
Abstract
Nucleoli, the largest subnuclear compartments, are formed around arrays of ribosomal gene repeats transcribed by RNA polymerase I. The primary function of nucleoli is ribosome biogenesis. Specific DNA damage response mechanisms exist to maintain the genomic stability of ribosomal repeats. Here, we provide a snapshot of our current understanding of processes involved in nucleolar DNA damage response. We discuss structure and function of ribosomal repeats, techniques developed for studying DNA damage response in nucleoli, as well as molecular mechanisms of DNA damage-induced repression of nucleolar transcription and nucleoli reorganization.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
DNA DAMAGE RESPONSE
Despite of its high chemical stability, the DNA molecule is still sensitive to different stress factors. Various DNA lesions are induced by aggressive environmental factors, such as ionizing radiation, UV light, and xenobiotics, and are classified as exogenous lesions. At the same time, endogenous lesions arise spontaneously during normal cell metabolism, for example, when replication forks collapse [1], transcription and replication machineries interfere with each other [2], sister chromatids are incompletely segregated during mitosis [3], etc. Tens or even hundreds of thousands of lesions daily arise in genomic DNA as a result of the total effect of exogenous and endogenous factors [4]. Only 10–50 of the DNA lesions are double-strand breaks (DSBs) according to rough estimates [5, 6]. Although occurring in minor amounts, DSBs are the most cytotoxic DNA lesions. DSB-associated genome instability leads to various pathologies, as is evident from the fact that defects in DSB repair systems directly correlate with the incidence of immune and neurological disorders [7] and cell malignant transformation [8].
Various molecular mechanisms have developed in evolution to maintain the DNA integrity and stability in the cell and are collectively known as the DNA damage response (DDR). The DDR is not restricted to DNA repair, but includes damage detection systems, amplification of the damage signal, recruitment of necessary repair proteins to the lesion, repair per se, the regulation of the cell cycle, and the mechanisms that trigger programmed cell death when repair is impossible [9]. Two main mechanisms are used to eliminate DSBs in higher eukaryotic cells. One is homologous recombination repair (HRR), which restores both physical integrity of the DNA molecule and its nucleotide sequence at the site of damage. DNA end joining is another mechanism utilized by the cell. This mechanism is less accurate and restores the DNA integrity, while the nucleotide sequence is not always restored. The relevant mechanisms include classic nonhomologous end joining (c-NHEJ) and alternative NHEJ (alt-HNEJ). While the NHEJ system is active throughout the cell cycle, HRR works predominantly in the S/G2 phases because the sister chromatid is utilized as a homology donor [10]. The choice of the repair pathway does not always depend on the cell cycle phase alone, but is determined in certain cases by the type of the DNA lesion [11] and the epigenetic context and transcriptional status of the region [12]. Thus, the response to damage is not uniform throughout nuclear DNA, and certain specialization is required in particular regions of the genome. This is clearly seen in the regions that are far more often affected by lesions as compared with other regions and are known as the hotspots. The set includes various genome regions enriched in repetitive sequences, such as telomeres and centromeres [13]; fragile sites, such as late-replicating regions [14]; and intensely transcribed regions [15–17]. Ribosomal genes (rDNA) are among the major sources of genome instability from this viewpoint [18, 19], because of several factors. First, ribosomal genes are the most intensely transcribed genes in the cell nucleus, and a high density of RNA polymerase I (pol I) in rDNA determines a manifold increase in the risk of collisions between transcription and replication machineries. The formation of R-loops is another vulnerability factor typical of rDNA. Newly synthesized RNA can hybridize with the coding DNA in cis to produce a RNA:DNA hybrid structure, which is known as the R-loop [20]. Single-stranded DNA displaced as a result of R-loop formation provides a preferential target for mutation processes, such as deamination and depurination [21]. Being rather stable, R-loops form barriers for the progress of RNA and DNA polymerases and eventually lead to DSBs [22]. Second, replication is complicated in the genome regions that have high contents of GC or simple sequence repeats [23], and rDNA is among these regions. When replicative stress persists for a long period of time, structure-specific endonucleases generate single-strand nicks and DSBs in an attempt to restart the progress of stalled replication forks [24]. Third, GC-rich regions are prone to forming the noncanonical DNA forms that consist of four strands each and are known as the G-quadruplexes. G-quadruplexes may be formed by the nontemplate DNA strand during the progress of pol I and provide a barrier to the further progress of replication forks [25].
