s method [2], potassium chlorate was mixed with concentrated nitric and sulfuric acids (KClO3 + conc. HNO3 + conc. H2SO4). The Hummers method [3] uses another oxidant—potassium permanganate in concentrated sulfuric acid in the presence of sodium nitrate (KMnO4 + conc. H2SO4 + NaNO3). As noted in [4], it is more correct to call it the Charpy–Hummers method as Charpy used the same mixture of potassium permanganate and sulfuric acid half a century before Hummers’ study [5]. In one of the rational modifications of the Charpy–Hummers method (which we conventionally call the Tour method), the oxidizing mixture was KMnO4 + 9 : 1 mixture of conc. H2SO4 and H3PO4 [6].
According to the data of [7], the C/O ratio in various graphite oxide specimens ranges from 3.5 (Brodie’s method) to 2.9 (Charpy–Hummers’ method), including the intermediate value 3.3 in Staudenmaier’s method. Thus, the most highly oxidized graphite specimens can be obtained using potassium permanganate and sulfuric acid in a faster (using small initial graphite particles) and environmentally friendly (without evolving nitrogen and chlorine oxides) process.
To separate graphite oxide from any of the indicated media, it is necessary to wash it for a long time in water; that is why it is almost impossible to obtain anhydrous (dry) graphite oxide: the specimens dried at room temperature over P2O5 contain at least 5–10% water. According to the recommendation given in [7], it is reasonable to use specimens with 10–17% water, which corresponds to the normal water vapor pressure in laboratory conditions. The presence of water affects both the estimated relative amount of carbon and oxygen-containing functional groups (when determining the composition of the substance) and the properties of the specimen. This complicates the determination of the types and relative number of functional groups in graphite oxide and introduces an uncertainty in the results of subsequent modification, whose mechanism evidently depends on the nature, number, and mutual arrangement (and influence) of functional groups and their availability (including in the structure with interfragment bonds and solvation shells).
The composition and structure of graphite oxide layers are generally determined by X-ray photoelectron spectroscopy (XPS), solid-state nuclear magnetic resonance spectroscopy (primarily, 13C SSNMR), and infrared (IR) absorption spectroscopy. Each of these methods has advantages and disadvantages in identification of the structural features and functional groups in graphite oxide specimens. However, none of them gives full information about the structure of graphite oxide, which has not yet been determined to a degree sufficient for correctly choosing the procedure for its purposeful modification.
XPS gives the general idea about the element composition of the specimen with details on different degrees of oxidation of atoms of each type under study, i.e., actually the variants of the nearest environment of atoms. An analysis of the surface (on average ~5 nm thick) layers of the specimen in the case of graphite oxide gives reliable information due to the layered structure of the material and presumably similar chemical modification of the outer and inner graphite layers during oxidation. The survey C 1s spectrum of almost any graphite oxide specimen has a characteristic structure with two main peaks, at 286.5 eV (dominant) and 284.5 eV (of lower integral intensity), and a shoulder at 288.2 eV. The main peak is attributed to epoxides and tertiary alcohols; the peak of lower intensity, to nonoxidized carbon; and the shoulder, to carboxyl groups, although it is not excluded that it is due to the superposition of the signals of ketones and carboxyls [4].
Several studies used a larger number of basis functions for deconvolution of the survey spectrum, especially in the range of the high-frequency shoulder, relating the responding peaks to the presence, of e.g., various carbonyl-containing functional groups (carboxyls, ketones, and carbonyls). This procedure has no strict criteria for selecting the necessary and sufficient number of basis functions; therefore, it is difficult to assess the correctness of interpretation variants. It is only clear that graphite oxide specimens always have fragments of nonoxidized graphite, dominant carbon atoms that form single bonds with oxygen atoms (C–O–C or C–OH), and atoms (in small numbers) that form C=O bonds. The nature of these groups is difficult to determine because, in the case of a fragment of the oxidized graphite sheet, their closest environment is certainly different from that in individual small organic molecules, for which the signals in the C 1s spectra are found in the range between 287.7 and 289.2 eV [8, 9].
