
Abstract
MicroRNAs epigenetically regulate physiological and pathological processes. Previously, we found that miR-204-5p is expressed at low levels in melanoma cells, and an increase in its level leads to a change in proliferation, migration, and invasion of these cancer cells. Now, using bioinformatics analysis, it has been shown that the target of miR-204-5p is FOXC1 transcription factor, which is implicated in carcinogenesis. Using the luciferase reporter assay, it was found that miR-204-5p suppresses expression of the FOXC1 gene by binding to its 3' non-coding region. Transfection of small interfering RNA (siRNA) targeting FOXC1 into melanoma cells caused a decrease in miR-204-5p levels, which is consistent with the generally accepted concept of feedback regulation of miRNA expression by target genes. According to the results of the MTT test and fluorescence microscopy, the proliferation level of melanoma cells under the influence of siRNA to FOXC1 decreased 72 h after transfection. Changes in the ratio of cells by cell cycle phase were analyzed using flow cytometry. Regulatory relationships between FOXC1 and miR-204-5p, and an inhibitory effect of FOXC1 knockdown on melanoma cell proliferation were revealed. Based on the results, it can be assumed that miR-204-5p regulates proliferation of melanoma cells by affecting FOXC1 expression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Cancer cell resistance to therapeutic agents is one of the main problems in modern oncology. This is caused by many factors, including the heterogeneity of tumor tissue and the possible presence of a pool of dormant (quiescent, G0) cells within the same tumor. It is not known at what stage of carcinogenesis dormant cells are formed [1], but they are able to persist in the body after antitumor therapy, circulate in the bloodstream and settle in other organs and tissues, while participating in the development of metastasis [2]. The molecular mechanisms of activation (transformation into proliferating tumor cells) of dormant cells, and the transition from G0 to G1, are not fully understood. It is assumed that the components of the immune surveillance system, the tumor microenvironment, and epigenetic regulators, including microRNA, are involved in this process.
MicroRNAs are short (20‒24 nt) noncoding RNAs that are involved in the post-transcriptional regulation of gene expression. MicroRNAs transcribed from genomic DNA and subjected to further processing and export to the cytoplasm [3, 4] play an important role in the regulation of many physiological and pathological processes [5]. We have previously shown that miR-204-5p is one of the lowest expressed microRNAs in melanoma cells, but not in melanocytic nevi [6]. It was found that restoration of the miR-204-5p level in tumor cells leads to a change in the rate of their proliferation, migration, and invasion [7]. In addition, recently Díaz-Martínez et al. [8] found that restoration of miR-204-5p expression in melanoma cells causes a loss of resistance to the targeted antitumor drug vemurafenib. The authors proposed various possible mechanisms of this phenomenon, including the effect of miR-204-5p on regulators of the cell cycle and apoptosis. We hypothesized that miR-204-5p not only directly participates in the regulation of the cell cycle, but also affects the transition of cells to the dormant state, G0. One of the causes for the resistance of cells to antitumor agents is associated precisely with tumor cells that are in the G0 phase and are therefore not sensitive to the action of agents that suppress proliferation.
We investigated the regulation mechanisms of melanoma cell proliferation under the influence of miR-204-5p.
EXPERIMENTAL
Cultivation of melanoma cells. Experiments were carried out on the human melanoma cell lines BRO (provided by the Research Institute of Fundamental and Clinical Immunology, Novosibirsk, Russia) and SK-MEL-2 (Biolot, Russia). The cells were cultured in RPMI-1640 medium with L-glutamine (PanEco, Russia) with 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific, United States) at 37°C and 5% CO2 in a Sanyo MSO-5AC CO2 incubator (Sanyo Electric Co., Ltd., Japan).
