
Abstract
Reverse Transcription quantitative Polymerase Chain Reaction (RT-qPCR) is a method of choice for quantifying micro RNAs (miRNAs). Typically, RT-qPCR data are normalized to reference genes. While miRNAs are used for diagnosing and subtyping breast cancer, various studies show their deregulation in this condition, thus, undermining miRNAs’ utility as a reference. This review examines the expression pattern of miR-16 and suggests normalization approaches for breast cancer. We analyzed the data from selected peer-reviewed studies to calculate the standardized mean difference (SMD) with subsequent Chi-square testing and identified the difference in miR-16 expression between breast cancer patients and healthy controls. With a negative SMD value of ‒0.56 and Chi-square of 62.62 (p-value = 0.05), the deregulation of miR-16 in breast cancer was confirmed. High variance in the stability value (SV) of miR-16 expression levels confirmed its inappropriateness as a control gene in breast cancer. The combination of miR-16 and miR-425 was confirmed as an accurate endogenous control.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Over the past few years, breast cancer (BC) is reported to be the second leading cause of death in women among all types of cancer. The death rate of breast cancer in women is significant, estimated at about 1 in 38 (2.6%) [1]. The chance that a woman is diagnosed with breast cancer is 1 in 8 (13%), while 1 in 39 women (3%) will die from this disease [2]. Even if the overall percentage of women getting breast cancer has not changed over the last decades, for African-American, Asian and Pacific Islander women, the rate has surged [3]. Improvement of the early diagnosis of breast cancer may aid in planning the treatment. miRNAs levels may be employed as biomarkers for the early diagnosis of breast cancer.
The history of microRNA (miRNA) began when it was first discovered as part of a group of regulatory genes in Caenorhabditis elegans in 1993 by Ambros and Ruvkun groups [4]. Years later, studies of miRNA went into blossom. From the DNA sequences in the nucleus, primary miRNAs are transcribed and then processed into precursor miRNAs and mature miRNAs. The mature versions of miRNAs are short, non-coding, and single-stranded RNA sequences, usually 19–23 nucleotides in length. To perform the regulatory function, they incorporate into the effector complex, RNA-induced silencing complex (RISC) [5]. By binding to the 3′ untranslated region of certain mRNAs, the complex consequently causes either mRNA degradation or translational inhibition. One miRNA can lead to the silencing of hundreds of genes. Thereby, as a class of molecules, miRNAs regulate cellular proliferation, differentiation, apoptosis, and glucose, cholesterol, and iron homeostasis [4].
As miRNAs play an important role in the normal development of animals, abnormal expression of miRNAs provides a window into pathophysiology of many human diseases, including cancer [6]. Most examined tumors have shown the unusual miRNAs profiles compared to healthy people, which suggests that miRNAs could be used as biomarkers for cancer diagnosis, prognosis, and therapy [6]. One of the earliest discoveries of miRNAs in relation to cancer is the loss of miR-15a/16-1 gene observed in B-cell chronic leukemia [6].
The utility of miRNAs in studying cancer and certainly breast cancer depends on reliable quantifying of their expressions. Methods to detect the expression of miRNAs include Northern blot [7], bead-based flow-cytometry [8], and microarray technology [9]. Reverse transcription quantitative PCR (RT-qPCR) has been the first choice and used most commonly in miRNAs quantification because of its high sensitivity, wide dynamic range, and low template requirements [10].