More detailed information on the presence of various structural motifs in graphite oxide is obtained by SSNMR spectroscopy. The main signals at chemical shifts of 132 (130), 71.2 (70), and 60.2 (60) ppm were attributed to the nonoxidized carbon atoms and the atoms involved in the C–OH and C–O–C (1,2-ether or epoxy) fragments, respectively [10, 11]. According to [10, 11], the assignment to epoxides, but not 1,3‑ethers, is confirmed by the decreased intensity of the signals after treatment with potassium iodide, which reacts with epoxides, but not with 1,3-ethers. The signal of carboxyl carbon atoms (which was expected near 190 ppm) was not recorded; this is possible in the case when similar functional groups are absent or present in very small numbers. The total composition was determined to be C8(O)1.7(OH)1.7 [10]. In higher resolution spectra, three peaks were distinguished in addition to the indicated signals [12, 13]. These peaks were of lower intensity (by more than an order of magnitude) and were attributed to ketones (191 ppm), carboxyls (167 ppm), and lactols or geminal diols (101 ppm) [12]. The two-dimensional correlation SSNMR spectrum made it possible to identify the most probable, close-lying groups, namely, the sequences COH···COC···COC and COH···COH···COC; the number of COC···COC and COH···COH fragments directly bonded to nonoxidized carbon was approximately three times smaller [13]. There is still no direct evidence for the presence of the terminal phenol and carboxyl groups and ketones and for the relative number of defects and aromatic fragments that remained after oxidation.
IR spectroscopy is the third main source of information about the presence of certain functional groups in the structure of graphite oxide. The spectra of graphite oxide specimens are generally similar. They contain high-frequency (3600–2200 cm–1), medium-frequency (1730–1615 cm–1), and characteristic (1500–700 cm–1) ranges. In the high-frequency range the signals of the stretching vibrations of OH bonds should be present; the main contribution is from vibrations of water molecules because the use of deuterated water leads to a drastic decrease in the signal intensity in this range and the signal shifts to 2700–1900 cm–1 [14]. The peak around 1725 cm–1 is attributed to the stretching vibrations of carbonyl groups. The type of these groups (ketones, aldehydes, or carboxyls) cannot be determined. The signal at 1620 cm–1 is often related to the bending vibrations of water tightly bonded to the structure of graphite oxide. Finally, the characteristic range of the spectrum is the result of a superposition of a large number of vibrations of various functional groups, among which the signals of hydroxyl and epoxy groups are supposed to be dominant. As shown in our previous work [15], however, the intensity and frequency of signals in the absorption spectrum in the case of these two types of groups strongly depends on their mutual arrangement, the presence of hydrogen bonds between them, and the presence and amount of hydration water. Therefore, it is impossible to draw conclusions about the relative number of epoxy and hydroxyl groups in the structure of graphite oxide based on the intensity of certain signals in the characteristic range.
Thus, it is still almost impossible to accurately determine the types of functional groups and their mutual arrangement and relative number in the structure of graphite oxide despite the large amount of experimental information. One can only consider the most likely versions under certain assumptions. This is exactly what was done by the majority of researchers, who offered various structural models of graphite oxide for almost 90 years.
The first model proposed by Thiele [16] assumed the presence of exclusively hydroxyl groups located in pairs above and below the plane of the graphite sheet, which corresponded to the molecular formula C6(OH)3. A few years later, Hofmann et al. [17] proposed a radically different version: based on the average C/O ratio, which was close to three, and the moderate C–O bond energy, it was concluded that only epoxy groups were present in the structure of graphite oxide, and the presence of hydrogen was explained by tightly bound water. Later [18], these authors admitted that hydroxyl groups could appear during the hydrolysis of some epoxy groups, which were thus also included in graphite oxide. A decade later, Ruess [19] indicated that the ring opening reaction typical of epoxides did not occur in the case of graphite oxide and proposed that all epoxides be replaced by 1,3‑ether groups, while a small number of hydroxyl groups (tertiary alcohols) should be retained. As the exclusively tertiary alcohols could not explain the high acidity of graphite oxide specimens, Hofmann, Boehm, and Clauss [20] expanded the model; their model admits that the system has enol fragments, which is possible only if some C=C bonds in the carbon plane are broken, leading to ketones (resulting from the keto-enol rearrangement). A decade later, Scholz and Boehm [21] combined some previous ideas in fragments, admitting that graphite oxide contained relatively regularly alternating tertiary alcohols, ketones, as well as reduced and nonoxidized portions of the carbon plane. Two decades later, Nakajima et al. [22, 23] proposed a new model, which actually turned out to be an insignificant modification of the Thiele model: it admitted that a generally irregular structure contained predominantly hydroxyl groups, including partially deprotonated ones. Finally, after publishing the relatively reliable experimental NMR and IR spectral data of graphite oxide, Lerf and Klinowski [24] proposed a model that suggested the presence of irregularly arranged epoxy and hydroxyl groups, alternating with the nonoxidized regions of a graphite sheet, with carboxyl groups located on its periphery.