Bioinformatics analysis. The search and analysis of miR-204-5p target genes was carried out using four computational tools for microRNA target prediction TargetScan (version 7.0; http://www.targetscan.org), miRDB (version 5.0; http://mirdb.org/miRDB), miRWalk 2.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/), and miRTarBase (version 4.5; http://mirtarbase.mbc.nctu.edu.tw/). Target genes of miR-204-5p, which were found in all four databases, were considered. At the next stage, target genes, whose TargetScore values (the probability deduced from the distribution of microRNA target genes using the Bayes-Gauss logarithmic model) in the miRDB database were at least 80, were identified. This is associated with the fact that all target genes are distributed in a special algorithm according to prediction scores from 50 to 100. To assess the role of miR-204-5p in the regulation of internal signaling using the PANTHER v10.0 program (www.pantherdb.org), analysis of the metabolic and signaling cascades changes in the cells was carried out.
Design of siRNA for FOXC1 knockdown. The selection of siRNA and scramble RNA sequences for negative control was performed using the siDirect 2 program (http://sidirect2.rnai.jp); siRNAs were selected using Wizard Software v.3.1. (Invitrogen, Thermo Fisher Scientific, United States).
The following parameters served as selection criteria: the content of G/C nucleotides is 35–55%, a sequence of 20–25 nt, the absence of transcripts of other genes that coincide with the sequences of the selected siRNAs, according to the results of the analysis using the BLAST system (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The selected sequences did not contain more than three identical nucleotides in a row nor nucleotide repeats. To assess the knockdown specificity in the cells, scramble siRNAs were used as the negative control.
Synthesis of sense scramble siRNAs was carried out by the Syntol Company (Russia):
FOXC1_sense siRNA: 5′-GGGAAUAGUAGCUGUCAAATTdTdT-3′;
FOXC1_scramble siRNA: 5′-GGAATGGTAGCGACATATATT-3′.
Transfection of siRNA into BRO and SK-MEL-2 melanoma cells was performed when the cell density reached 70%.
Assessment of melanoma cell viability. The MTT test was used to assess proliferation of BRO and SK-MEL-2 melanoma cells. The cells were cultured in a 96-well plate without intervention for 24 h, and upon reaching a concentration of 1 × 104 cells in 100 μL of culture medium, they were transfected with siRNA to FOXC1. Transfection was performed using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. After 24 h, the culture medium was removed, a solution of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) (Invitrogen, Thermo Fisher Scientific) was added to each well at a concentration of 5 mg/mL, in the amount of 15 μL MTT per 135 μL culture medium. The cells with the MTT solution were incubated for 4 h at 37°C and 5% CO2 in a CO2 incubator. The rate of formazan accumulation was used to assess the metabolic activity of the cells, corresponding to the rate of proliferation, one, two, and three days after transfection. The measurements were carried out on an Efos-9305 spectrophotometer (Shvabe Photosystems, Russia) at a wavelength of 560 nm. Cells transfected with scramble siRNA were used as the negative control. The experiment was carried out in three technological replicates. The data were normalized to the corresponding values in the control and expressed as a percentage.
Fluorescence microscopy. For fluorescence microscopy, BRO and SK-MEL-2 cells at a concentration of 5 × 104 cells/mL were plated into a 96-well plate, transfected with siRNA and incubated for 24 h at 37°C and 5% CO2. After 24, 48, and 72 h, cells were stained using the CyQUANT Direct Cell Proliferation Assay (Thermo Fisher Scientific), which contains a green fluorescent dye that directly binds to DNA. After a 30‑minute incubation at room temperature, fluorescence microscopy was performed using a Floid® Cell Imaging Station (Floid Software, version 22809; Thermo Fisher Scientific) at a magnification of ×460. Cells were counted in 10 fields. The nuclei of proliferating cells were stained with green, and the nuclei of living nonproliferating cells remained unstained. The experiment was carried out in three technological replicates.