Finally, ribosomal genes occur as long tandem repeats and have a higher recombination potential, which may lead to major rearrangements in the case of DNA damage. This factor explains why rDNA instability often accompanies many cancers [26], neurodegenerative disorders [27], and premature aging [28].
Data accumulated over recent years indicate that unique features are characteristic of the response to ribosomal gene damage [29–32]. The features are a main focus of this review. We start with characterizing the structure and function of ribosomal repeats and their organization in the nucleolar compartment. Then we consider the technical aspects of studying DNA damage in the nucleolus; analyze the unique features of the response to rDNA damage, including transcriptional repression and nucleolar reorganization; and consider the mechanisms of rDNA repair and their specifics.
STRUCTURAL AND FUNCTIONAL ORGANIZATION OF THE NUCLEOLUS
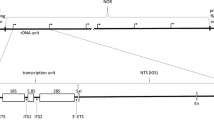
The nucleolus is a large membraneless subnuclear compartment that is found in all types of eukaryotic cells. The main functions of the nucleolus are transcription of the ribosomal genes, rRNA processing, and assembly of ribosome subunits. The ribosomal genes are organized in tandem repeats, which are known as the nucleolar organization region (NOR). The human haploid genome harbors, on average, 300 copies of these 43-kb repeats. Their clusters are in the short arms of all acrocentric chromosomes (chromosomes 13, 14, 15, 21, and 22) (Fig. 1) [33, 34]. The length of a ribosomal gene cluster is surprisingly variable and differs not only between different individuals, but also between different cells of one individual, varying from 50 kb to several megabases [35]. The rDNA karyotype is unique to each individual human as a result of this high variability. The established maximal repeat number is approximately 400 per human genome, while the minimal known repeat number is only 14 [36]. The majority of rDNA repeats are arranged head to tail and are canonical repeats. However, approximately one-third of the repeats form palindromic, or noncanonical, repeats. Each 43-kb repeat unit includes a 13-kb rRNA-coding segment and a 30-kb intergenic spacer. The spacer harbors various regulatory elements, such as a promoter, enhancer, and terminator sequences [37] (Fig. 1).

Schematic organization of the ribosomal repeat. Designations: 18S, 5.8S, and 28S are the sequences coding for the respective RNAs; AsiSI is a recognition site for AsiSI restriction enzyme; CORE is the minimal (core) promoter; ETS is the external transcribed spacer; IGS, is the intergenic spacer; I-PpoI is a recognition site for I-PpoI restriction endonuclease; ORI is an origin of replication; Pol1 is a RNA polymerase I promoter; Pol2 is a RNA polymerase II promoter; T0 is a terminator-like element; T1–T10 are transcription terminators; UCE (upstream control element) is a 5′-terminal regulatory element.
Two forms are known for rDNA repeats: intensely transcribed repeats occur in the form of open euchromatin, while inactive repeats are organized in heterochromatin. Repeats of either form are localized in the nucleolus. Thus, the intensity of rRNA synthesis can be regulated not only by changing the total ribosomal repeat number, but also by epigenetically changing the balance between active and repressed repeat copies [38].