This model evidently did not add any novelty in the gradually emerging concept of the structure of graphite oxide. It turned out to be, in a sense, merely rationalization of preceding models: less probable functional fragments or those that were present in much smaller amount were excluded and those that were confirmed by almost all experimental methods at high probability were preserved. At the same time, some questions remain unanswered, for example, the question of why the epoxides whose signals were observed in both NMR and IR spectra were not involved in typical ring opening reactions. The same concerns the probability of structural motifs with enols or ketones in the inner part of a graphite sheet, i.e., the probability of local cleavages of individual C=C bonds or the formation of layer defects corresponding to the loss of one or more carbon atoms. These problems, together with the questions of which functional groups and in what relative amount are present at the outer edge of a carbon sheet, constitute the group of problems that are critical for correct determination of the structural motifs of graphite oxide and reliable prediction of conditions of its chemical modification.
It is impossible to solve this problem based on experimental data alone for reasons mentioned above (in the discussion of the physical methods of investigation), namely, it is impossible to unambiguously assign the spectral signals to definite functional groups because the signals can shift within the overlapping spectral ranges depending on the local environment and the presence of solvating water molecules. Moreover, any signal assignment is based on a comparison with the existing databases for known compounds, but all these compounds are relatively small and none of them has sufficiently long oxidized or nonoxidized areas of graphite sheet, which inevitably and in different ways affect the electron density distribution and, accordingly, the elasticity of bonds in the functional groups of interest. The degree and nature of this effect can be judged only from theoretical ab initio calculations of model systems, whose size makes it possible to take into account the main expected effects of the real structure. In the case of calculations, however, we also encounter some problems. It is almost impossible to calculate the mechanism and preferable routes of oxidation of graphite treated with a certain mixture (based on potassium chlorate or permanganate in the presence of concentrated sulfuric or nitric acid). It is necessary to construct very large models, which include significant areas of at least two graphite sheets, a large number of acid molecules that provide graphite exfoliation and reagent and water molecules. Moreover, this should be a dynamic simulation with imitation of certain physical (temperature) conditions. The latter is readily feasible in classical molecular dynamics of the canonical ensemble of molecules, which, however, does not allow the modeling of chemical transformations. To describe the electron density redistribution and cleavage/formation of chemical bonds, the variant of ab initio molecular dynamics is needed, in which the forces acting on the nuclei are determined by solving quantum-chemical equations, and which is thus applicable only to systems of limited size. As a result, when using the theoretical approach, researchers again have to admit (as an initial approximation) that certain functional groups are present on the surface of a carbon sheet without specifying the routes and conditions of their formation. The nature of these groups is inferred (as in the case of the creation of graphite oxide models) from the available experimental data and organic chemistry concepts. The above uncertainty of both sources of information necessitates the inclusion of various combinations of functional groups. The most probable combinations can be inferred only by comparing the theoretical results to similar data obtained in experiment and revealing similar motifs. This approach was just used in our study and made it possible to clarify some of the above problems of the structure of graphite oxide.
EXPERIMENT AND METHODS OF THEORETICAL MODELING
As mentioned in the previous section, when constructing a reliable model of graphite oxide, it is necessary to take into account not only the presence of certain functional groups, but also their possible combinations; when studying individual groups, it is necessary to consider different variants of the close neighborhood, and the boundary effects should not introduce noticeable perturbation in the simulated properties. This is possible for a sufficiently large model system. Therefore, as a basic structural unit we chose a fragment of 54 carbon atoms in the graphite sheet, which was a central hexagon surrounded by two full circular rows of structural hexagons. When studying the properties of functional groups on the inner surface of a carbon sheet, the external ring of 12 hexagons plays the role of a neighborhood, and the missing carbon atoms are replaced by the terminal hydrogen atoms. When studying the boundaries of a carbon layer, some of these hydrogen atoms are replaced by the functional groups of interest. The epoxy and hydroxyl groups can be located on the inner surface of a defect-free carbon sheet. Both the hydroxyl groups and ketones or carboxyls can be localized at the periphery of the layer. To find out which groups and in which combinations are present in real graphite oxide specimens, the calculated absorption spectra of model systems were compared to the recorded experimental spectrum of the graphite oxide specimen.