Regardless of the technique employed, data normalization is a key for obtaining an accurate and reliable quantification of gene expression levels [11]. Variations including differences in initial sample amount, RNA recovery, RNA integrity, and efficiency of cDNA synthesis are likely to be generated through the multi-step process of qRT-PCR [11]. Internal reference genes is among the most preferred normalization approaches [12, 13]. These target sequences should experience the same variations during the quantification process. Hence, the success of the whole quantification assay is highly dependent on the choice of the appropriate control gene [11].
miR-16 is widely used as a reference gene in cancer studies, including breast cancer. It has also being disturbed in various types of cancers. In miR-16, a 9-nucleotide seed region targets the 3'UTR of various genes, including the anti-apoptotic protein BCL2. Furthermore, genes included in the G1-S checkpoint such as CCND1 (cyclin D1), CCND3 (cyclin D3), CCNE1 (cyclin E1), and CDK6 (cyclin-dependent kinase 6) [14, 15]; and in the Wnt signaling pathway, such as WNT3A (wingless-type MMTV integration site family, member 3A) [15] are also miR-16’s targets. Therefore, miR-16 regulates the cell cycle, elevates cells apoptosis, and suppresses tumorigenicity [16]. While various studies have demonstrated the highly stable expression of miR-16 in human miRNAs profile across a wide range of cancers types as well as sample sources, which emphasizes their role as reference miRNA, miR-16 has also been used as a biomarker in detecting various human cancers. Therefore, a controversy exists, and has to be sorted out.
In this review, we will discuss in detail the expression profile of miR-16 in breast cancer, evaluate its suitability as an internal control gene and further suggest a potential normalization approach using reference genes.
EXPERIMENTAL
Publication search. Relevant studies, published up to 2020, were identified in PubMed, Embase, and ScienceDirect databases. The article searching was performed using the following keywords: “microRNA 16” OR “miRNA-16” OR “mir-16” OR “mir16” AND “expression” OR “reference gene” OR “control gene” OR “internal control” OR “housekeeping” OR “deregulation” OR “biomarker” AND “breast cancer” OR “breast neoplasm.” The last search was performed on 11 April 2020.
Inclusion and exclusion criteria. The following criteria are required for studies to be included in this meta-analysis: (1) evaluation of miR-16 expression in breast cancer; (2) a case-control design; (3) written in English; (4) availability of sample size (AND) Cq value (OR) stability value. Studies were excluded when they are: (1) review articles, meta-analysis, abstracts or conference papers; (2) duplicate publications; (3) studies on animals; (4) studies without a case-control design; BIOCROSS score is smaller than 7.
Data extraction. Data were extracted from each eligible study and manually checked. Then, items were recorded for each eligible study: the first author, publication year, country, ethnicity, sample size and mean age of subjects in the case and the control groups, type of cancer, source of controls, Cq value or stability counts in the case and the control groups.
Quality assessment. The review question checklist of BIOCROSS was applied to evaluate the quality of each study involved in this meta-analysis. BIOCROSS is the method of choice to examine the quality of biomarker cross-sectional studies [17]. As our analysis also contained the studies of endogenous control, we only obtained the first seven items in the list, which could be applied for all kinds of cross-sectional analyses. Three “Issues to consider” (IC) are provided, if they all are, the study will have a score of “2.” If one or two issues are missing, a reduced score of “1” should be awarded. Not mentioning any of the issues should lead to a score of “0”. Thus, the highest score is 14.
Statistical analysis. The expressions of miR-16 in terms of Cq value, SD, and sample size were obtained and used to determine the differences between patients with (cases), and without (controls) breast cancer. An inverse-variance random-effects model was applied to determine the standardized mean difference (SMD), Chi-square test and Inter-study heterogeneity via the I2 index. All statistical analyses were performed using RevMan v5.3 software: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014, and normal excel software.
RESULTS AND DISCUSSION
Study Characteristics
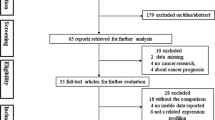
Eligible studies were selected after a process of searching and screening, which is illustrated in Fig. 1. Throughout various sources of data, a total of 350 articles were found by reading titles and abstracts. 296 articles then were excluded as they did not meet the criteria, in which 121 articles were duplicates, 65 articles were abstracts, 2 articles were meta-analysis, 31 were review articles, 9 articles were related to other diseases, 47 articles were related to other miRNAs, and 21 more articles were not conducted in a case-control design. Remaining 54 articles were examined in detail, with 35 more articles excluded since they did not provide sufficient data. Finally, a total of 19 eligible articles remained for final analysis [14, 15, 18‒34].

