
Abstract
Phosphorescence data on boron difluoride β-diketonates of various structure have been systematized. Nonplanar boron difluoride molecules are characterized by the inversion of the S1 and T2 levels, which promotes efficient population of triplet levels and intense phosphorescence or delayed fluorescence of crystals. Planar molecules are characterized by a classical sequence of singlet and triplet levels and a coplanar arrangement of antiparallel molecules, which contributes to excimer delayed fluorescence.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Organic dyes exhibiting room-temperature phosphorescence and delayed fluorescence have been actively studied [1–5]. Such materials are candidates for use in design of organic light emitting diodes (OLEDs), chemosensors, temperature sensors, bioimaging, etc. Organic photoluminescent materials are usually considered non-phosphorescent due to extremely weak spin–orbit coupling and high sensitivity to temperature and oxygen access [6]. Boron difluoride β-diketonates, as a rule, exhibit phosphorescence only at low temperatures, when the nonradiative relaxation of the triplet state is inhibited by the low mobility of the dye molecule [7, 8]. For the development of optical functional materials with intense phosphorescence at room temperature, it is important to reveal the relationship between the electronic and geometric structure of phosphors and the efficiency of population of the triplet states of molecules [9, 10].
Recently, the room-temperature phosphorescence of boron β-diketonates has been reported [11, 12]. Boron difluoride β-diketonates, unlike most organic compounds, brightly luminesce not only in solution, but also in crystals [13, 14]. Due to their bright luminescence and good photostability in the crystalline state, boron difluoride β-diketonates can be used as a phosphor in OLEDs [15]. Among boron difluoride β-diketonates, there are compounds that have luminescence from bright blue to infrared in the crystalline state. A characteristic feature of boron complexes is the formation of molecularly organized systems in crystals, including dimers, trimers, and aggregates of conjugated molecules, the supramolecular architecture of which leads to the emergence of a unique complex of intermolecular interactions, including dimeric (excimeric), interdimeric, and stacking interactions. In this regard, to understand the features of the luminescence of crystalline boron difluoride β-diketonates, it is necessary to compare their spectral and structural characteristics.
This paper presents the results of a comparative analysis of the structure and luminescent properties of some boron difluoride β-diketonates.
EXPERIMENTAL
All compounds were isolated as single crystals, and their unit cell parameters corresponded to the literature data cited in Table 1. The complexes were synthesized and purified in accordance with known procedures [16–22]: by the reaction of the corresponding β-diketones with boron trifluoride etherate (complexes 2 and 9) [16]; by acylation of aromatic compounds with acetic anhydride and gaseous boron trifluoride (complexes 1 and 3–5) [17]; by acylation of α- and β-naphthols with acetic anhydride and boron trifluoride bis-acetate (complexes 6 and 7) [18]; by acylation of para-acetoxyacetophenone with acetic anhydride and boron trifluoride α-etherate (complex 8) [19]; by acylation of α- and β-naphthols with acetic anhydride and boron trifluoride etherate (complexes 10 and 11) [20]; by the reaction of ortho-hydroxydibenzoylmethane with boron trifluoride etherate and butyl borate (complex 12) [21]; by acylation of anthracene with acetic anhydride and gaseous boron trifluoride (complex 13) [22].

1: Colorless crystals, mp 103–104°С. IR (KBr), ν, cm–1: 1611, 1590 (С6Н4), 1554, 1541 (С=О, С=С), 1156, 1138 (B–F), 1072, 1050 (В–О). For С13H15BF2O2 anal. calcd., %: С, 61.94; Н, 6.00. Found, %: С, 61.36; Н, 6.09.
2: Colorless crystals, mp 155–156°C. IR (KBr), ν, cm–1: 3138 (OHas), 2928 (C–HPh), 1597 (C=O), 1547(C=C), 1439 (CH3), 1367, 1358 (δs(B–O)), 1156, 1138, 1113 (δ(B–F)), 1090, 1057 (δas(B–O)). For C10H9BF2O2 anal. calcd., %: С, 55.72; Н, 4.78. Found, %: С, 55.96; H, 4.82.