Luciferase reporter assay for assessing the effect of microRNA. To confirm FOXC1 as a functional miR-204-5p target gene in melanoma cells, the Ambion® pMIR-REPORT Luciferase miRNA Expression Reporter Vector System (Invitrogen™, Applied Biosystems, United States) was used. The nucleotide sequence of the 3' non-coding region (3'-UTR) of the FOXC1 gene, containing the predicted target site for miR-204-5p, was synthesized. Then, the synthesized DNA fragment was cloned into the 3'-UTR of the firefly luciferase gene (Rluc) in the pMIR-REPORT reporter plasmid according to the manufacturer’s instructions. The correctness of the obtained construct was confirmed by Sanger sequencing. In the resulting plasmid, expression of the luciferase reporter undergoes a regulation that mimics the regulation of FOXC1 through the analyzed region with the target site of microRNA. The second normalizing plasmid of the pMIR-REPORT-β-gal system, containing the β‑galactosidase (GALB) gene, was used as an internal standard for signal normalization in order to eliminate possible differences in the transfection efficiency of individual samples. The pMIR-REPORT-β-gal plasmid was added during transfection in a ratio of 1 : 10. The resulting construct was transfected into BRO and SK-MEL-2 melanoma cells using Lipofectamine 3000 (Thermo Fisher Scientific). Nontransfected BRO and SK-MEL-2 melanoma cells, plasmid DNA without the 3'-UTR sequence of the FOXC1 gene, cells transfected only with a synthetic analogue (mimic) of miR-204-5p, and mirVana®miRNA (Ambion, Thermo Fisher Scientific), with the final mimic content of 30 nM were used as negative controls.
The efficiency of transfection was assessed by direct counting of fluorescent cells under a microscope after transfection with the control pcDNA3m3-cgreGFP2 plasmid (carrying the gfp gene from Clytia gregaria, mutated for maturation at 37°C) at a concentration equal to that for the studied plasmid (GFP vector provided by the Laboratory of Biophobiology, Biophysics Institute of the Siberian Branch of the RAS - Division of Federal Research Center “Krasnoyarsk Scientific Center of the Siberian Branch of the RAS,” Krasnoyarsk, Russia).
The luciferase activity of melanoma cells was analyzed 24 h after transfection using a Dual-Light® Systems kit (Applied Biosystems). The cells were lysed and, according to the manufacturer’s instructions, buffer A, Galacton-Plus® β-galactosidase substrate at a dilution of 1 : 100 in buffer B, containing the luciferase substrate luciferin, was added to the lysate. The enzymatic activity of luciferase and β-galactosidase was measured on a Mithras LB 940 Multimode Reader plate luminometer (Berthold Technologies GmbH&Co, Germany). The relative luciferase activity, reflecting the level of FOXC1 expression, was calculated from the ratio of the luminescence intensity of the luciferase reaction product to that for the product of the β-galactosidase reaction in each well.
Evaluation of the vemurafenib effect on melanoma cell proliferation. The concentration of 50% inhibition (IC50) of melanoma cell proliferation was determined using the MTT test. BRO and SK-MEL-2 melanoma cells were cultured in a 96-well plate for 24 h until a concentration of 2 × 105 cells was reached in 500 μL of RPMI-1640 culture medium with L-glutamine and 10% FBS, after which vemurafenib (Carbosynth Ltd, England), dissolved in DMSO (Panreac quimica SA, Spain), at a final concentration of 0.75, 1.25, 2.5, 5.0, and 10.0 μM was added to the wells. The cells with DMSO at the concentration corresponding to that in 5.0 μM vemurafenib were used as a control. The cells were cultured for 72 h, after which the culture medium was removed and 135 μL of fresh culture medium and 15 μL of MTT solution with a concentration of 5 mg/mL were added to each well. The plate was placed in a CO2 incubator for 4 h at 37°C and 5% CO2. The metabolic activity of the cells was assessed spectrophotometrically and was calculated using the formula: (ODs/ODm) × 100%, where the values ODs and ODm correspond to the optical density of the cells of the experimental and control samples at 560 nm. Optical density was determined on an Anthos 2010 ELISA spectrophotometer (Biochrom Ltd, England). IC50 values were calculated using the Graphpad PRISM 8.0 software. The experiment was carried out in three technological replicates.