Flowchart of the identification of eligible studies for meta-analysis of miR-16 expression in breast cancer. 350 articles were initially obtained through databases and other sources searching. 331 articles were then excluded during the process of screening with various criteria, finally giving a number of 19 articles that were included in the meta-analysis.
miR-16 as Reference Gene in Breast Cancer Studies
miR-16 was recommended as a suitable reference gene in various breast cancer studies as it stably expressed and showed no difference between cancer and control groups [14]. A total of 8 studies examined in this review had employed miR-16 in this manner (Table 1).
Davoren et al. [14] in 2008 identified miR-16 as the single most stably expressed miRNA, with a GeNorm M-value of 1.191. miR-16 was also selected in the top 15 most stably expressed miRNAs across 40 normal human tissue types [14]. Moreover, the expression of miR-16 indicated no difference between the primary site and metastatic one [23]. It was reported that the expression and reproducibility of miR-16 were stable across 226 peripheral blood samples [19]. In 2013 McDermott et al. [15] compared miR-16 as endogenous control to other candidates, including U6, a common endogenous control in breast cancer. The result has shown significant fluctuation in the expression levels of U6 while the expression of miR-16 was relatively stable. Most serum miRNAs were unstable after repeated freezing and thawing; yet, the levels of miR-16 were not affected [21]. In another study [22], four commonly-used controls (miR-16, miR-223, let-7a, and RNU6B) were tested for their expression levels in breast specimens. While let-7a and RNU6B failed to pass the criterion, miR-16 displayed an abundant expression and high stability.
Six studies reported the stability values (SV) of miR-16 using the same NormFinder software, which provides a direct measure for evaluating the systematic error introduced when using the gene for normalization. This software takes into account intra- and inter- group variations and calculates the stability value for the best combination of two genes. The lower SV indicates the more stable expression pattern of reference genes. Although these studies all indicated a small SV for miR-16, it is necessary to examine how the data vary. From the included six studies, we built a graph to show the mean and standard deviation of miR-16’s SV (Fig. 2). It is clear that the data obtained varied with the standard deviation of 0.479, which is 71% of the mean (0.6776). In each study, miR-16 has demonstrated a stable expression; yet, when we compared between different studies, high variation was observed.

SV of miR-16 from included studies with calculation of the mean and SD. SV data were obtained from 6 studies. The mean and the standard deviation of SV were calculated and displayed as a number above each bar. “Total” bar depicts the mean and the standard deviation of “across all studies” (0.6776 ± 0.479).
miR-16 Is Deregulated in Breast Cancer Studies
Aberrant expression of miR-16 was evident in many studies of numerous types of cancers as well as non-aligmant diseases. Breast cancer is among the most reported cancer with dysregulated miR-16. The following eight studies displayed this cancer-related expression pattern of miR-16 (Table 2).
In a nutshell, miR-16 was significantly up-regulated in breast cancer patients in comparison to healthy people. There were higher levels of exosomal miR-16 in estrogen receptor (ER) and progesterone receptor (PR) positive BC patients as compared to ER‒, PR‒ and triple-negative BC patients [18]. The high expression of miR-16 in breast cancer patients was affected by stages of the disease as ones suffering from stage III invasive intraductal carcinoma show a significant up-leveled pattern [25]. This microRNA was showed to be significantly increased in lymph node-negative patients while decreased to normal levels when lymph node metastases occur. Unlike other tested miRNAs, the increased levels of miR-16 were not affected by chemotherapy in plasma of the BC patients [30]. One study carried out on 30 plasma samples suggested that their expressions are non-gender specific [27]. Interestingly, Appaiah et al. [29] in 2011 showed a significant upregulation of miR-16 in the serum of disease-free patients, who previously were diagnosed with breast cancer, when compared with healthy subjects. The elevated level of miR-16 was also observed in patients with active metastatic process [29]. Furthermore, the deregulation of miR-16 in a triple-negative breast cancer (TNBC) was shown. The result confirmed the low level of miR-16 associated with tumor load, which may potentially be used as a diagnostic marker for TNBC. Also, in post-surgery patients’ plasma, the level of miR-16 was significantly restored [26].