To exclude the contributions of terminal hydrogen atoms in the calculated spectrum and imitate the overall rigidity of the carbon framework at the stage of solving the vibrational problem, the corresponding atoms were replaced by effective particles with masses of 100 au. To take into account the error of the numerical estimates of frequencies in the harmonic approximation, a scaling factor of 0.98 was used, which seemed reasonable exactly for the characteristic frequency range. The signals of the calculated spectrum were broadened during visualization with Lorentzian functions with a half-height width of 50 cm–1 to simulate the real spectrum of a complex system at normal temperature.
The quantum-chemical calculations were carried out at the density functional approximation using the B3LYP exchange-correlation functional and the double-zeta basis of Gaussian atomic functions (6‑31G) augmented with polarization functions on all the atoms of the functional groups (6-31G(d,p)). This approach provides a correct description of molecular systems built of the atoms of the elements of the first and second rows, including the case with interfragment hydrogen bonds. The calculations were carried out with the Firefly [25] and GAMESS US [26] software packages using the computing resources of the supercomputer complex of the Moscow State University [27] and visualized using the Chemcraft software package [28].
Graphite oxide was obtained by the procedure proposed in [6]. Pure graphite (2 g) was added to a 9 : 1 mixture of concentrated sulfuric (240 mL, H2SO4, 95%) and phosphoric (26 mL, H3PO4, 85%) acids. Then potassium permanganate (12 g) was added in small portions to the mixture. The process lasted 24 h. The product was washed with water by centrifuging at a frequency of 10 000 rpm and dried at reduced pressure. The graphite oxide product was a greenish yellow-brown layered material.
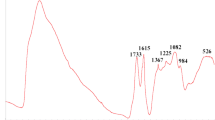
According to the XPS analysis, the specimen contained (at %) 67.9 C, 31.3 O, 0.4 N, 0.3 S, and 0.1 Mn and was characterized by the C/O ratio = 2.17, which corresponds to a rather high oxidation degree. The IR absorption spectrum of this specimen was recorded using a Nicolet IR200 Fourier transform spectrometer (Thermo Scientific) in the internal reflection mode (KBr) at an incidence angle of 45° (20 scans at a resolution of 4 cm–1). The spectrum is presented in Fig. 1.

(Color online) Experimental IR absorption spectrum of the synthesized graphite oxide specimen.
The recorded spectrum of graphite oxide contains all the three typical absorption ranges: high-frequency (3600–2400 cm–1), medium-frequency (with distinct peaks at 1733 and 1615 cm–1), and characteristic range, in which the most distinct peaks are those at 1367, 1225, 1082, and 984 cm–1. The nature of the peaks in the medium and characteristic ranges was of primary interest.
AB INITIO MODELING OF THE FRAGMENTS OF GRAPHITE OXIDE
The quantum-chemical calculations were carried out for model graphite oxide fragments constructed based on the experimental data on the composition of the specimens (C/O ratio) assuming that the fragments contain hydroxyl, epoxy, keto, and carboxyl groups and water molecules.