3: Colorless acicular crystals, mp 158–159°С. IR (KBr), ν, cm–1: 1610, 1592 (С6Н4), 1550, 1510 (С=О, С=С), 1164, 1143 (B–F), 1085, 1050 (В–О). For С11H11BF2O2 anal. calcd., %: С, 58.02; Н, 4.95. Found, %: С, 59.32; Н, 4.89.
4: Pale yellow translucent crystals, mp 147–149°С. IR (KBr), ν, cm–1: 1606 (С6Н4), 1561, 1511 (С=О, С=С), 1222, 1189 (B–F), 1085, 1053 (В–О). For С11H11BF2O3 anal. calcd., %: С, 54.82; Н, 4.62. Found, %: С, 54.55, Н, 4.70.
5: Yellow-orange crystals, mp 260–261°С. IR (KBr), ν, cm–1: 1560, 1543 (С=О, С=С), 1152, 1128 (B–F), 1059, 1024 (В–О). For С17H13BF2O2 anal. calcd., %: С, 68.50; Н, 4.40. Found, %: С, 68.63; Н, 4.36.
6: Pale yellow translucent crystals, mp 153–155°C. IR (KBr), ν, cm–1: 3145, 3062 (С–HAr), 1627 (C10H7), 1598, 1541 (C=O, C=C), 1373 (B–O), 1184, 1155 (B–F), 1078, 1053 (B–O). For C14H11BF2O2 anal. calcd., %: C, 64.66; H, 4.26. Found, %: C, 64.45; H, 4.22.
7: Bright yellow crystals, mp 182–183°C. IR (KBr), ν, cm–1: 3145, 3064 (С–HAr), 1627 (C10H7), 1598, 1542 (C=O, C=C), 1373 (B–O), 1184, 1153 (B–F), 1078, 1049 (B–O). For C14H11BF2O2 anal. calcd., %: C, 64.66; H, 4.26. Found, %: C, 64.40; H, 4.28.
8: Pale yellow crystals, mp 160–161°C. IR (KBr), ν, cm–1: 1756 (СH3C=O), 1586 (C6H4), 1543, 1506 (C=O, C=C), 1350 (B–O), 1197, 1172 (B–F), 1047, 1012 (B–O). For C12H11BF2O4 anal. calcd., %: С, 53.78; Н, 4.14. Found, %: С, 53.74; Н, 4.12.
9: Pale yellow translucent crystals, mp 190–191°C. IR (KBr), ν, cm–1: 1595 С6H5), 1547, 1488 (C=O, C=C), 1373 (B–O), 1247, 1153 (B–F), 1103, 1045 (B–O). For C15H11BF2O2 anal. calcd., %: С, 66.22; Н, 4.08. Found, %: С, 66.28; Н, 4.11.
10: Orange crystals, mp 193–194°C. IR (KBr), ν, cm–1: 1619, 1599 (C10H7), 1565, 1530, 1459 (С=О, С=С), 1200–1170 (B–F), 1062–1044 (B–O). For C12H9BF2O2 anal. calcd., %: С, 61.59; Н, 3.88. Found, %: С, 61.61; Н, 3.92.
11: Pale yellow translucent crystals, mp 156–157°C. IR (KBr), ν, cm–1: 1627, 1580 (C10H7), 1545, 1490, 1474 (С=О, С=С), 1200–1190, 1062 (B–F, B–O). For C12H9BF2O2 anal. calcd., %: С, 61.59; Н, 3.88. Found, %: С, 61.56; Н, 3.90.
12: Pale yellow crystals, mp 195–196°C. IR (KBr), ν, cm–1: 3459 (O–H), 1624 (C6H5, C6H4), 1566, 1539 (C=O, C=C), 1157, 1134 (B–F), 1083, 1049 (B–O). For C15H11BF2O3 anal. calcd., %: C, 62.54; H, 3.85. Found, %: C, 62.36; H, 3.74.
13: Brown crystals, mp 228–229°С. IR (KBr), ν, cm–1: 1626 (С10Н7), 1571, 1541 (С=О, С=С), 1190, 1167 (B–F), 1086, 1065 (В–О). For С18H13BF2O2 anal. calcd., %: С,69.23; Н, 4.26. Found, %: С, 69.27; Н, 4.23.