Aberrant expression patterns of miR-16 were reported in breast cancer. An inverse-variance random-effects model was applied (using Cq value, SD, and sample size) to investigate the SMD value and chi-square test. The SMD is sometimes used interchangeably with the term “effect size.” Generally, the comparator is a placebo or an alternative active treatment. An SMD of zero means that the new treatment and the placebo have equivalent effects. If improvement is associated with higher scores on the outcome measure, SMDs greater than zero indicate the degree to which treatment is more efficacious than placebo, and SMDs less than zero indicates the degree to which treatment is less efficacious than placebo [35]. Additionally, the chi-square test was used to examine the difference between classes in population. If the observed chi-square test statistic is greater than the critical value, the null hypothesis will be rejected, which means there is a difference between the investigated groups.
With the SMD result of –0.52 [–1.25, 0.22], it indicated that there was a significant difference in the expression of miR-16 between breast cancer patient samples and healthy control samples. In terms of the Chi-square test, the obtained F-value is 62.62, which was larger than the F critical value of 12.59 (degrees of freedom = 6, p-value= 0.05), and the p-value was smaller than 0.00001 (Table 3). This confirmed that there was a statically significant difference in miR-16 expression between breast cancer patients and healthy people. It supports the idea that miR-16 deregulated in breast cancers and could not be used as an internal control gene for this type of cancer.
Potential Endogenous Control Approaches
Apart from its deregulation in breast cancer, there was other evidence suggesting that miR-16 is unsuitable to be a normalizer. Erythrocytes are the plentiful source of miR-16. Errors made in course of extraction easily lead to hemolysis, which sends miR-16 levels in plasma into artificial increase [36]. McDonald et al. [37] reported that the level of circulating endogenous miR-16 was higher in plasma samples than in serum samples due to hemolysis, increasing in Cq of 1–2 (i.e., a 2- to 4-fold increase in copy number). The result emphasized that the errors in normalization are likely to occur if endogenous miR-16 used as an internal control. Thereby, this miRNA may not be an ideal internal control without being strictly monitored for hemolysis [34].
Shen J. et al. [32] examined the efficiency of five microRNAs (miR-93, miR-103, miR-191, miR-423-3p, and miR-425) as candidate reference microRNAs for normalization. It turned out that miR-93 was the most stably expressed, thus was chosen as reference microRNA. Early Stückrath et al. [30] showed that miR-16 expressed aberrantly and chose miR-1207 as a reference to normalize the RT-qPCR data. This is because this miR had a constant expression with no difference between all testing cohorts as well as the smallest coefficient variation of the populations [30]. Nevertheless, these miRNAs were shown in other studies to be deregulated [18, 38]. Thus, choosing a housekeeping gene, which could be ideal, is hard to achieve.
More accurate approach for normalizing RT-qPCR data is needed. Use of combined reference genes was proposed to improve the accuracy of the assay (Table 4). In 2013 McDermott et al. [15] presented that the combination of 2 miRNAs, (miR-16 and miR-425) as endogenous control generated more precise and reliable results than any methods using miRNA alone or U6. This approach combined was able to detect the deregulation pattern of target miRNA expression differentiated between cancers and controls in human blood specimens [16]. MiR-16 continued to be used in combination with other miRNAs as, for example, in the study by Davoren et al. [14] who, based on the results of NormFinder, indicated let-7a and miR-16 as the most stable pair of endogenous controls.