To allow transcription of the rRNA genes, a preinitiation complex is assembled on the rDNA promoter to include pol I, the UBF and SL1 transcription factors. When RNA polymerase I starts synthesizing rRNA, UBF and SL1 remain associated with the promoter region, thus allowing continuous recruitment of pol I molecules [38, 39]. Each rRNA gene can therefore be transcribed several times simultaneously. Such transcription creates a Christmas tree-like structure, which is possible to visualize by electron microscopy [40]. An initial 47S rRNA precursor transcript is processed to produce the mature 18S, 5.8S, and 28S rRNAs, which then undergo posttranscriptional modification with small nucleolar RNPs and additional processing factors. Finally, the processed mature rRNAs bind with ribosomal proteins and are exported into the cytoplasm [41].
Ribosomal repeats of higher eukaryotes are organized in three intranucleolar compartments: a fibrillar center (FC), a dense fibrillar component (DFC), and a granular component (GC). Each of the components is easily identifiable by specific morphology via electron or light microscopy [41]. Many FCs can occur in a nucleolus, each being organized around one to three ribosomal genes [42, 43]. FCs store extra transcription components, including pol I and transcription factors, such as UBF, Treacle, SL1, and others. However, transcription does not occur in FCs, but takes place at the boundary between FCs and the DFC [43, 44]. The DFC surrounds each FC and harbors mostly early rRNA processing factors. In turn, the DFC is surrounded by the GC, where rRNA moves after transcription for further maturation and ribosome assembly. Thus, the organization and structure of the intranucleolar compartments is tightly associated with steps of ribosome biogenesis and rRNA transport.
Properties of liquid drops (condensates) are characteristic of many membraneless subnuclear compartments, such as splicing centers (nuclear speckles) and Cajal bodies. The phenomenon that underlies the formation of these organelles is known as the liquid–liquid phase separation (LLPS). LLPS is now increasingly recognized as a basis of the internal organization of the entire cell [45, 46]. Nucleoli also display certain liquid properties [47]. Intrinsically disordered domains of fibrillarin, nucleophosmin, and pol I (which are key structures of the DFC, GC, and FC, respectively) proved to be essential for drop formation, and their RNA-binding motifs are necessary for maintaining LLPS [48, 49]. The structure of the nucleolus is highly dynamic owing to its liquid properties. For example, a large-scale reorganization of the nucleolus and the formation of the so-called nucleolar caps are observed when ribosomal gene transcription is inhibited with actinomycin D (ACD) [50]. To accompany the cap formation, FC and DFC proteins together with rDNA move to the periphery of the nucleolus so that the FC faces the nucleoplasm and the DFC faces the interior of the nucleolus. Many ribosomal proteins are transferred from the nucleolus into the nucleoplasm simultaneously with the above events. Once released, ribosomal proteins sequester MDM2 ubiquitin ligase in the nucleoplasm and thus facilitate an increase in p53, which leads to a cell cycle arrest, senescence, or apoptosis [51]. Nucleolar reorganization follows this scenario not only upon direct inhibition of ribosomal gene transcription, but also in many other types of stress, including heat shock [52, 53], oxidative stress, osmotic stress [31], and DNA damage arising within rDNA [29, 30] or in the rest of the genome [32, 54]. Thus, changes in the transcription level of ribosomal genes and, in particular, their repression are a universal cell response to stress, and the nucleolus is thought to act as a sensor and coordination center of this response [55].