Dormant cells were isolated according to the method described previously [9] using the antitumor agent vemurafenib at the concentration of 10 IC50. The cells were cultured for 3 days, the culture medium was replaced with a fresh one and cultured for another 48 h, after which siRNA was transfected using Lipofectamine 3000 to suppress FOXC1 expression.
Determination of the proportion of cells in the G0 phase by flow cytometry. After incubation with vemurafenib, BRO and SK-MEL-2 melanoma cells at a concentration of 3 × 105 cells/well were removed from a 6-well plate with trypsin-EDTA buffer (PanEco), centrifuged at 10 000g in a miniSpin centrifuge (Eppendorf, Germany) for 5 min, and the supernatant was removed. The cell suspension was fixed with methyl alcohol (70–100%). For this, 0.5 mL of methyl alcohol was added to the resulting precipitate and incubated for 30 min. Treatment with Triton X-100 for 15 min was used to permeabilize the fixed cells. After each step, the cells were washed twice with phosphate buffered saline (PBS). The cells were stained with anti-Ki-67 monoclonal antibodies conjugated with FITC (#11-5698-82, eBioscienceTM, Thermo Fisher Scientific) at a 1: 200 dilution in FBS. Upon completion of the staining procedure, the samples were supplemented with the DNA-binding dye, propidium iodide (PI) (Thermo Fisher Scientific, Inc., Netherlands), to a concentration of 100 μg/mL and incubated for 15 min.
The stained cells were analyzed on a Navios flow cytometer (Beckman Coulter, Inc., United States) of the Center for Collective Use of the Krasnoyarsk Scientific Center of the Siberian Branch of the RAS using a blue laser (488 nm) and detector filters. The passband for FITC was 530/30 nm, and for PI it was 610/20 nm.
The results were analyzed using Navios Software v. 1.2 and Kaluza v. 2.1.1 (Beckman Coulter, Inc.); at the same time, at least 50 000 cells were examined in each sample. Gating of cells at different stages of the cell cycle was performed in the logarithmic mode. The cells in the G0 phase are characterized by Ki-67 negativity and low levels of the PI signal. In this regard, gating of cells in the G0 phase was assessed in the range of up to 10 on the Ki-67-FITC fluorescence scale (negative result) and in the range of 0.7–1.3 relative fluorescence units of PI.
Determination of miR-204-5p expression level in melanoma cells. The reverse transcription reaction was performed using the MMLV RT kit (Evrogen, Russia). For the synthesis of cDNA, a set of random primers from the MMLV RT kit (Evrogen) and specific primers for microRNA, TaqMan Assays hsa-miR-204-5p (No. A25576, Applied BiosystemsTM, Thermo Fisher Scientific), were used.
The level of miR-204-5p expression was determined by real-time PCR using a StepOneTM Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific) with 40 amplification cycles. The small nuclear RNAs (snRNAs) RNU6A and RNU6B (Applied Biosystems) were used as endogenous controls. The experiment was carried out in three replicates. The ∆∆CT method was used to analyze the data.
Statistical processing. The statistical significance of the obtained differences was calculated using the Student’s t test and the Mann–Whitney U test in the Statistica 12.0 software package for statistical analysis (StatSoft, Russia). The results were considered significant at p < 0.05. The data are presented as the mean and standard deviation (SD).
RESULTS
Identification of FOXC1 as a Possible miR-204-5p Target
As a result of the bioinformatics analysis for miR-204-5p, 235 target genes were identified. For the four databases used, 36 common genes were identified, 13 of which are involved in the regulation of cell proliferation. The subsequent analysis of gene ontologies of biological processes and molecular functions using the PANTHER v10.0 system allowed us to identify genes among the above 13 genes, whose biological functions are associated with the process of cell proliferation: SIRT1 (sirtuin 1), FOXC1 (forkheadbox C1), TGFBR1 (transforming growth factor beta receptor 1) and USP47 (ubiquitin specific peptidase 47).