Melting points were determined on a Buchi Melting Point B-540 instrument. IR spectra were recorded on a Perkin Elmer Spectrum BX 400 FT IR spectrophotometer. Luminescence and luminescence excitation spectra were recorded on a Shimadzu RF 5301 spectrofluorimeter. Phosphorescence spectra and lifetimes at room temperature were recorded on a Agilent Technologies Varian Cary Eclipse spectrometer and at 77 K, on a Fluorolog 3 spectrometer.
X-ray single-crystal diffraction study of crystal 8 recrystallized from acetonitrile was performed on a Bruker SMART-1000 CCD diffractometer with МоKα radiation at 296(2) K. The collection of experimental data from the sample was carried out in three groups of 906 frames each at angles φ = 0°, 90°, and 180° by ω-scanning with a step of 0.2° and a counting time of 20 s per frame. The unit cell parameters were refined, and the integrated intensities were recalculated into structural amplitudes using the APEX2 software package [23].
The structure of 8 was determined by direct methods with subsequent refinement of the positional and thermal parameters in the anisotropic approximation for all non-hydrogen atoms using the SHELXTL software [24]. The positions of hydrogen atoms were determined and refined as riding on their parent atoms with a C–H distance of 0.93 Å.
Crystallographic data for C12H11O4BF2: a prism (0.35 × 0.16 × 0.10 mm), monoclinic system, space group Cc, a = 11.6171(6) Å, b = 11.5684(6) Å, c = 10.4190(5) Å, β = 122.097(2)°, V = 122.097(2) Å3, Z = 4, ρcalc = 1.501 g/cm3, μ = 0.130 mm–1, MoKα radiation (λ = 0.71073 Å), data were collected in θ range 2.717°–31.103°, the number of measured reflections (Rint) was 13780, the number of reflections with I ≥ 2σ(I) was 3630, the number of refined parameters was 174; for I > 2σ(I), R1 = 0.0274, wR2 = 0.0762; for all reflections, R1 = 0.0291, wR2 = 0.0776.
The crystallographic information was deposited with the Cambridge Crystallographic Data Center under the number CCDC 2225753, from where it can be obtained upon request at the website: www.ccdc.cam.ac.uk/data_request/cif.
RESULTS AND DISCUSSION
To study the relationship between the crystal structure and luminescent properties, 13 brightly luminescent crystalline compounds were selected. The crystal structures of twelve of them have been studied earlier (references to the corresponding papers are given in Table. 1).
Figure 1 shows the molecular structure of complex 8 studied in this work. On the one hand, this compound can be considered as an acetate and, on the other hand, as boron difluoride β-diketonate with an acceptor substituent. The following features should be noted: the molecule is nonplanar, the boron atom and the central carbon atom of the diketonate ring are above the plane of the molecule to give a boat-type conformation. The acetate group is in the para-position of the phenyl ring, the angle between the planes of the phenyl ring and the acetate group is 60°; with this inclination, the n electrons of the O(3) atom weakly interact with the π electrons of the phenyl ring. Molecule 8 contains three atoms that can form hydrogen bonds: fluorine atoms and an oxygen atom of the carboxyl group, which contributes to efficient intermolecular interaction. In the crystal structure of 8, the molecules are arranged in stacks parallel to the a axis. Inside the stack, a π–π stacking interaction is observed between the phenyl ring of one molecule and the chelate ring of the other (Fig. 1b). Interacting molecules are also connected with neighboring ones by π–π stacking interactions to give a structure of the “skew ladder” type, which is characteristic of J-aggregates. The stacks are interconnected by С–F···H and C=O···H bonds (Fig. 1b), which are typical of crystals of organofluorine compounds [25–27].

Crystal structure of complex 8: (a) molecular structure and (b) stack fragment of the crystal.
For the studied crystals 1–13, phosphorescence was detected, and for some of them, delayed fluorescence was also observed. Two pairs of isomers (6 and 7, 10 and 11) and compounds 1 and 2, which have the same structure of the π-system of the molecule, stand out among the studied compounds (Fig. 2). With the same number of π electrons at identical atoms, the compounds differ in geometric structure: in molecules 1, 6, and 11, the aromatic substituent is at an angle to the chelate ring, while molecules 2, 7, and 10 of their analogs are flat.

Structures of complexes 1 and 2, 6 and 7, and 10 and 11.