SYSTEMS TO INDUCE DAMAGE TO rDNA
The repair systems that eliminate lesions from rDNA are experimentally studied by inducing damage to rDNA by various methods. Each method has its advantages and drawbacks. The choice of the method depends on the problem to be addressed. Cells were exposed to γ irradiation or UV light in early studies of the cell response to rDNA damage [54, 56, 57]. However, these factors act nonspecifically and damage not only ribosomal genes, but total genomic DNA as well. Laser microirradiation or ion microbeams produce more local damage because they are possible to focus directly on the nucleolus [54, 58]. Certain limitations and drawbacks are still characteristic of the methods. First, the methods lack site specificity. Second, many other DNA lesions (so-called clustered DNA damage) are induced together with DSBs by laser and ion beams, thus substantially complicating the interpretation of findings related to the recognition and repair of the lesions. The above drawbacks are absent in the approaches that utilize specific endonucleases to introduce lesions of a known type into known rDNA sites. Cells with ectopic expression of Physarum polycephalum I-PpoI homing endonuclease provide a system that is commonly used to induce DSBs in rDNA in order to study the response to ribosomal gene damage [29–31, 59–61]. An I-PpoI recognition site of 13–15 bp occurs in the 28S rRNA-coding region of the ribosomal repeat (Fig. 1) and is absent from the rest of the genome; the site shows 100% identity among all eukaryotes [62]. Expressed in human cells, I-PpoI induces lesions in approximately 10% of its target sites in rDNA; i.e., approximately 30 DSBs are introduced in ribosomal genes [63]. I-PpoI additionally targets approximately 13 other genomic sites, the majority of which belong to 28S rRNA pseudogenes [64].
Ectopic expression of Arthrobacter endonuclease AsiSI provides another system to induce DSBs in rDNA [29, 65]. An 8-bp recognition site of the enzyme occurs in the 47S RNA-coding region (Fig. 1). However, AsiSI induces lesions not only in the ribosomal repeat, but also in 174 other sites of the human genome [66]. AsiSI is therefore far less commonly used to study rDNA damage as compared with I-PpoI. A quantitative analysis has not been performed yet to characterize the AsiSI-induced rDNA lesions. It should also be noted that AsiSI fails to cleave the recognition sites that contain methylated CpG dinucleotides. Because methylation is responsible for inactivating part of the rDNA repeats [38], AsiSI most likely cleaves only active repeats, which are demethylated.
Finally, CRISPR/Cas9 technology provides a highly efficient means to introduce DSBs in rDNA in a targeted manner. A DSB is possible to induce in almost every part of the rDNA repeat by CRISPR/Cas9 technology when a proper protospacer adjacent motif (PAM) is in place. DSBs were successfully induced in both transcribed region and the nontranscribed intergenic spacer of the ribosomal repeat with the CRISPR/Cas9 system in order to study the response to rDNA damage [29, 60]. However, it should be noted that long-term expression of I-PpoI, AsiSI, or Cas9 in cells leads to repetitive cleavage–restoration cycles at target DNA sites until repair errors change the target sequence [63]. This limitation of the method is not substantial, but still important to consider in long-term experiments.
RIBOSOMAL DNA DAMAGE INDUCES PIKK-DEPENDENT REPRESSION OF NUCLEOLAR TRANSCRIPTION
The team headed by R. Casellas was the first to report the data on the response to DNA damage in the nucleolus in 2007 [54]. It was shown that large-scale repression of ribosomal gene transcription arises in mouse embryonic fibroblasts exposed to genotoxic stress, which was induced using γ irradiation, laser microirradiation, or etoposide (a DNA topoisomerase II inhibitor). The repression is accompanied by a nucleolar reorganization and, in particular, the formation of nucleolar caps, which are identical to those forming when ribosomal gene transcription is inhibited with ACD [54]. An amazing finding was made in further studies of the mechanisms of the observed phenomenon. Nucleolar transcription was not passively blocked by DNA damage. Transcriptional repression proved to be a regulated process and depended on activation of ATM kinase, which is one of the main proteins of phosphatidylinositol 3-kinase-like kinases (PIKKs) responsible for DSB recognition and repair [67, 68]. Recently, the role of ATM in DSB-induced repression of nucleolar transcription was confirmed in studies with I-PpoI [29, 31, 59–61], AsiSI [65], and CRISPR/Cas9 [29, 60]. As was found in studies of CRISPR/Cas9-induced breaks, ATM-dependent transcriptional repression arises regardless of whether the coding or noncoding regions of a ribosomal repeat are affected by lesions [60]. The finding excluded the possibility of a nonregulated physical stop of pol I by breaks in the template DNA strand.