Analysis of the human FOXC1 sequence (NM_001453) showed that the 3'-UTR of its mRNA contains regions with a high degree of nucleotide complementarity to the miR-204-5p sequence. Based on these data, it could be assumed that the identified sequence in FOXC1 mRNA is the miR-204-5p binding site with a strong regulatory effect (Fig. 1).

Predicted binding of miR-204-5p to 3'-UTR of FOXC1 mRNA (Homo sapiens forkhead box C1; Gene ID 2296).
Study of FOXC1 as a Direct miR-204-5p Target
To confirm FOXC1 as a functionally significant miR-204-5p target gene, we evaluated the regulatory activity of this microRNA using a luciferase reporter construct containing the 3'-UTR of the FOXC1 gene with the target site for miR-204-5p. It was found that miR-204-5p inhibited the luciferase activity of the reporter system by 72% in BRO melanoma cells and by 34% in SK-MEL-2 melanoma cells (Fig. 2). From this, we can conclude that miR-204-5p suppresses FOXC1 expression.

Relative luciferase activity in SK-MEL-2 and BRO melanoma cells 48 h after transfection with miR-204-5p mimic. *р = 0.005.
In addition, we studied the effect of siRNA to FOXC1, transfected into melanoma cells, on miR-204-5p expression. It was found that in this case the miR-204-5p level decreased to 0 in BRO cells, and from 1.23 to 0.64 in SK-MEL-2 cells (Fig. 3).

Expression of miR-204-5p in SK-MEL-2 (a) and BRO (b) melanoma cells transfected with FOXC1 siRNA. The expression level of miR-204-5p was calculated relative to expression of RNU6A and RNU6B before and after treatment with FOXC1 siRNA. Here and in other figures: siRNAsc—scramble siRNA; *p < 0.05.
Decreasing FOXC1 Level in Melanoma Cells Is Accompanied by a Decrease in Proliferative Activity
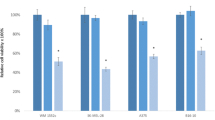
We studied the effect of FOXC1 siRNA transfection into melanoma cells on the level of their proliferation. According to the MTT test, 72 h after transfection, the viability of BRO and SK-MEL-2 cells decreased by 39 and 40%, respectively, compared with control cells transfected with scramble siRNA (Fig. 4).

The viability of BRO (a) and SK-MEL-2 (b) melanoma cells 24, 48, and 72 h after transfection with FOXC1 siRNA. Cell viability was determined using the MTT test; the proliferative activity of control cells was taken as 100%.
Similar data were also obtained by immunofluorescence microscopy: 72 h after transfection, proliferation of BRO and SK-MEL-2 cells decreased by 2.7 and 1.5 times, respectively (Fig. 5). Thus, it is logical to assume that the FOXC1 gene is the target of miR-204-5p, and the decrease in the level of the FOXC1 transcription factor induced by this interaction leads to a change in the rate of proliferation of melanoma cells.

The proliferative activity of BRO and SK-MEL-2 melanoma cells 72 h after transfection with FOXC1 siRNA according to the results of fluorescence microscopy based on visualization of living, proliferating, undamaged cells using DNA-binding dye according to CyQUANT Direct Cell Proliferation Assay technique.
Effect of Vemurafenib and FOXC1 siRNA on miR-204-5p Expression
We analyzed the effect of vemurafenib on proliferation of melanoma cells. This compound belongs to the group of targeted drugs targeting the mutant BRAF protein, a component of the MAPK signaling cascade [10]. It is known that tumor cells within the same tumor can be heterogeneous in terms of the mutational status. By inducing apoptosis in BRAF-positive cells, BRAF-negative cells can maintain viability, including being in or transferring into the dormant state, the G0 phase. It was shown that the use of cytostatic agents can promote the transition of tumor cells to the G0 phase [11]. Considering these data, we analyzed the effect of the BRAF inhibitor, vemurafenib (at a 10 IC50 concentration ), on SK-MEL-2 cells. It was found by flow cytometry that after treatment with vemurafenib, the proportion of Ki-67-negative cells increased from 7% to 35%, while the level of miR-204-5p decreased by 65%. As can be seen from the data shown in Fig. 6, transfection of FOXC1 siRNA into these cells did not prevent a decrease in the level of miR-204-5p (Fig. 6), and 72 h after its transfection, a decrease in the cell proliferative activity was recorded (Table 1).