In the simplest C54H18O3 model (Fig. 2), where the basic C54 graphite fragment was modified by introducing three epoxy groups in the central part, the geometrical parameters of these –C–O–C– groups were close to those typical of small epoxides: the internuclear distance between the carbon nuclei was slightly shorter than the length of the single bond (1.53–1.54 Å), and the two C–O distances in each triangle were similar, 1.463 Å on the average. In the spectrum of this model system, all the lines have relatively low intensity, and the most noticeable signals are those at 1158, 1200, 1340, and 1661 cm–1. Note that around 980 cm–1, where the experimental spectrum contains a rather intense peak, which is often attributed precisely to the epoxy groups, the predicted probability of absorption of the model system is minimum. The signal at 1158 cm–1 corresponds to the breathing vibrations of the (mostly nonoxidized) carbon framework, in which its hexagonal rings are distorted, and to the similar distortions of the epoxy triangles. The bending vibrations of epoxides, caused by the distortion of the framework, are responsible for the noticeable signal intensity due to the displacements of oxygen nuclei, which lead to a change in the dipole moment of the structure. The nature of the signals at 1200 and 1340 cm–1 is similar. The former signal corresponds to the distortions of the carbon framework promoted by the rocking vibrations of the epoxy groups attached to it. The latter signal corresponds to the distortion of the framework, which correlates with the asymmetric deformation of the epoxy triangles. However, the resulting signal intensity is markedly lower than that of the 1158 cm–1 peak in both cases due to the predominantly tangential displacement of oxygen nuclei relative to the surface. Finally, the signal at 1661 cm–1 corresponds exclusively to the stretching vibrations of isolated C–C bonds. Note that of all the above signals, only the peak at 1340 cm–1 lies in the vicinity of the experimental signal of noticeable intensity, and all the peaks, including this one, are almost an order of magnitude weaker than the experimental ones in the medium-frequency and characteristic spectral ranges. This means that individual epoxy groups that modify the graphite plane cannot lead to the appearance of the characteristic lines in the absorption spectrum of graphite oxide.

(Color online) Structure of the C54H18O3 model system and its calculated absorption spectrum in the range 2000–500 cm–1.
Another (in a certain sense limiting) variant of the model structure is a graphite fragment modified exclusively with hydroxyl groups in such a way that the groups are arranged in pairs above and below its plane and do not form hydrogen bonds with one another because of being far apart, with only one exception (С54H18(OH)8, Fig. 3). If two OH groups are attached to the neighboring carbon nuclei, while being located on the opposite sides of the carbon plane, they have the same geometrical parameters: the C–O distances are 1.46 Å, and the O–H distances are 0.97 Å. In those situations when the OH group is either removed to a greater distance from the neighboring groups or involved in the hydrogen bond with the group closest to it, its configuration is distorted more significantly: the C–O distance increases to 1.49–1.50 Å, which inevitably affects the stretching vibration frequency.

(Color online) Structure of the C54H18(OH)8 model system and its calculated absorption spectrum in the range 2000–500 cm–1.
The signal intensities are expected to be significantly higher if the model spectrum contains the signals of hydroxyl groups: they increase almost by half an order compared to the intensities in the C54H18O3 system. An intense peak appears at 987–942 cm–1, whose intensity is half of the integral intensity of the 1176 cm–1 signal; the strongest signal appears at 1372 cm–1; and, finally, there is a weak peak at 1596 cm–1 (its intensity is comparable to that in the spectrum of the system containing epoxides). Note that the latter peak is of the same nature as the signal in the medium-frequency range of the C54H18O3 system; i.e., it corresponds to the vibrations of individual C=C bonds, and, as in the latter system, its intensity is markedly lower than the characteristic intensity of the experimental spectrum. This confirms the hypothesis that the band in the medium-frequency range of the spectrum may also be due to the stretching vibrations of C=C bonds, but their contribution is not dominant. The signals in the characteristic range are of the following nature. The most intense signal at 1372 cm–1 corresponds to the breathing vibrations of the carbon skeleton and the related bending vibrations of hydroxyl groups, which greatly change the dipole moment, thus leading to high probability of the corresponding vibrational excitation. Finally, the signal in the range 987–942 cm–1 that is close to it in intensity is also due to the bending vibrations of hydroxyl groups, but these are coupled to the local distortions of the pyramidal fragments of the C4 surface.
The experimental absorption peak around 980 cm–1, assigned usually to epoxides, possibly has a completely different nature; therefore, its presence cannot be regarded as evidence for the presence of a significant number of epoxy groups in the structure of graphite oxide. The experimental band with a peak at 1367 cm–1 can also have a significant contribution from the vibration excitations of hydroxyl groups, when their concerted deformation is promoted by the corresponding distortion of the carbon surface between them.