Using the example of compound 2, which has planar molecules, and compound 1, the phenyl ring of which is turned by 55° relative to the chelate ring, let us consider the influence of the geometric factor on the deactivation of the excited state in crystals. The presence of methyl substituents in the ortho positions of the phenyl ring in 1 leads to a different type of crystal packing compared to 2 and, as a result, to different types of intermolecular interactions [28].
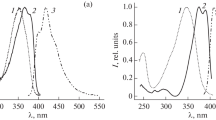
In crystal structure 1, the phenyl rings of neighboring molecules are arranged to each other in a T-shape, and there is no overlap of the π-systems of neighboring molecules. In crystal structure 2, molecules are stacked parallel to the c axis, with an intermolecular distance of 3.44 Å, and there is an efficient π–π stacking interaction. Different types of interactions in crystals 1 and 2 lead to the formation of luminescent centers of different nature. Figure 3 shows the fluorescence and phosphorescence spectra of crystals 1 and 2. For crystals 1, monomer fluorescence (λmaх = 395 nm) and phosphorescence (λmaх = 535 nm, τ = 0.20 µs) are observed (Fig. 3). In the phosphorescence spectrum of 2, two bands are observed with maxima at 480 and 600 nm, which are related to delayed excimer fluorescence and phosphorescence, respectively.

Luminescence spectra of crystals (1, 3) 1 and (2, 4) 2: (1, 2) fluorescence and (3, 4) phosphorescence.
The geometry of isomers 10 and 11 is also different. In crystal structure 10, molecules are planar and stacked with efficient π-stacking interaction resulting in excimer luminescence peaking at 523 nm. Molecules in structure 11 have a bend along the line boron–central carbon atom of the chelate ring and can form only dimers [20]; the maximum of the luminescence spectrum of crystals at 450 nm corresponds to monomer fluorescence. For 10, phosphorescence and delayed fluorescence are absent. For 11, phosphorescence is observed with a maximum at 590 nm (Fig. 4).

Luminescence spectra of crystals (1, 2) 11 and (3) 10: (1, 3) fluorescence and (2) phosphorescence.
To reveal the origin of the different ways of deactivation of the electronic excitation energy in the studied isomers, quantum-chemical simulation of molecules 10 and 11 has been performed [29]. For 11, in contrast to 10, an inversion of the S1 and T2 levels is observed during vibrational relaxation from the geometry S0 to the optimal geometry S1, which is determined by the intersection of the potential energy surfaces in the states S1 and T2. Then, internal conversion T2→T1 follows, and emission is observed during the T1→S0 transition (Fig. 5a).

Diagram of photophysical processes in crystals of (a) nonplanar and (b) planar molecules of boron difluoride β-diketonates.
For crystals 6 and 7, in addition to phosphorescence, delayed fluorescence is observed. For crystals 7, low-intensity delayed fluorescence and bright phosphorescence are observed, while for crystal 6, on the contrary, the spectrum is dominated by a delayed fluorescence band coinciding with the fluorescence band of a dilute solution [18] (Fig. 6).

Luminescence spectra of crystals: (1) fluorescence of 6, (2) delayed fluorescence of 6, (3) fluorescence of 7, and (4) phosphorescence of 7.
To reveal the origin of the different ways of deactivation of the electronic excitation energy in the studied isomers, quantum-chemical simulation of molecules 6 and 7 has been performed [18]. For 6, in contrast to 7, an inversion of the S1 and T2 levels is observed during vibrational relaxation from the S0 geometry to the optimal S1 geometry, which causes the intersection of the potential energy surfaces in the S1 and T2 states (Fig. 5a). For 6, the inversion of the S1 and T2 levels upon relaxation from the S0 geometry to the optimal S1 geometry contributes to the population of the T2 level, phosphorescence through the T2→T1→S0 channel, and delayed fluorescence through the T2→S1→S0 channel. The delayed fluorescence of crystals 6, like the fluorescence of 7, is monomer one [18] (Fig. 6).
For 7, the classical arrangement of singlet and triplet levels is observed (Fig. 5b). The T2 level is populated from S1 (T2→T1→S0). The fluorescence of crystals 7 (530 nm) coincides with the excimer fluorescence of solutions with a high concentration of 7 and is of excimer origin. The delayed fluorescence of crystals (531 nm) is also excimer one [18]. The significantly higher intensity and duration of the delayed fluorescence of crystals 6 compared to 7 is worth noting.