The level of G0-positive SK-MEL-2 cells before (a) and after (b) treatment with vemurafenib: positive cells are localized in the lower square of the diagrams. The miR-204-5p level in SK-MEL-2 cells after treatment with vemurafenib (c) and transfection with FOXC1 siRNA (d).
DISCUSSION
Previously, it was shown that in melanoma, miR-204-5p expression is strongly reduced compared with benign melanocytic nevi [5, 12]. Based on data on the participation of miR-204-5p in the regulation of proliferation of squamous cell carcinoma and breast cancer cells [13, 14], we hypothesized that this microRNA is also involved in the regulation of melanoma cell proliferation.
As a result of the bioinformatics analysis, we identified possible miR-204-5p target genes involved in the regulation of cell proliferation. One of these genes is FOXC1. The FOXC1 protein encoded by this gene is a transcription factor involved in the development of tissues and organs, including skin. Altered expression of FOXC1 was described for breast [14], endometrium [15], and pancreas cancer cells [16]. FOXC1 belongs to the Forkhead box (Fox) family of transcription factors, which is a group of evolutionarily conserved transcriptional regulators with a common DNA-binding domain [17]. Members of this family play an important role in both physiological processes and carcinogenesis, affecting metabolism, development, differentiation, proliferation, apoptosis, migration, and invasion of tumor cells [18]. It was also shown that the MST1R/PI3K/AKT signaling cascade in melanoma cells is activated by FOXC1 overexpression, which leads to an increase in their proliferation, migration, and invasion [19]. Moreover, FOXC1 is known to regulate the formation of mesenchymal niches for hematopoietic stem cells [20].
To prove that FOXC1 functions as a regulatory target of miR-204-5p, we created a genetic construct with a fragment of the FOXC1 gene inserted. Analysis of the luciferase activity of cells expressing the reporter gene and miR-204-5p revealed that under the action of miR-204-5p, expression of the reporter protein decreases. Therefore, miR-204-5p apparently regulates FOXC1 expression by binding to the complementary FOXC1 mRNA sequence. It should be noted that the decrease in luciferase activity differed in the two studied types of melanoma cells, but it was significantly not less than 25%, and based on this we can consider the specificity of the miR-204-5p action. Thus, the FOXC1 gene can be a target of and be regulated by miR-204-5p. This conclusion is also confirmed by the results of our analysis of the FOXC1 siRNA effect on miR-204-5p expression. It was shown that, upon transfection of melanoma cells with siRNA to FOXC1, the miR-204-5p level decreased, which may be caused by the inherent ability of target genes to regulate the level of microRNA expression according to the feedback principle [21]. Thus, FOXC1 regulates proliferation of melanoma cells, and is in turn a functional target of miR-204-5p. It is known that FOXC1, acting on the cyclins CDK1, CDK2, CDK4, and CDK6, affects the PI3K/AKT signaling cascade and thus progression trough the cell cycle [22].
It was found that a decrease in the FOXC1 level causes inhibition of proliferation, and ectopic expression of FOXC1 in tumor cells induces arrest of the transition from the G0 to G1 phase [23]. To increase the proportion of cells in the G0 phase, we used BRAF-negative cells treated with vemurafenib. It was shown that vemurafenib induced an increase in the proportion of Ki-67 negative cells, which was accompanied by a decrease in miR-204-5p expression. This result is consistent with the data of Vitiello et al. [24] on the effect of vemurafenib on miR-204-5p expression. The authors showed that the treatment of the BRAF-negative cell lines MeWo and SK-Mel-197 with vemurafenib did not lead to an increase in miR-204-5p expression.