When both types of functional group (hydroxyl and epoxy groups) are included in the model system, the result (the absorption spectrum) significantly depends on the extent of interaction of these groups. The С54H18O3(OH)6 system (Fig. 4) was constructed in such a way that all the epoxy and hydroxyl groups were distant from one another, with three hydroxyl groups located on the edge of the carbon fragment and three in its inner part; two epoxy triangles were located on the common sides of the peripheral carbon hexagons, and one on the common side of the hexagons of the outer and inner rings. It is noteworthy that the first two epoxy groups are highly asymmetric: one C–O distance is 1.45 Å, and the other is 1.48–1.51 Å; due to this they are closer in shape to the deprotonated hydroxyl groups suggested in the model of [22, 23]. The third –C–O–C group is similar to 1,2-ether rather than epoxide because of the increased C–C distance of 1.56 Å at C–O distances still equal to 1.46 Å. The calculated absorption spectrum of this system comprises four most intense peaks in the characteristic range (1370, 1320, 1037, and 886 cm–1) and two peaks of much lower intensity around 1595 and 1171 cm–1. As in the above model systems, the latter peaks correspond to the stretching vibrations of individual C–C bonds and bonded hexagonal rings. The peaks at 1370 and 1320 cm–1 are due to the excitation of the bending vibrations of the peripheral and internal hydroxyl groups, respectively, strongly coupled to the asymmetric distortions of the C3 or C4 pyramids in both cases. The excitation of similar vibrations of the peripheral hydroxyl groups, but coupled to the symmetric distortion of the C3 or C4 carbon pyramids, is responsible for the signals at 1037 and 886 cm–1, which fall in the range of noticeable absorption in the experimental spectrum, but are shifted by 50 and 100 cm–1 to the low-frequency range relative to the main peaks. In the latter case, there is also a small contribution of the distortions of epoxy (1,2-ether) triangles, which cannot determine the high integral intensity of the spectral signal.

(Color online) Structure of the C54H18O3(OH)6 model system and its calculated absorption spectrum in the range 2000–500 cm–1.
The С54H8O2(OH)14(H2O) system (Fig. 5) was constructed in a radically different way. The number of hydroxyl groups in it is significantly increased, and many of them are involved in hydrogen bonds. Of the eight hydroxyl groups located on the periphery of the carbon sheet, two pairs are complemented with close-lying keto groups, and four pairs form a hydrogen-bonded chain. Another six hydroxyl groups are localized in the inner part of the carbon fragment, three on each side of its middle plane; they form an H-bonded triangle in one case and an H-bonded chain with an additional water molecule in the other. The calculated spectrum of this model system is radically different from the spectra discussed above. The intensities of all the signals are higher by almost an order of magnitude. The highest peaks are those at 1403 and 1340 cm–1; the intensity of the 1611 cm–1 signal is half the intensity of these peaks, but still much higher than previously; and the peak at 1136 cm–1 is lower, but still of noticeable height. The significantly increased signal at 1611 cm–1 is due to the excitation of not only the stretching vibrations of C=C bonds, but also of the C=O stretching vibrations of ketones and, to a lesser extent, the bending vibrations of hydroxyl groups directly H-bonded to ketones.

(Color online) Structure of the C54H8O2(OH)14(H2O) model system and its calculated absorption spectrum in the range 2000–500 cm–1.
It is the vibrations of the keto and hydroxyl groups that determine the multiply increased intensity of the spectral signal due to noticeable changes in the dipole moment of the structure. As for the most intense peaks at 1403 and 1340 cm–1 of the calculated spectrum, both are due to the bending vibrations of hydroxyl groups that form hydrogen bonds with neighbors. Thus, the peak at 1403 cm–1 corresponds to the concerted vibrations of three close-lying hydroxyl groups, which form a hydrogen-bonded triangle in the inner part of the structure. The peak at 1340 cm–1 corresponds to the vibrations of peripheral hydroxyl groups involved in hydrogen bonds either with each other or with the keto group. In all cases, the chains of hydrogen bonds meet the criterion of conjugation [29], when covalent (O–H) and hydrogen (H···O) bonds regularly alternate. This conjugation facilitates the concerted displacements of the protons of hydrogen bonds, increasing the probability of the corresponding excitation due to the multiply increasing local change in the dipole moment of the structure. In view of the comparable intensities of lines in the medium and characteristic ranges of the experimental spectrum and the fact that the number of keto groups located predominantly on the boundaries of carbon sheets should be noticeably smaller than the number of hydroxyl groups per unit surface, it seems reasonable to draw the following conclusion: it is relatively unlikely that the structure of graphite oxide include fragments with hydroxyl groups involved in the conjugated chains of hydrogen bonds. Otherwise, the intensity of the signals in the characteristic range would be significantly higher than that observed in experiment.