Further studies showed that ATM is not the only PIKK that regulates rDNA transcription in response to DNA damage. ATR was also found to play a role in the process. Although ATR is to a lesser extent involved in DSB repair and is mostly responsible for maintaining the stability of DNA replication, several studies showed that ATR acts to ensure nucleolar transcriptional repression in response to DSBs induced by various agents [29, 30]. Our team demonstrated that ATR acts together with ATM to repress nucleolar transcription in response to co-transcriptional R-loops induced by hypoosmotic stress [31]. However, it remained unclear whether conversion of R-loops to DSBs or the mere R-loop structure acts to trigger the ATR/ATM-dependent signaling pathway.
DNA-PKcs is another PIKK that is primarily involved in DSB recognition and repair, like ATM. There are only two studies reported the possibility of DNA-PKcs-dependent repression of polI. One was carried out using in vitro systems [69], and the other implicated DNA-PKcs in rDNA transcriptional repression in response to cisplatin- and UV-induced damage [70]. A role of DNA-PKcs in repression of rDNA transcription was not confirmed in the majority of further studies, which were carried out using CRISPR/Cas9, I-PpoI, or ionizing radiation [31, 54, 59]. The prevailing opinion now is therefore that the role of DNA-PKcs in the response to rDNA damage is limited to repair. It still cannot be excluded that ATM, ATR, and DNA-PKcs eventually trigger the same mechanisms of transcriptional repression by acting as effectors of different lesions.
MECHANISMS OF RIBOSOMAL DNA TRANSCRIPTIONAL REPRESSION
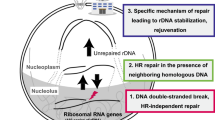
On exposure to ionizing radiation, the repair factors Nbs1, MDC1 and ATM are necessary for efficient repression of nucleolar transcription, while several other repair proteins, including Ku80, BRCA1, 53BP1, and histone H2AX, are not essential. Studies with FRAP microscopy showed that ATM-dependent repression of nucleolar transcription is due to inhibited initiation of transcription driven by pol I and the removal of the enzyme from chromatin at elongation [54]. However, the exact mechanism responsible for DSB-induced rDNA repression is still the focus of research. A search for and identification of ATM targets in the nucleolus may provide crucial data for understanding the mechanism. Several targets were identified in phosphoproteome studies [71, 72]. The set included the RPA34 subunit of pol I, the TAF1C component of the SL1-initiator complex, transcription termination factor 1 (TTF1), the UBF transcription factor, and its interaction partner Treacle. A direct role in DSB-induced repression of ribosomal genes was demonstrated only for Treacle in further studies (Fig. 2).

Generalized scheme of the mechanisms that sustain repression of RNA polymerase I-dependent transcription in response to DNA damage in ribosomal repeats. Damage to rDNA induces methylation of histone H3 and phosphorylation of histone H2B, the effects facilitating chromatin compaction and transcriptional silencing. In addition, ATM/ATR-dependent phosphorylation of Treacle accompanies rDNA damage and stimulates the Treacle interaction with the MRN complex and TOPBP1. This interaction is also necessary for transcriptional repression of ribosomal genes.
Treacle (also known as TCOF1) is a nucleolar phosphoprotein; mutations of its gene are found in Treacher–Collins syndrome, which is a congenital disorder characterized by craniofacial deformities [73]. Treacle harbors 17 S/T-Q motifs, which act as specific phosphorylation sites for ATM and other PIKKs. Studies by S.J. Elledge, M. Stucki, and D.H. Larsen showed that ATM-dependent phosphorylation of Ser1199 in one of the S/T-Q motifs of Treacle is necessary for the Treacle interaction with Nbs1 when DSBs are induced in ribosomal genes with the use of CRISPR/Cas9 or ionizing radiation. In turn, the interaction is necessary for rDNA transcriptional repression in response to damage [29, 32, 54, 74].