Raychaudhuri et al. [33] explored potential reference genes in various samples of primary breast cancer and lymph node metastases and demonstrated that, across the use of miR-16, let-7a, U48, U44 alone, and combined use of various miRNA pairs, the combination of U48 and let-7a had the lowest SV of 0.21. As Hu Z. et al. [31] show, the combination of miRNA-484 and miRNA-191 was the best choice as an endogenous control for serum miRNA detection, at least for most common cancers. In another study, the combination of miR-103a and miR-132 had the highest stability [34].
The comparison between miR-16 and the new combined control genes was performed by investigating the SV (Fig. 3). Combining miR-16 with other control genes helped decreasing the SV. For example, SV of miR-16 and miR-425 calculated by Davoren et al. [14] was 0.185, or 85% lower than that for using miR-16 only. The combination of miR-16 and let-7a also reduced the SV to 0.221, or 42% lower than for miR-16 alone [15]. Other studies employed other miRNA combinations, including U48 and let-7a, miR-484 and miR-191, as well as miR-103a and miR-132 [31, 33]. In all cases, obtained SV were lower than for miR-16 alone.

Comparison of SV between miR-16 and combined control genes strategy. The stability in expression of miR-16 was compared with those of combined control genes in terms of SV. The higher SV of miR-16 confirmed its lower stability to the combined strategy.
Sources of Heterogeneity
The heterogeneity test was described in terms of I2 statistic, which quantifies the amount of dispersion and illustrates to what extent the results of studies are consistent (range from 0 to 100%). The range of I2 is independent of effect size and unaffected by the number of studies in the meta-analysis. I2 statistic describes the percentage of the variability in effect estimation, which is due to heterogeneity rather than sampling error [39]. A low p-value provides evidence of heterogeneity of intervention effects. The higher I2 is, the more heterogeneity the analysis gets. Regarding to Table 3, a high I2 value (90%) illustrated a significant heterogeneity in the meta-analysis.
The studies included in this review varied in many aspects. The first reason could be due to differences of the techniques employed to measure miR-16 expression. Most studies used plasma/serum samples, but the others extracted miRNAs from tissue samples. The stages of breast cancer could also be a source of heterogeneity. The samples were obtained in different stages of breast cancer, which have been stated to have different miRNA profiles. Most studies lack information about the follow-up time, and a history of previous treatments. The outcomes of these studies were demonstrated in various ways. Some reported the Ct and SD values but other studies use fold change or illustrate their findings in term of quartile range, which can lead to the difficulty in calculating the Ct for meta-analysis.
CONCLUSIONS
This study provides an overall view of the expression pattern of miR-16 and clarifies its ability as an endogenous control in breast cancer. Although various studies have used miR-16 to normalize their RT-qPCR data, it showed deregulation in breast cancer as well as its subtypes. Moreover, as miR-16 is likely to be released from erythrocytes via hemolysis, its levels in the plasma may be prone to artificial elevation during the extraction. Combining miRNAs into panels may help, for example, use of the combination of miR-16 and miR-425 as in the study of McDermott A.M. et al. [15]. The accuracy of this combined endogenous control has been proven by other subsequent studies. While finding an ideal housekeeping gene for internal control may not be possible, this strategy allows improvement in the accuracy of the RT-qPCR assays for the diagnosis of breast cancer.
REFERENCES
American Cancer Society. 2020. How common is breast cancer? https://www.cancer.org/cancer/breast-cancer/about/how-common-is-breast-cancer.html.
Centers for Disease Control and Prevention. 2019. Basic information about breast cancer. https://www.cdc.gov/ cancer/breast/basic_info/index.htm.
