
Abstract
The new complexes [M(NA)2(H2O)4]SiF6 ⋅ 2H2O, where M2+ = Co, Ni, Zn (complexes I, II, and III, respectively), and NA = C6H6N2O is nicotinamide, [Cu(NA)2(SiF6)(H2O)2] ⋅ 2H2O (IV), and (HNA)2SiF6 (V) have been synthesized from aqueous solutions and studied by chemical, IR spectroscopic, and X-ray diffraction analyses. Their unit cell parameters are a = 16.2448(18) Å, b = 6.8834(8) Å, c = 10.0767(11) Å, β = 102.765(3)°, V = 1098.92 Å3, space group C2 for I; a = 16.1591(7) Å, b = 6.8777(3) Å, c = 10.0314(5) Å, β = 102.410(1)°, V = 1088.82 Å3, space group C2 for II; a = 16.2265(6) Å, b = 6.8965(3) Å, c = 10.0696(5) Å, β = 102.390(1)°, V = 1100.60 Å3, space group C2 for III; a = 6.5915(4) Å, b = 7.7670(4) Å, c = 10.1506(5) Å, α = 110.7390(10)°, β = 105.824(2)°, γ = 95.448(2)°, V = 456.868 Å3, space group P\(\overline{1}\) for IV; and a = 14.1904(7) Å, b = 9.0468(4) Å, c = 11.8160(7) Å, β = 106.277(2)°, V = 1456.1 Å3, space group C2/c for V. Complexes I–III are isostructural and represent ionic compounds formed by hexafluorosilicate anions and complex cations. The coordination polyhedron of the metal is a slightly distorted octahedron built of the O atoms of four coordinated water molecules and the two N atoms of two pyridine rings of two nicotinamide molecules in trans positions of the polyhedron. Complex IV has a polymeric chain structure. The coordination polyhedron of the copper cation represents an octahedron, which is elongated along its axis and built of the two N atoms of two pyridine rings of two nicotinamide molecules, the two O atoms of coordinated water molecules, and the two F atoms of hexafluorosilicate anions acting as bridges between neighboring cations. In the structure of complex V, the nitrogen atom of the pyridine ring HNA+ is protonated. The geometry of hexafluorosilicate anions is identical in all the five complexes. The structures have branched networks of hydrogen bonds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTON
Heterocyclic nitrogen-containing compounds, which are components of some vitamins and medicines, are important for the life of living systems [1, 2]. In particular, nicotinamide (NA) participates in metabolic processes of a human organism [3–6], is a component of some enzymes, e.g., the coenzyme nicotineamide-adenine dinucleotide, and is often used in the synthesis of metal complexes for the creation of medicinal preparations [7]. The NA-based complexes are promising compounds with a high bioactivity [8]. Nicotinamide exhibits anti-inflammatory properties and is used as a hydrotropic agent in pharmaceutical chemistry [9]. For example, it is used to increase the solubility of nifedipine and indomethacin. The antibacterial and antifungal activity of some nicotinamide based complexes of transition metals were reported [10–16]. The NA coordination compounds of metal bioelements are of interest not only as potential bioactive substances, but also as complexes with ambidentate ligands. An NA molecule has three potential donating atoms, namely, the nitrogen atom of the pyridine ring, the nitrogen atom of the amino group, and the oxygen atom of the carbonyl group. Nicotinamide is preferably coordinated through the nitrogen atom of the pyridine ring [9]. The structures of coordination polymers were described [17–21], and the structure of the complex with NA as a bridging ligand coordinated through the nitrogen atom of the pyridine ring and the oxygen atom of the amide group was considered [22].
The objectives of this work are to synthesize the complexes [M(NA)2(H2O)4]SiF6 ⋅ 2H2O, where M2+ = Co, Ni, Zn (complexes I, II, and III, respectively), NA = C6H6N2O is nicotinamide, [Cu(NA)2(SiF6)(H2O)2] ⋅ 2H2O (IV), and (HNA)2SiF6 (V), and perform their IR spectroscopic study and structural characterization.
EXPERIMENTAL
Synthesis of the complexes. The initial compounds used were CoCO3, NiO, 2CuCO3 · Cu(OH)2, ZnCO3 of chemically pure grade, a H2[SiF6] solution with ω = 45% (pure for analysis grade), and NA (chemically pure grade).
Metal hexafluorosilicates МSiF6 · 6H2O (M2+ = Co, Ni, Cu, Zn) were synthesized by the reaction between the initial compounds and H2[SiF6] with further crystallization at room temperature [23].