Recently, two research teams showed independently that the ATR activator TOPBP1 also binds with Treacle in response to rDNA damage [30, 31]. Stuki and colleagues [30] found that the Treacle–TOPBP1 interaction is necessary for repressing ribosomal transcription in the case of damage induced using I-PpoI and that the interaction is mediated by both ATR and ATM kinase activities and Nbs1 [30]. Because Nbs1 interacts with the N-terminal domain of Treacle and TOPBP1 interacts with its C-terminal domain, the interactions do not interfere with each other and can occur simultaneously. Treacle binding with TOPBP1 is most likely also regulated by ATM/ATR-dependent phosphorylation of S/T-Q motifs of Treacle. Our team showed that stabilization of R-loops in hypoosmotic stress similarly leads to ATR-dependent Treacle–TOPBP1 interaction, which is necessary for ATM activation and nucleolar transcriptional repression [31]. As was observed by another team, TOPBP1 overexpression is sufficient for ATR-dependent repression of ribosomal genes in the absence of their damage [75]. It is therefore possible to assume that Treacle temporarily loses its transcription function as a result of its ATM/ATR-dependent modification and direct interaction with the repair proteins TOPBP1 and Nbs1 and that this loss facilitates repression of ribosomal genes (Fig. 2).
Although a lower nucleosome density is characteristic of ribosomal genes, histone modifications contribute to the regulation of rDNA transcription in response to damage. Histone H2B is phosphorylated at Ser14 (H2BS14p) when rDNA damage is induced by ionizing radiation or I-PpoI, and H2BS14p is a repressive modification. MST2 kinase is responsible for histone H2B phosphorylation in this case and is activated by ATM with the involvement of the adaptor protein RASSF1A. Lack of any component in the ATM–RASSF1A–MST2 axis leads to a loss of H2BS14p and prevents DSB-induced repression of ribosomal genes [76]. Legube and colleagues [65] showed that the level of histone H3 trimethylated at Lys9 (H3K9Me3) increases upon AsiSI-induced rDNA damage, and H3K9Me3 is also a factor in inactive chromatin formation. Methylation is due to activities of the HUSH repressor complex and SUV39H1 histone methyltransferase. A knockdown in either protein decreases the extent of DSB-induced repression and prevents the formation of nucleolar caps [65]. Thus, a set of mechanisms governs DNA damage-induced transcriptional repression of ribosomal genes and includes not only modification of transcription components, but also changes in the epigenetic status of nucleolar chromatin.
LONG-DISTANCE EFFECTS OF rDNA TRANSCRIPTIONAL REPRESSION
The effect of DNA damage on transcription has recently come to be studied not only for the genes that directly contain a DSB, but also for intact adjacent genes. DSB induction in genes intensely transcribed by RNA polymerase II was shown to lead to their ATM/DNA-PK-dependent repression [77, 78]. A decrease in transcription or an accumulation of repressive chromatin marks was observed in genes located up to several kilobases away from a specific break site [77, 79]. Ribonucleotide incorporation and the active RNA polymerase II form were lacking in γH2AX-marked regions when DSBs were induced with ionizing or laser irradiation [80]. Thus, the range of the transcription-repressing effect is most likely limited to a repair compartment. The question is whether similar long-distance effects occur in the case of rDNA damage. This issue is poorly understood. How long away from the rDNA damage site can the signaling cascades that repress transcription range? Do the cascades act only within the ribosomal repeat where an induced lesion occurs? Or does transcriptional repression additionally act in cis to involve the adjacent intact repeats?
Kruhlak et al. [54] used laser microbeams to induce DSBs in a single nucleolus. The microbeam power ensured the introduction of one DSB per 1 Mb DNA. The results obtained using this approach showed, first, that transcriptional repression occurs exclusively in the exposed nucleolus and does not extend to the other nucleoli of the same cell nucleus. Second, nucleolar transcriptional repression, nucleolar reorganization, and cap formation can occur at an extremely low DSB density, approximately one DSB per 23 ribosomal repeats. However, the DSB frequency was only roughly estimated and most likely underestimated in [54] because the DSB amount was not measured directly, but was inferred from the intensity of H2AX phosphorylation in the nucleolus, which has a lower nucleosome density [81].