O’Brien J., Hayder H., Zayed Y., Peng C. 2018. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 9, 402. https://doi.org/10.3389/fendo.2018.00402
MacFarlane L.A., Murphy P.R. 2010. MicroRNA: Biogenesis, function and role in cancer. Curr. Genomics. 11 (7), 537–561. https://doi.org/10.2174/138920210793175895
Tan W., Liu B., Qu S., Liang G., Luo W., Gong C. 2018. MicroRNAs and cancer: Key paradigms in molecular therapy. Oncol. Lett. 15 (3), 2735‒2742. https://doi.org/10.3892/ol.2017.7638
Lu J., Getz G., Miska E.A., Alvarez-Saavedra E., Lamb J., Peck D., Sweet-Cordero A., Ebert B.L., Mak R.H., Ferrando A.A., Downing J.R., Jacks T., Horvitz H.R., Golub T.R. 2005. MicroRNA expression profiles classify human cancers. Nature. 435 (7043), 834–838. https://doi.org/10.1038/nature03702
Miska E.A., Alvarez-Saavedra E., Townsend M., Yoshii A., Šestan N., Rakic P., Constantine-Paton M., Horvitz H.R. 2004. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 5 (9), R68. https://doi.org/10.1186/gb-2004-5-9-r68
Chen C. 2005. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 33 (20), e179–e179. https://doi.org/10.1093/nar/gni178
Li W., Ruan K. 2009. MicroRNA detection by microarray. Anal. Bioanal. Chem. 394 (4), 1117‒1124. https://doi.org/10.1007/s00216-008-2570-2
Bustin S. 2002. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): Trends and problems. J. Mol. Endocrinol. 29 (1), 23–39. https://doi.org/10.1677/jme.0.0290023
Nolan T., Hands R.E., Bustin S.A. 2006. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 1 (3), 1559–1582. https://doi.org/10.1038/nprot.2006.236
Aqeilan R.I., Calin G.A., Croce C.M. (2010) miR-15a and miR-16-1 in cancer: Discovery, function and future perspectives. Cell Death Differ. 17 (2), 215–220. https://doi.org/10.1038/cdd.2009.69
Linsley P.S., Schelter J., Burchard J., Kibukawa M., Martin M.M., Bartz S.R., Johnson J.M., Cummins J.M., Raymond C.K., Dai H.Y., Chau N., Cleary M., Jackson A.L., Carleton M., Lim L. 2007. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol. Cell. Biol. 27 (6), 2240–2252. https://doi.org/10.1128/MCB.02005-06
Davoren P.A., McNeill R.E., Lowery A.J., Kerin M.J., Miller N. 2008. Identification of suitable endogenous control genes for microRNA gene expression analysis in human breast cancer. BMC Mol. Biol. 9 (1), 76. https://doi.org/10.1186/1471-2199-9-76
McDermott A.M., Kerin M.J., Miller N. 2013. Identification and validation of miRNAs as endogenous controls for RQ-PCR in blood specimens for breast cancer studies. PLoS One. 8 (12), e83718. https://doi.org/10.1371/journal.pone.0083718
Liu Q., Fu H., Sun F., Zhang H., Tie Y., Zhu J., Xing R., Sun Z., Zhenget X. 2008. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 36 (16), 5391.