It should be noted that the combined effect of vemurafenib and FOXC1 siRNA on melanoma cells resulted in a decrease in both the miR-204-5p level and the proliferative activity of the cells. This indirectly confirms the presence of a regulatory relationship between miR-204-5p and its target, FOXC1. It is possible that in skin melanoma, miR-204-5p, affecting the PI3K/AKT signaling cascade through FOXC1 expression, modulates the activity of cyclins and the progression trough the cell cycle, is involved in the functioning of cells in the dormant state, preventing an effective response to an anticancer drug.
REFERENCES
Schardt J.A., Meyer M., Hartmann C.H., Schubert F., Schmidt-Kittler O., Fuhrmann C., Polzer B., Petronio M., Eils R., Klein C.A. 2005. Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell. 8, 227–239.
Lim P.K., Bliss S.A., Patel S.A., Taborga M., Dave M.A., Gregory L.A., Greco S.J., Bryan M., Patel P.S., Rameshwar P. 2011. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 71, 1550–1560.
Wang Z. 2010. MicroRNA: A matter of life or death. World J. Biol. Chem. 1, 41–54.
Chen Z., Li Z., Soutto M., Wang W., Piazuelo M.B., Zhu S., Guo Y., Maturana M.J., Corvalan A.H., Chen X., Xu Z., El-Rifai W. 2018. Integrated analysis of mouse and human gastric neoplasms identifies conserved microRNA networks in gastric carcinogenesis. Gastroenterology. 28, 46–52.
Aushev V.N. 2015. MicroRNA: Small molecular of great significance. Klin. Onkogematol. 8, 1‒12.
Palkina N., Komina A., Aksenenko M., Moshev A., Savchenko A., Ruksha T. 2018. MiR-204 and miR-3065 exert antitumor effect on melanoma cells. Oncol. Lett. 15, 8269–8828.
Toda H., Kurozumi S., Kijima Y., Idichi T., Shinden Y., Yamada Y., Arai T., Maemura K., Fujii T., Horiguchi J., Natsugoe S., Seki N. 2018. Molecular pathogenesis of triple-negative breast cancer based on microRNA expression signatures: Antitumor miR-204-5p targets AP1S3. Eur. J. Hum. Genet. 63, 1197–1210.
Díaz-Martínez M., Benito-Jardón L., Alonso L., Koetz-Ploch L., Hernando E., Teixidó J. 2018. miR-204-5p and miR-211-5p contribute to BRAF inhibitor resistance in melanoma. Cancer Res. 78, 1017–1030.
Li S., Kennedy M., Payne S., Kennedy K., Seewaldt V., Pizzo S., Bachelder E. 2014. Model of tumor dormancy/recurrence after short-term chemotherapy. PLoS One. 9 (5), e98021.
Ravnan M.C., Matalka M.S. 2012. Vemurafenib in patients with BRAF V600E mutation-positive advanced melanoma. Clin. Ther. 34 (7), 1474–1486.
Kudryavtsev I.V., Khaidukov S.V., Zurochka A.V., Chereshnev V.A. 2012. Protochnaya tsitometriya v eksperimental’noi biologii (Flow Cytometry in Experimental Biology). Yekaterinburg: Ural. Otd. Ross. Akad. Nauk.
Shellman M., Shellman Y. 2020. Human against machine? Machine learning identifies microRNA ratios as biomarkers for melanoma. J. Invest. Dermatol. 140, 18–20.
Tang J., Li Z., Zhu Q., Wen W., Wang J., Xu J., Wu W., Zhu Y., Xu H., Chen L. 2020. miR-204-5p regulates cell proliferation, invasion, and apoptosis by targeting IL-11 in esophageal squamous cell carcinoma. J. Cell. Physiol. 235, 3043–3055.