Transcriptional repression throughout the total nucleolus is provoked by I-PpoI endonuclease [59], although I-PpoI induces DSBs only in 10% of ribosomal repeats [63]. The DSB rate might be underestimated again because the efficiency of cell transfection with I-PpoI and the DSB distribution between active and inactive repeats are disregarded in the estimation method.
Larsen et al. [29] more accurately estimated the DSB amount in rDNA. DSBs were induced in the rDNA intergenic spacer with CRISPR/Cas9 and led to transcriptional repression and cap formation. Quantitative PCR showed that approximately 30% of rDNA repeats were damaged, corresponding to one DSB per three repeats or one DSB per a FC [29]. Given that actively transcribed regions are predominantly affected by DSBs induced with CRISPR/Cas9 [82], it is possible to assume that the repressive potential of one DSB is limited to one FC.
Lesions induced in part of ribosomal repeats were shown to provoke local repression of the damaged region rather than global repression of the total nucleolus. Relevant studies were carried out using AsiSI endonuclease, which induces breaks in fewer ribosomal repeats as compared with I-PpoI and CRISPR/Cas9. It was observed that only a certain part of the ribosomal repeats is repressed and moves to caps in a nucleolus, while the remaining intact repeats are still transcribed within the nucleolus [65]. An attempt was recently made to study the response of the nucleolus to local rDNA lesions induced with carbon ion microbeams [58]. The method makes it possible to focus the radiation beam to a dot of no more than 1 μm in diameter and to irradiate a small intranucleolar area rather than the total nucleolus. DSBs are possible to induce at a rate of 0.2–20 DSBs/μm by varying the ion content in a beam. As was demonstrated with this approach, transcriptional repression occurs exclusively in the exposed area regardless of the DSB number and fully colocalizes with phosphorylated histone H2AX, which is a DSB marker. Large-scale transcriptional repression and nucleolar reorganization were not observed in the study [58].
The likelihood of global nucleolar transcriptional repression most likely correlates with the amount of lesions induced in the nucleolus. It cannot be excluded that repression extends in cis from one DSB to several neighbor ribosomal repeats. However, the effect is spatially limited, to one FC presumably, and this limitation prevents a local lesion from provoking drastic transcriptional repression of the total rDNA pool.
TRANSCRIPTIONAL REPRESSION AND THE FORMATION OF NUCLEOLAR CAPS
A reorganization of nucleoli with the formation of nuclear caps usually correlates with inhibition of nucleolar transcription. Caps form both when pol I is directly inhibited with ACD and when ATM/ATR-dependent repression develops in response to rDNA damage. Active forms of ATM and ATR are located directly in caps, and inhibition of the PIKKs fully blocks their generation while the amount of induced lesions remains constant in rDNA [29–31, 59]. The cap formation mechanism is a matter of discussion. Two hypotheses were advanced to explain the mechanism. One assumes a passive process that is based on the changes arising in liquid phase-properties of the nucleolus upon transcriptional repression. The FC and DFC occur as drops in an actively transcribing nucleolus and are prevented from fusion by intense transcription and the presence of RNA. A transcription arrest leads to local depletion of the RNA component, drop properties of FC components consequently start to prevail, leading to fusion, aggregation, and passive translocation of the components to the periphery of the nucleolus [49, 83]. Transcriptional inhibition is therefore responsible for cap formation according to this hypothesis. The other hypothesis suggests active rDNA movement with the involvement of motor mechanisms of the cell. The LINK complex, which connects the nuclear lamina with the cytoskeleton, and actin filaments are responsible for the translocation of part of damaged rDNA repeats in response to rDNA damage induced with AsiSI. That is, the role is played by the same factors that ensure DSB relocation and clustering in other genomic loci according to the available data [84–86]. However, this LINK/actin-dependent translocation was observed only in the S and G2 phases of the cell cycle, indicating that the process depends on the specifics of AsiSI-induced break repair [65].