Wirsching J., Graßmann S., Eichelmann F., Harms L.M., Schenk M., Barth E., Berndzen A., Olalekan M., Sarmini L., Zubere H., Aleksandrova K. 2018. Development and reliability assessment of a new quality appraisal tool for cross-sectional studies using biomarker data (BIOCROSS). BMC Med. Res. Methodol. 18 (1), 122. https://doi.org/10.1186/s12874-018-0583-x
Ni Q., Stevic I., Pan C., Müller V., Oliveira-Ferrer L., Pantel K., Schwarzenbach H. 2018. Different signatures of miR-16, miR-30b and miR-93 in exosomes from breast cancer and DCIS patients. Sci. Rep. 8 (1), 12974. https://doi.org/10.1038/s41598-018-31108-y
Heneghan H.M., Miller N., Kelly R., Newell J., Kerin M.J. 2010. Systemic miRNA-195 differentiates breast cancer from other malignancies and is a potential biomarker for detecting noninvasive and early stage disease. Oncologist. 15 (7), 673–682. https://doi.org/10.1634/theoncologist.2010-0103
Zhu W., Qin W., Atasoy U., Sauter E.R. 2009. Circulating microRNAs in breast cancer and healthy subjects. BMC Res. Notes. 2 (1), 89. https://doi.org/10.1186/1756-0500-2-89
Xiang M., Zeng Y., Yang R., Xu H., Chen Z., Zhong J., Xie H., Xu Y., Zeng X. 2014. U6 is not a suitable endogenous control for the quantification of circulating microRNAs. Biochem. Biophys. Res. Commun. 454 (1), 210–214. https://doi.org/10.1016/j.bbrc.2014.10.064
Hu J., Wang Z., Liao B.Y., Yu L., Gao X., Lu S., Wang S., Dai Z., Zhang X., Chen Q., Qiu S.-J., Wu Y., Zhu H., Fan J., Zhou J., Wang J. 2014. Human miR-1228 as a stable endogenous control for the quantification of circulating microRNAs in cancer patients: miR-1228 as a control for quantifying circulating miRNAs. Int. J. Cancer. 135 (5), 1187–1194. https://doi.org/10.1002/ijc.28757
Rinnerthaler G., Hackl H., Gampenrieder S., Hamacher F., Hufnagl C., Hauser-Kronberger C., Zehentmayr F., Fastner G., Sedlmayer F., Mlineritsch B., Greil R. 2016. miR-16-5p is a stably-expressed housekeeping microRNA in breast cancer tissues from primary tumors and from metastatic sites. Int. J. Mol. Sci. 17 (2), 156. https://doi.org/10.3390/ijms17020156
Rice J., Roberts H., Rai S.N., Galandiuk S. 2015. Housekeeping genes for studies of plasma microRNA: A need for more precise standardization. Surgery. 158 (5), 1345–1351. https://doi.org/10.1016/j.surg.2015.04.025
Usmani A., Shoro A.A., Shirazi B., Memon Z., Hussain M. 2017. miR-16: A novel hereditary marker in breast cancer and their offspring. J. Pak. Med. Assoc. 67 (3), 5.
Shin V.Y., Siu J.M., Cheuk I., Ng E.K.O., Kwong A. 2015. Circulating cell-free miRNAs as biomarker for triple-negative breast cancer. Br. J. Cancer. 112 (11), 1751–1759. https://doi.org/10.1038/bjc.2015.143
Ng E.K.O., Li R., Shin V.Y., Jin H.C., Leung C.P.H., Ma E.S.K., Pang R., Chua D., Chu K.-M., Law W.L., Law S.Y.K., Poon R.T.P., Kwong A. 2013. Circulating microRNAs as specific biomarkers for breast cancer detection. PLoS One. 8 (1), e53141. https://doi.org/10.1371/journal.pone.0053141
Zhao Q., Deng S., Wang G., Liu C., Meng L., Qiao S., Shen L., Zhang Y., Lü J., Li W., Zhang Y., Wang M., Pestell R.G., Liang C., Yu Z. 2016. A direct quantification method for measuring plasma microRNAs identified potential biomarkers for detecting metastatic breast cancer. Oncotarget. 7 (16), 21865–21874. https://doi.org/10.18632/oncotarget.7990
Appaiah H.N., Goswami C.P., Mina L.A., Badve S., Sledge G.W., Liu Y., Nakshatri H. 2011. Persistent upregulation of U6:SNORD44 small RNA ratio in the serum of breast cancer patients. Breast Cancer Res. 13 (5), R86. https://doi.org/10.1186/bcr2943
Stückrath I., Rack B., Janni W., Jäger B., Pantel K., Schwarzenbach H. 2015. Aberrant plasma levels of circulating miR-16, miR-107, miR-130a and miR-146a are associated with lymph node metastasis and receptor status of breast cancer patients. Oncotarget. 6 (15), 13387.