Thus, the analysis of the luminescent properties of three pairs of compounds—1 and 2, 6 and 7, and 10 and 11—makes it possible to reveal some regularities. The nonplanar structure of the boron difluoride β-diketonate molecule (1, 6, and 10) contributes to the inversion of the S1 and T2 levels, and intense phosphorescence is observed, which is determined by the intersection of the potential energy surfaces in the S1 and T2 states (Fig. 5a). For structures 3, 8, 12, and 13, in which molecules do not form dimers of antiparallel molecules characteristic of boron difluoride β-diketonates, only phosphorescence is observed (Table 1).
At the same time, the planar structure of molecules, their coplanar arrangement, the classical sequence of singlet and triplet levels, and a small energy gap between S1, T1, and T2 contribute to excimer delayed fluorescence. For crystals 2, 4, 7, and 9, in which the molecules are planar and have an antiparallel arrangement in stacks favorable for the formation of excimers, intense excimer delayed P-type fluorescence is observed, which occurs because of triplet–triplet annihilation with the formation of an excimer (Fig. 5b).
CONCLUSIONS
Data on the phosphorescent properties of boron difluoride β-diketonates of various structures have been systematized: (1) with planar molecules when the entire molecule lies in the same plane, and (2) with nonplanar molecules when, due to steric hindrance, the aromatic substituent is turned at an angle to the chelate ring. The nonplanar structure of the boron difluoride β-diketonate molecule promotes the inversion of the S1 and T2 levels, efficient population of the triplet levels, and intense phosphorescence of the crystals. At the same time, molecules with a planar structure are characterized by a classical sequence of singlet and triplet levels, and a small energy gap between S1, T1, and T2 contributes to the population of the triplet level. For planar molecules with antiparallel arrangement in stacks favorable for the formation of excimers, intense excimer delayed P-type fluorescence is observed, which is accompanied by triplet–triplet annihilation to form an excimer. At an arrangement unfavorable for the formation of excimers, phosphorescence of the crystals occurs. The data obtained in this work can be useful for the development of promising luminescent materials with long-lived emission.
REFERENCES
N. Gan and H. Shi, Z. An, et al., Adv. Funct. Mater 28, 1802657 (2018). https://doi.org/10.1002/adfm.201802657
T. Zhang, X. Ma, and H. Wu, Angew. Chem., Int. Ed. Engl. 28, 11206 (2020). https://doi.org/10.1002/anie.201915433
X. Wang, M. Dong, Z. Li, et al., Dyes Pigm. 204, 110400 (2022). https://doi.org/10.1016/j.dyepig.2022.110400
Z. Wu, J. Nitsch, and T. B. Marder, Adv. Opt. Mater 9, 2100411 (2021). https://doi.org/10.1002/adom.202100411
T. Y. Chikineva, D. S. Koshelev, A. V. Medved’ko, et al., Russ. J. Inorg. Chem. 66, 170 (2021). https://doi.org/10.1134/S0036023621020054