Complexes I–IV were synthesized via the separate mixing of aqueous solutions of CoSiF6 · 6H2O (0.309 g, 0.001 mol), NiSiF6 · 6H2O (0.309 g, 0.001 mol), ZnSiF6 · 6H2O (0.316 g, 0.001 mol), CuSiF6 · 6H2O (0.314 g, 0.001 mol), and NA (0.244 g, 0.002 mol). The resulting solutions were allowed to stand at room temperature for slow crystallization. The crystals formed in several days were separated from the mother solution by filtration and dried in a desiccator over CaCl2. Their yield was 70 (I), 75 (II), 67 (III), and 90% (IV).
When developing the synthesis conditions, it was established that the addition of NA (0.244 g, 0.002 mol) to a hexafluorosilicic acid solution with ω = 9% under stirring leads to the formation of colorless (HNA)2SiF6 (V) crystals in several days; these crystals were separated from the mother solution by filtration and dried in a desiccator over CaCl2. The yield was 56%.
Chemical analysis was performed gravimetrically for \({\text{SiF}}_{{\text{6}}}^{{{\text{2}}-}}\) in the form of BaSiF6 [24], for cobalt and nickel in the form of sulfates, and zinc in the form of 8-oxyquinolinate [25]. The content of copper in complex IV was determined photocolorimetrically [26]. Elemental C,H,N-analysis was performed on a ThermoFlash 2000 analyzer.
IR spectra of complexes I–V were recorded on an Agilent Cary 630 FTIR IR Fourier-transform spectrometer in the region of 4000–400 cm–1 in a KBr matrix. Major absorption frequencies of complexes I–V (ν, cm–1): 3621 w, 3409 m, 3258 m, 1667 m, 1623 m, 1567 m, 1439 m, 1215 w, 1065 w 744 s, 707 s for I; 3621 w, 3409 m, 3264 m, 1673 m, 1623 m, 1567 m, 1439 m, 1221 w, 1070 w, 743 s, 707 s for II; 3621 w, 3403 m, 3319 m, 1673 m, 1623 m, 1567 m, 1439 m, 1221 w, 1065 w 746 s, 707 s for III; 3532 w, 3493 w, 3370 m, 1684 m, 1634 m, 1606 m, 1450 w, 1411 m, 1210 w, 1070 w, 753 s, 685 s for IV; 3392 m, 1679 m, 1623 m, 1561 m, 1427 m, 1204 m, 696 s for V.
X-ray diffraction analysis of complexes I–V was performed on a Bruker APEX DUO diffractometer equipped with a 4K CCD detector in φ and ω scanning modes at 150(2) K. Absorption corrections were applied using the SADABS software [27], which uses the repeated measurements of the same reflections for different crystal orientations. The structures were solved by direct methods and refined by the full-matrix least-squares technique on F 2 in the anisotropic approximation for non-hydrogen atoms using the SHELXTL software suite [28]. The hydrogen atoms of organic ligands were located from difference electron density maps and refined as rigid bodies, and the hydrogen atoms of water molecules were refined freely in the isotropic approximation, limiting the O–H bond length by 0.96 Å. In the structures of complexes I–III, the \({\text{SiF}}_{{\text{6}}}^{{{\text{2}}-}}\) anion is disordered over two symmetry-related positions with equal populations.
Crystallographic data and details of X-ray diffraction experiment are given in Table 1. The structural data for complexes I–V were deposited with the Cambridge Structure Database (ССDС nos. 1983435–1983439) and can be freely downloaded by request from the website www.ccdc.cam.ac.uk/data_reguest/cif.
RESULTS AND DISCUSSION
The synthesized complexes represent fine-crystalline powders stable in the air. Crystals of complexes III and V are colorless, complex I is pink, complex II is light blue, and complex IV is blue. The complexes decompose in strong solutions of sulfuric and nitric acids, DMSO, DMF, and ethylenediamine and are insoluble in acetone, ethanol, diethyl ether, toluene, n-hydrocarbons, and alcohols (ethanol, butanol, isopropanol, isobutanol). The solubility in water at 25.0 ± 0.5°C is 0.08, 0.03, 0.15, 0.02, and 0.22 mol/L for complexes I, II, III, IV, and V, respectively.