Finally, for more correct understanding of the localization and mutual arrangement of various oxygen-containing functional groups in graphite oxide specimens, we considered С54H10O(OH)5(COOH)5 model system (Fig. 6). In addition to the hydroxyl groups located both in the inner part of the structure and on the boundary of the carbon sheet (and forming only local hydrogen bonds), the model included peripheral carboxyl groups, lying either near each other or near the keto or hydroxyl group. The calculated spectrum of this system involves the following peaks: 1733 and 1672 cm–1 in the medium range and 1539, 1448, 1380, 1244, and 1141 cm–1 in the characteristic range.

(Color online) Structure of the C54H10O(OH)5(COOH)5 model system and its calculated absorption spectrum in the range 2000–500 cm–1.
As would be expected, the vibrations of carboxyl groups make a significant contribution to the signals in the medium part of the spectrum. The combined vibrations of carboxyl groups are observed at a higher frequency, when the stretching of the C=O fragment (not involved in hydrogen bonds) is coupled to the change in the C–O–H angle regardless of whether the OH fragment is involved as a donor or acceptor in the hydrogen bonds with the neighboring groups or not. At a lower frequency (1672 cm–1) the spectrum again contains the signal of the strongly coupled stretching vibrations of the C=C bonds of the framework and the bending vibrations of the peripheral hydroxyl groups and the carboxyl group, in which the C=O fragment as a proton acceptor forms a bond with the neighboring hydroxyl group.
The signal in the vicinity of 1540 cm–1 is due to the bending vibrations of the peripheral H-bonded C–O–H fragments of either hydroxyl or carboxyl groups, strongly coupled to the distortions of the carbon framework. The weaker 1450 cm–1 signal is similar in nature, but it is largely due to the bending of hydroxyl groups on the inner surface of the carbon sheet. The deformations of the C–O–H fragments of carboxyl groups also make a significant contribution to the 1380 cm–1 signal, but in this case, these movements are strongly coupled not only to the delocalized distortions of the carbon framework, but also to the local stretching of the corresponding C‒C(O)(OH) bonds. Finally, the signals in the vicinity of 1140 cm–1 correspond to the breathing vibrations of the C6 carbon rings, when the expansion of the rings connected in one band is accompanied by a compression of the similar band of adjacent rings, and this alternation occurs within the whole carbon frame, initiating the distortion of hydroxyl fragments on its periphery.
Thus, in the absence of epoxy groups and H‑bonded hydroxyl groups, the calculated spectrum of the model system has no peaks in the range 980–1080 cm–1. The inclusion of carboxyl groups in the system leads to the appearance of more distinct signals in the range 1240–1380 cm–1, where the deformation of their C–O–H fragments strongly couples to the more or less local distortions of the carbon framework, and at 1730 cm–1, where the stretching of the C=O fragment of these groups that is not involved in hydrogen bonds is coupled to a change in the corresponding C–O–H angle.
CONCLUSIONS
Quantum-chemical modeling of СnHp(O)k(OH)l(COOH)m systems that imitate the structure of the local fragments of the oxidized graphite sheet, including epoxy and hydroxyl groups in its inner part and hydroxyl, carboxyl, and keto groups on the boundary, allowed us to clarify the nature of the majority of signals in the experimental absorption spectrum of graphite oxide in its medium and characteristic ranges.
In the characteristic range of the spectrum, the peak in the vicinity of 980 cm–1 cannot be caused by excitation of the vibrations of individual epoxy groups. A considerable contribution to this signal is made by the bending vibrations of hydroxyl groups located in the inner part of the carbon sheet, which are strongly coupled to the distortions of the corresponding C4 pyramids. In the vicinity of 1100 cm–1, the excitation of the breathing vibrations of the nonoxidized carbon framework and the distortion of the peripheral hydroxyl groups initiated by it can show itself. The signal with a peak at 1370 cm–1 should have a considerable contribution from the bending vibrations of the C–O–H fragments of the carboxyl groups, which interact with the stretching vibrations of the corresponding C–C(O)(OH) bonds and the distortions of the carbon framework, both local (individual C=C bonds) and those that embrace relatively large areas of the nonoxidized carbon surface.