Hu Z., Dong J., Wang L.E., Ma H., Liu J., Zhao Y., Tang J., Chen X., Dai J., Wei Q., Zhang C., Shen H. 2012. Serum microRNA profiling and breast cancer risk: The use of miR-484/191 as endogenous controls. Carcinogenesis. 33 (4), 828–834. https://doi.org/10.1093/carcin/bgs030
Shen J., Hu Q., Schrauder M., Yan L., Wang D., Medico L., Medico L., Guo Y., Yao S., Zhu Q., Liu B., Qin M., Beckmann M.W., Fasching P.A., Strick R., et al. 2014. Circulating miR-148b and miR-133a as biomarkers for breast cancer detection. Oncotarget. 5 (14), 5284–5294. https://doi.org/10.18632/oncotarget.2014
Raychaudhuri M., Schuster T., Buchner T., Malinowsky K., Bronger H., Schwarz-Boeger U., Höfler H., Avril S. 2012. Intratumoral heterogeneity of microRNA expression in breast cancer. J. Mol. Diagn. 14 (4), 376–384. https://doi.org/10.1016/j.jmoldx.2012.01.016
Zhang L., Xu Y., Jin X., Wang Z., Wu Y., Zhao D., Chen G., Li D., Wang X., Cao H., Xie Y., Liang Z. 2015. A circulating miRNA signature as a diagnostic biomarker for non-invasive early detection of breast cancer. Breast Cancer Res. Treat. 154 (2), 423–434. https://doi.org/10.1007/s10549-015-3591-0
Faraone S.V. 2008. Interpreting estimates of treatment effects: implications for managed care. Pharm. Ther. 33 (12), 700.
Leidner R.S., Li L., Thompson C.L. 2013. Dampening enthusiasm for circulating microRNA in breast cancer. PLoS One. 8 (3), e57841. https://doi.org/10.1371/journal.pone.0057841
McDonald J.S., Milosevic D., Reddi H.V., Grebe S.K., Algeciras-Schimnich A. 2011. Analysis of circulating microRNA: Preanalytical and analytical challenges. Clin. Chem. 57 (6), 833–840. https://doi.org/10.1373/clinchem.2010.157198
Hamam R., Hamam D., Alsaleh K., Kassem M., Zaher W., Alfayez M., Aldahmash A., Alajez N.M. 2017. Circulating microRNAs in breast cancer: novel diagnostic and prognostic biomarkers. Cell Death Dis. 8 (9), e3045. https://doi.org/10.1038/cddis.2017.440
Higgins J.P., Thompson S.G. 2002. Quantifying heterogeneity in a meta-analysis. Stat. Med. 21 (11), 1539–1558.
ACKNOWLEDGMENTS
The authors would like to thank the Oncology Hospital HCMC for their contribution to collecting samples.
Funding
This research is funded by Vietnam National University, Ho Chi Minh City (VNU-HCM) under grant number 562-2020-18-02.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
COMPLIANCE WITH ETHICAL STANDARDS
There is no conflict of interest to disclose. This article does not contain any studies involving animals or human participants performed by any of the authors.
ADDITIONAL INFORMATION
The text was submitted by the author(s) in English.
Additional information
Abbreviations: miRNAs, microRNAs; BC, breast cancer; Cq, quantitative cycle; RT-qPCR, reverse transcription quantitative polymerase chain reaction; SMD, standard mean difference; I2, inter-study heterogeneity; SV, stability value.
Rights and permissions
About this article

Cite this article
Thu, H.N., Vy, H.T., Thanh, T.N. et al. miRNA-16 as an Internal Control in Breast Cancer Studies: A Systematic Review and Meta-Analysis. Mol Biol 55, 941–949 (2021). https://doi.org/10.1134/S0026893321050137
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026893321050137