Since, according to the concept of “hard–soft acids and bases”, Co2+ ions are classified as soft Lewis acids, Ni2+ and Cu2+ ions are intermediate acids, and Si4+ ions are hard acceptors of electron pairs, both ionic and polymeric complexes can be formed [29, 30]. The assignment of absorption bands in the IR spectra of the complexes was performed in compliance with [31–35]. The water molecules in the structures of complexes I–IV are characterized by the bands of ν(OH) stretching vibrations in the region 3621–3403 cm–1. The bands of ν(NH) primary amide stretching vibrations are observed at 3359 and 3152 cm–1 for NA and 3258 (I), 3264 (II), 3319 (III), 3370 and 3180 (IV), 3392, and 3219 cm–1 (V) for the complexes; i.e., the bands slightly shift toward lower frequencies for complexes I–III and to ward higher frequencies for complexes IV and V. The values of δ(NH2) shift toward higher frequencies to 1623 cm–1 for complexes I–III and 1634 cm–1 for complex IV in comparison with δ(NH2) = 1617 cm–1 for NA. The frequency ν(CO)NA = 1679 cm–1 slightly shifts toward lower values to 1667, 1673, and 1673 cm–1 for complexes I, II, and III, respectively, and increases in complex IV (1684 cm–1), thus it may serve as an evidence for the absence of bond between the oxygen atom of the amide group and the copper atom. The presented data are more likely to be indicative of the fact that the amide group does not participate in coordination and the structural dissimilarity of complex IV to the other compounds. In most complexes with nicotinamide, the metal ion is coordinated through the nitrogen atom of the pyridine ring [9, 17–21], but there are some known complexes in which it is coordinated through the amide group [22]. It is likely that NA is coordinated to complexing ions through the nitrogen heteroatom of the pyridine ring, as confirmed by the shift of νring in the IR spectra of these complexes toward higher frequencies in the region of 1600–1030 cm–1 in comparison with the spectrum of uncomplexed NA.
The band of stretching vibrations from the octahedral \({\text{SiF}}_{{\text{6}}}^{{{\text{2}}-}}\) anion at 741 cm–1 almost does not shiftfor complexes I–III (744, 743, and 744 cm–1), but changes for complex IV (756 cm–1), thus indicating the change of the structural role of the hexafluorosilicate ion in this compound.
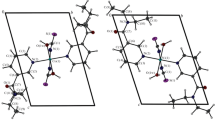
Single crystals of the complexes were separated by means of long-term isothermal crystallization from their aqueous solutions. Crystals of complexes I–III are isostructural. The analysis of the array of reflections has shown the presence of a centrosymmetric group in this series of crystals. The possible variants have proven to be space groups C2 and Сm. As a result, the structures were solved in monoclinic group C2 as it showed the best refinement parameters. The complexes have an island structure (Fig. 1). The independent part contains a half of the complex cation, a half of the hexafluorosilicate anion, and one crystallization water molecule. The coordination polyhedron of the metal cation is a slightly distorted octahedron (bond angles differ from the ideal ones by 3°) built of the O atoms of four coordination water molecules and the two N atoms of pyridine rings of nicotinamide in the trans positions of the polyhedron. The hexafluorosilicate anion acts as a counterion without being included into the inner coordination sphere of the metal. The metal cation and the Si atom occupy the positions on different axes 2. The structure contains a branched network of hydrogen bonds between the NA amino group and the coordinated water molecules as an H donor, and the crystallization water molecules, the hexafluorosilicate anions, and the O atom of the NA carbonyl group as a hydrogen bond acceptor (d(D···A), 2.71–2.99 Å).

Structural fragment of complexes I–III with disordered hexafluorosilicate ion part (dashed).
Cationic and anionic layers alternate in packing along axis с (Fig. 2). Inside the cationic layers, the π‑systems of NA ligands sustain parallel ordering by the “head-to-tail” principle with a distance of ∼3.5 Å between the planes of ligands from neighboring complex units (the turn angle for the ligands of neighboring cations is less than 2°). The centers of cations are arranged by the hexagonal law distorted in compliance with the shape of a particle in plane ab. The layers are arranged over each other almost without shift. The anionic layers also form a one-layer hexagonal packing strictly over and under the centers of cations.

Structural fragment of a polymeric chain in complex IV.
Complex IV crystallizes in a triclinic system and has a polymeric chain structure. The coordination polyhedron of the copper cation (Fig. 2) represents an octahedron, which is elongated along its axis and built of the two N atoms of two pyridine rings of nicotinamide, the two O atoms of coordinated water molecules, and the two F atoms of hexafluorosilicate anions acting as bridges between neighboring cations. The Cu and Si atoms occupy two systems of inversion centers in the space group. Polymeric chains are arranged in a unit cell in parallel to axis b to form a hexagonal packing of rods in the same direction. Ths structure also contains a branched network of hydrogen bonds between the NA amino group and the coordinated water molecules as an H-donor and the crystallization water molecules, the hexafluorosilicate anions, and the O atom of the NA carbonyl group as a hydrogen bond acceptor (d(D···A), 2.77–2.92 Å).