H. Ma, A. Lv, L. Fu, et al., Ann. Phys. 531, 1800482 (2019). https://doi.org/10.1002/andp.201800482
Y. L. Chow, C. I. Johansson, Y. Zhang, et al., J. Phys. Org. Chem. 9, 7 (1996).
P. Xu, H. Chen, H. Duan, et al., Russ. J. Gen. Chem. 92, 1814 (2022). https://doi.org/10.1134/S1070363222090225
V. M. Kozenkov, A. A. Spakhov, V. V. Belyaev, et al., Liq. Cryst. 16, 9 (2016). https://doi.org/10.18083/LCAppl.2016.4.9
V. A. Zhinzhilo and I. E. Uflyand, Russ. J. Gen. Chem. 92, 1937 (2022). https://doi.org/10.1134/S1070363222100097
G. Zhang, J. Chen, S. J. Payn, et al., J. Am. Chem. Soc. 129, 8942 (2007). https://doi.org/10.1021/ja0720255
J. Li, X. Wang, X. Zhao, et al., Chin. J. Chem. 40, 2507 (2022). https://doi.org/10.1002/cjoc.202200354
A. Sakai, M. Tanaka, E. Ohta, et al., Tetrahedron Lett. 53, 4138 (2012). https://doi.org/10.1016/j.tetlet.2012.05.122
B. Poggi and E. Lopez, et al., Macromol. Rapid Commun. 43, 2200134 (2022). https://doi.org/10.1002/marc.202200134
B. Domercq, C. Grasso, J.-L. Maldnado, et al., J. Phys. Chem. B 108, 8647. https://doi.org/10.1021/jp036779r
V. E. Karasev and O. A. Korotkikh, Zh. Neorg. Khim. 31, 869 (1986).
A. G. Mirochnik, Z. N. Puzyrkov, E. V. Fedorenko, et al., Russ. J. Inorg. Chem 67, 1425 (2022). https://doi.org/10.1134/S003602362209008X
E. V. Fedorenko, A. G. Mirochnik, A. V. Gerasimenko, et al., J. Photochem. Photobiol. Chem. 412, 113220 (2021). https://doi.org/10.1016/j.jphotochem.2021.113220
US Pat. 004846; 16.20.2003 Publ.
B. V. Bukvetskii, E. V. Fedorenko, A. G. Mirochnik, et al., J. Struct. Chem. 47, 56 (2006). .https://doi.org/10.1007/s10947-006-0265-0
E. V. Fedorenko, A. G. Mirochnik, A. V. Gerasimenko, et al., Dyes Pigm. 159, 557 (2018). https://doi.org/10.1016/j.dyepig.2018.07.022
E. V. Fedorenko, B. V. Bukvetskii, A. G. Mirochnik, et al., J. Lumin. 130, 756 (2010). https://doi.org/10.1016/j.jlumin.2009.11.027
Bruker. APEX2. Bruker AXS Inc., Madison, 2012.
Sheldrick G.M. SHELXTL/PC, Versions 5.10, Bruker AXS Inc., Madison, Wisconsin, 1998.
V. R. Thalladi, H.-C. Weiss, D. Bläser, et al., J. Am. Chem. Soc. 12, 8702 (1998). https://doi.org/10.1021/ja981198e
A. Brammer, E. Bruton, and P. Sherwood, Cryst. Growth Des. 1, 277 (2001). https://doi.org/10.1021/cg015522k
D. Rohde, C.-J. Yan, and L.-J. Wan, Langmuir 22, 4750 (2006). https://doi.org/10.1021/la053138+
E. V. Fedorenko, B. V. Bukvetskii, A. G. Mirochnik, et al., Russ. Chem. Bull. 58, 2240 (2009). https://doi.org/10.1007/s11172-009-0312
S. A. Tikhonov, E. V. Fedorenko, A. G. Mirochnik, et al., Spectrochim. Acta A 214, 67 (2019). https://doi.org/10.1016/j.saa.2019.02.002
A. W. Hanson and E. W. Macaulay, Acta Crystallogr. 28, 1961 (1972).
A. G. Mirochnik, B. V. Bukvetskii, E. V. Gukhman, et al., J. Fluor. 13, 157 (2003). https://doi.org/10.1023/A:1022939209971
Y. Dromzee, J. Kossanyi, and V. Wintgens, Z. Kristallogr. 212, 372 (1997). https://doi.org/10.1524/zkri.1997.212.5.372
B. V. Bukvetskii, E. V. Fedorenko, A. G. Mirochnik, et al., J. Struct. Chem. 52, 221 (2011). https://doi.org/10.1134/S0022476611010331
B. V. Bukvetskii, E. V. Fedorenko, A. G. Mirochnik, et al., J. Struct. Chem. 51, 545 (2010). https://doi.org/10.1007/s10947-010-0079-y
Funding
The work was supported by the Ministry of Education and Science of the Russian Federation in the framework of State assignment no. FWFN (0205)-2022-0003.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflicts of interest.
Additional information
The article devoted to the 50th anniversary of Konstantin Yu. Zhizhin, Corresponding Member of the Russian Academy of Sciences
Translated by G. Kirakosyan
Rights and permissions
About this article

Cite this article
Mirochnik, A.G., Fedorenko, E.V. & Gerasimenko, A.V. Boron Difluoride β-Diketonates: Structure and Phosphorescence. Russ. J. Inorg. Chem. 68, 722–728 (2023). https://doi.org/10.1134/S0036023623600600
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036023623600600