Hong B.S., Ryu H.S., Kim N., Kim J., Lee E., Moon H., Kim K.H., Jin M.S., Kwon N.H., Kim S., Kim D., Chung D.H., Jeong K., Kim K., Kim K.Y., et al. 2019. Tumor suppressor miRNA-204-5p regulates growth, metastasis, and immune microenvironment remodeling in breast cancer. Cancer Res. 79, 1520–1534.
Chung T., Lau T., Cheung T., Yim S., Lo K., Siu N., Chan L., Yu M., Kwong J., Doran G., Barroilhet L., Ng A.S., Wong R., Wang V., Mok S., et al. 2012. Dysregulation of microRNA-204 mediates migration and invasion of endometrial cancer by regulating FOXC1. Int. J. Cancer. 130, 1036–1045.
Subramani R., Camacho F., Levin C., Flores K., Clift F., Galvez A., Terres M., Rivera S., Kolli S., Dodderer J., Miranda M., Rodriguez A., Pedroza D., Chatterjee A., Lakshmanaswamy R. 2018. FOXC1 plays a crucial role in the growth of pancreatic cancer. Oncogenesis. 7, 52–63.
Myatt S.S., Lam E.W. 2007. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer. 7, 847–859.
Lai E., Prezioso V.R., Smith E., Litvin O., Costa R.H., Darnell J.E. 1990. HNF-3A, a hepatocyte-enriched transcription factor of novel structure is regulated transcriptionally. Genes Dev. 4, 1427‒1436.
Wang J., Li L., Liu S., Zhao Y., Wang L., Du G. 2016. FOXC1 promotes melanoma by activating MST1R/PI3K/AKT pathway and is associated with poor prognosis in melanoma. Oncotarget. 7, 375‒387.
Omatsu Y., Seike M., Sugiyama T., Kume T., Nagasawa T. 2014. Foxc1 is a critical regulator of haematopoietic stem/progenitor cell niche formation. Nature. 508, 536–540.
Gulyaeva L.F., Kushlinskiy N.E. 2016. Regulatory mechanisms of microRNA expression. J. Transl. Med. 14, 143.
Liu Y., Miao Y., Gao X., Wang Y.Y., Wang H., Zheng Y.W., Zhao Z.Y. 2018. MicroRNA-200a affects the proliferation of airway smooth muscle cells and airway remodeling by targeting FOXC1 via the PI3K/AKT signaling pathway in ovalbumin-induced asthmatic mice. Cell. Physiol. Biochem. 50, 2365–2389.
Zhou Y., Kato H., Asanoma K., Kondo H., Arima T., Kato K., Matsuda T., Wake N. 2002. Identification of FOXC1 as a TGF-β1 responsive gene and its involvement in negative regulation of cell growth. Genomics. 80, 465–472.
Vitiello M., Tuccoli A., D′Aurizio R., Sarti S., Giannecchini L., Lubrano S., Marranci A., Evangelista M., Peppicelli S., Ippolito C., Barravecchia I., Guzzolino E., Montagnani V., Gowen M., Mercoledi E., et al. 2017. Context-dependent miR-204 and miR-211 affect the biological properties of amelanotic and melanotic melanoma cells. Oncotarget. 8, 25395–25417.
Funding
This work was financially supported by the Russian Science Foundation (project no. 19-15-00110).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests. The authors declare that they have no conflict of interest.
Statement on the welfare of animals. This article does not contain any studies involving animals performed by any of the authors.
Statement of compliance with standards of research involving humans as subjects. This article does not contain any studies involving humans as subjects.
Additional information
Translated by D. Novikova
Rights and permissions
About this article

Cite this article
Dubovtseva, I.Y., Aksenenko, M.B., Nikolaeva, E.D. et al. FOXC1-Mediated Effects of miR-204-5p on Melanoma Cell Proliferation. Mol Biol 55, 610–617 (2021). https://doi.org/10.1134/S0026893321020199
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026893321020199