Nicotinamidium hexafluorosilicate V crystallizes in monoclinic system. In the structure of this complex, it can clearly be seen that the nitrogen atom of the pyridine ring HNA+ is protonated. This is likely to destroy the overall conjugation of the ligand π-system in such a fashion that the rotation angle of the pyridine ring with respect to the amide group becomes equal to 38.1° (compare with 2.8° (I), 0.5° (II), 0.8° (III), and 2.7°(IV)). A two-layer hexagonal packing of combined cation–anion layers is formed in the structure along axis b. In this case, the plane of the pyridine ring HNA+ is turned almost perpendicularly (70.0°) to the hexagonal layer plane. The Si atoms of hexafluorosilicate anions occupy one of the two systems of axes 2, whereas the entire cation is located in the general position. The structure contains hydrogen bonds: the strong “acidic” hydrogen bond between the protonated pyridine ring HNA+ and the carbonyl group of the neighboring cation as a hydrogen-bond acceptor (d(D···A), 2.66 Å) alongside with the two medium-strength hydrogen bonds with the participation of an amino group like N–H···F (d(D···A), 2.85 and 2.92 Å).
The geometry of hexafluorosilicate anions in all the five complexes is identical.
CONCLUSIONS
Our studies of complexes I–III have shown that they have island structures built of isolated [M(NA)2(H2O)4]2+ cations, \({\text{SiF}}_{{\text{6}}}^{{{\text{2}}-}}\) anions, and crystal water molecules, as observed earlier in the hexafluorosilicate of cobalt(II) complexes with DMSO and DMF [36]. At the same time, complex IV [37] has an appreciably differing structure. Complex IV has a polymeric chain structure, in which hexafluorosilicate anions act as bridges between the neighboring cations, and the coordination polyhedron of the copper cation is an octahedron elongated along its axis. Hexafluorosilicate of the copper(II) complex with DMSO has an island structure built of neutral molecules, in which the hexafluorosilicate anion is coordinated through one fluorine atom to the copper(II) cation, and the coordination environment of the copper(II) cation is a square pyramid. Hence, the distinction between the structures is determined by the coordination mode of \({\text{SiF}}_{{\text{6}}}^{{{\text{2}}-}}\) anions.
In addition, nicotinamidium hexafluorosilicate (HNA)2SiF6 (V), in which the nitrogen atom of the pyridine ring HNA+ is protonates, has been synthesized and structurally characterized.
REFERENCES
D. A. Kose and H. Necefogly, Therm. Anal. Calorim. 93, 509 (2008).
T. V. Koksharova, I. S. Gritsenko, S. V. Kurando, et al., Visnik ONU 14, 91 (2009).
G. S. Kurkcuoglu and O. Z. Yesilel, et al., J. Mol. Struct. 1096, 84 (2015). https://doi.org/10.1016/j.molstruc.2015.04.046
M. Puchonova, Z. Repicka, J. Moncol, et al., J. Mol. Struct. 1092, 1 (2015). https://doi.org/10.1016/j.molstruc.2015.02.086
C. S. Dilip, V. Thangaraj, and RajA. P. Paul, Arab. J. Chem. 9, 731 (2016). https://doi.org/10.1016/j.arabjc.2011.07.016
A. C. Tella, S. O. Owalude, P. A. Ajibade, et al., J. Mol. Struct. 1125, 570 (2016). https://doi.org/10.1016/j.molstruc.2016.07.016