In the medium part of the spectrum, the 1615 cm–1 peak is due to the stretching vibrations of the carbon–carbon bonds, but its high intensity is due to the coupled stretching vibrations of the C=O fragments and the bending of the hydroxyl groups H-bonded to them. In the absence of carbonyl fragments in the system, the signal in this spectral range has a very low intensity. The integral absorption at higher frequencies (the 1733 cm–1 peak) is due to the vibrations of carboxyl groups, which can be represented by a superposition of C=O bond stretching and the change in the C–O–H angle.
REFERENCES
M. B. C. Brodie, Ann. Chim. Phys. 45, 351 (1855).
L. Staudenmaier, Ber. Dtsch. Chem. Ges. 31, 1481 (1898).
W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc. 8, 1339 (1958).
Graphene Oxide: Fundamentals and Applications, Ed. by A. M. Dimiev and S. Eigler (Wiley, Chichester, 2017).
G. Charpy, C. R. Hebd. Sêances Acad. Sci. 148, 920 (1909).
D. C. Marcano, D. V. Kosynkin, J. M. Berlin, et al., ACS Nano 4, 4806 (2010).
H. P. Boehm and W. Scholz, Liebigs Ann. Chem. 691, 1 (1966).
D. Briggs, in Handbook of X-ray Photoelectron Spectroscopy, Ed. by C. D. Wagner, W. M. Riggs, L. E. Davis, J. F. Moulder, and G. E. Muilenberg (Perkin-Elmer, Eden Prairie, 1979), Vol. 3.
G. B. Hofflund, in Handbook of Surface and Interface Analysis (Marcel Dekker, New York, 1998).
M. Mermoux, Y. Chabre, and A. Rousseau, Carbon 29, 469 (1991).
A. Lerf, H. He, M. Forster, and J. Klinowski, J. Phys. Chem. B 102, 4477 (1998).
W. Gao, L. B. Alemany, L. Ci, and P. M. Ajayan, Nat. Chem. 1, 403 (2009).
L. B. Casabianca, M. A. Shaibat, W. W. Cai, et al., J. Am. Chem. Soc. 132, 5672 (2010).
A. M. Dimiev, L. B. Alemany, and J. M. Tour, ACS Nano 7, 576 (2013).
E. A. Shilyaeva and Yu. V. Novakovskaya, Struct. Chem. 30 (2), 583 (2019).
H. Thiele, Z. Anorg. Allg. Chem. 190, 145 (1930).
U. Hofmann, A. Frenzel, and E. Csalan, Liebigs Ann. Chem. 510, 1 (1934).
U. Hofmann and E. Konig, Z. Anorg. Allg. Chem. 234, 311 (1937).
G. Ruess, Monatsh. Chem. 76, 381 (1947).
A. Clauss, R. Plass, H. P. Boehm, and U. Hofmann, Z. Anorg. Allg. Chem. 291, 205 (1957).
W. Scholz and H. P. Boehm, Z. Anorg. Allg. Chem. 369, 327 (1969).
T. Nakajima, A. Mabuchi, and R. Hagiwara, Carbon 26, 357 (1988).
T. Nakajima and Y. Matsuo, Carbon 32, 469 (1994).
A. Lerf, H. He, M. Forster, and J. Klinowski, J. Phys. Chem. B 102, 4477 (1998).
A. A. Granovsky, Firefly, Vers. 8. http://classic.chem.msu.su/gran/firefly/index.html.
M. W. Schmidt, K. K. Baldridge, J. A. Boatz, et al., J. Comput. Chem. 14, 1347 (1993).
V. Sadovnichy, A. Tikhonravov, Vl. Voevodin, and V. Opanasenko, in Contemporary High Performance Computing: From Petascale toward Exascale (CRC, Boca Raton, FL, 2013), p. 283.
Chemcraft–Graphical Software for Visualization of Quantum Chemistry Computations. https://www.chemcraftprog.com.
Yu. V. Novakovskaya, Struct. Chem. 23, 1253 (2012).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by L. Smolina
Rights and permissions
About this article

Cite this article
Shilyaeva, E.A., Novakovskaya, Y.V. Functional Groups of Graphite Oxide: Experimental Data and Ab Initio Modeling. Russ. J. Phys. Chem. 93, 1908–1917 (2019). https://doi.org/10.1134/S0036024419100273
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036024419100273