Z. Vaskova, N. Kitanovski, Z. Jaglicic, et al., Polyhedron 81, 555 (2014). https://doi.org/10.1016/j.poly.2014.07.017
F. A. Al-Saif and M. S. Refat, J. Mol. Struct. 1021, 40 (2012). https://doi.org/10.1016/j.molstruc.2012.04.057
A. Dziewulska-Kulaczkoska, L. Mazur, and W. Ferenc, J. Therm. Anal. Calorim. 96, 255 (2009).
N. A. Smith, P. Zhang, L. Salassa, et al., Inorg. Chim. Acta 454, 240 (2017). https://doi.org/10.1016/j.ica.2016.06.014
F. E. Ozbek, M. Sertcelik, M. Yuksek, et al., J. Mol. Struct. 1150, 112 (2017). https://doi.org/10.1016/j.molstruc.2017.08.074
G. S. Askin, H. Necefoglu, A. M. Tonbul, et al., Acta Crystallogr., Sect. E 71, 479 (2015). https://doi.org/10.1107/S2056989015006490
N. Bozkurt, N. Dilek, N. C. Delibas, et al., Acta Crystallogr., Sect. E 69, 389 (2013). https://doi.org/10.1107/S1600536813015948
V. A. Sharnin, S. V. Dushina, K. V. Grazhdan, et al., J. Chem. Thermodyn. 82, 116 (2015). https://doi.org/10.1016/j.jct.2014.11.004
O. Altun and M. Suozer, J. Mol. Struct. 1149, 307 (2017).
A. C. Tella, S. O. Owalude, P. A. Ajibade, et al., J. Mol. Struct. 1125, 570 (2016). https://doi.org/10.1016/j.molstruc.2017.07.069
R. B. N. Baig, B. R. Vaddula, M. N. Nadagouda, et al., J. Royal Soc. Chem. 17, 1243 (2015).
C. Li, F. Cui, H. Zhang, et al., Spectrochim. Acta A 134, 367 (2015). https://doi.org/10.1016/j.saa.2014.06.080
D. Chisca, L. Croitor, O. Petunov, et al., CrystEngComm 20, 432 (2018). https://doi.org/10.1039/c7ce01988b
M. Arici, O. Z. Yesilel, E. Acar, et al., Polyhedron 127, 293 (2017). https://doi.org/10.1016/j.poly.2017.02.013
K. Gor, G. S. Kurkcuogly, O. Z. Yesilel, et al., J. Mol. Struct. 1060, 166 (2014). https://doi.org/10.1016/j.molstruc.2013.12.024
J. Xue, X. Hua, W. Li, et al., J. Mol. Struct. 1059, 108 (2014). https://doi.org/10.1016/j.molstruc.2013.11.001
T. Yildirum, D. A. Kose, E. Avci, et al., J. Mol. Struct. 1176, 576 (2019). https://doi.org/10.1016/j.molstruc.2015.02.086
I. G. Ryss, Chemistry of Fluorine and Its Inorganic Compounds (Goskhimizdat, Moscow, 1956) [in Russian].
L. V. Myshlyaeva and V. V. Krasnoshchekov, Analytical Chemistry of Silicon (Nauka, Moscow, 1972) [in Russian].
G. Charlot, Les methodes de la chimie analytique: Analyse quantitative minerale (Masson, Paris, 1961.
G. M. Sheldrick, SADABS, Version 2.01 (Bruker, Madison (WI), 2004).
G. M. Sheldrick, Acta Crystallogr. 71, 3 (2015).
R. G. Pearson, J. Chem. Educ. 45, 581 (1985).
A. D. Garnovskii, A. P. Sadimenko, O. A. Osipov, et al., Hard-Soft Interactions in Coordination Chemistry (Izd-vo Rostovsk. Univ., Rostov-on-Don, 1986) [in Russian].
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds (Interscience, New York, 1986).
G. G. Sadikov, A. S. Antsyshkina, T. V. Koksharova, et al., Cryst. Repts 52, 819 (2007). https://doi.org/10.1134/S1063774507050112
S. C. Jain and R. Rivest, Can. J. Chem. 47, 2209 (1969).
A. Smith, Applied IR Spectroscopy (Wiley, New York, 1979; Mir, Moscow, 1982).
E. Pretsch, F. Buehlmann, and C. Affolter, Structure Determination of Organic Compounds: Tables of Spectral Data (Springer, Berlin, 2000; Mir, Moscow; BINOM, Laboratoriya Znanii, 2013).
T. G. Cherkasova, I. Yu. Bagryanskaya, N. V. Pervukhi-na, et al., Russ. J. Inorg. Chem. 62, 760 (2017). https://doi.org/10.1134/S003602361706002X
T. G. Cherkasova, N. V. Pervukhina, et al., Russ. J. Inorg. Chem. 63, 899 (2018). https://doi.org/10.1134/S0036023618070045
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by E. Glushachenkova
Supplementary material
Rights and permissions
About this article

Cite this article
Cherkasova, T.G., Pervukhina, N.V., Kuratieva, N.V. et al. Complexation of Cobalt(II), Nickel(II), Copper(II), and Zinc(II) Hexafluorosilicates with Nicotinamide in Aqueous Solution. Russ. J. Inorg. Chem. 64, 1120–1126 (2019). https://doi.org/10.1134/S0036023619090055
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036023619090055