Thus, several factors may be responsible for cap formation. For example, cap formation is a direct consequence of transcriptional repression and is driven only by LLPS forces when the majority of ribosomal repeats is repressed upon exposure to ACD, ionizing radiation, I-PpoI, or CRISPR/Cas9. When only a minor portion of repeats is damaged (as is the case with AsiSI), LLPS forces are not strong enough, and LINK and actin may contribute to the translocation. However, the translocation efficiency will depend on the type of lesions and, therefore, the type and rate of their repair in this case.
Two main roles are presumably played by cap formation. First, rDNAs of different chromosomes are spatially separated to prevent aberrant interchromosomal recombination. Second, rDNA repair is ensured by cap formation. Certain repair factors (including HRR factors) remain undetectable until nucleolar caps form in the case of rDNA breaks [87]. The factor set includes MDC1, 53BP1, and BRCA1 [29, 87]. The exact cause of the phenomenon is still unknown now. It is thought that the proteins may lack the domains that are necessary for their compatibility with the liquid phase within the nucleolus [49]. At the same time, some of the proteins are recruited to the damaged site via histone modifications and, in particular, H2AX phosphorylation [88]. As is known, a lower nucleosome density is characteristic of ribosomal genes, and the absence of the above repair proteins from the nucleolus may therefore reflect a weak response to DNA damage.
DNA REPAIR IN THE NUCLEOLUS
Two main pathways, NHEJ and HRR, are responsible for DSB repair in ribosomal genes, like in other regions of the genome. NHEJ repair starts with DSB recognition by the Ku70–Ku80 heterodimer. The heterodimer provides a platform for other repair factors, including nucleases, polymerases, and ligases. The Ku70–Ku80 complex binds with DNA-PKcs kinase, leading to its conformational modification and activation. Then the XLF/XRCC4/DNA ligase IV complex is recruited to the break to ensure ligation of the DNA ends. When I-PpoI is used to induce lesions, the Ku70–Ku80 heterodimer binds to rDNA within the nucleolus long before caps form [89]. Chemical inhibition or a knockdown of DNA-PK and knockdowns of certain NHEJ factors substantially increase the amount of DSBs induced in rDNA by endonucleases I-PpoI and AsiSI. In addition, lack of NHEJ factors aggravates the transcriptional repression induced by I‑PpoI and AsiSI [59, 65]. The findings clearly point to a functional dependence of rDNA repair on NHEJ.
NHEJ factors other than 53BP1 are absent from nucleolar caps [87]. In contrast, the majority of HRR factors, such as RPA, MDC1, Rad51, and BRCA1, are recruited predominantly to nucleolar caps. HRR is consequently thought to proceed most intensely in caps. Three main steps are possible to recognize in HRR. At the first step, the MRE11–RAD50–NBS1 (MRN) complex; Exo1, Dna2, and CtIP nucleases; and BLM helicase bind to the break. The proteins resect the DNA ends and produce single-stranded regions, which are immediately covered by RPA. At the second step, RPA is replaced with RAD51 recombinase and a nucleoprotein filament forms. RPA is known to have high affinity for ssDNA, and special mediator proteins are consequently required for its replacement with RAD51. BRCA1 acts as such a mediator. The events are followed by a homology search and invasion of the DNA strand to be repaired into the homologous duplex to produce a displacement loop and a Holliday junction. At the third step, the damaged DNA region is synthesized [88].
As is well known, HRR occurs predominantly in the S and G2 phases of the cell cycle because the sister chromatid is present. When rDNA lesions are induced with I-PpoI and AsiSI, HRR factors are, in fact, localized in nucleolar caps of S/G2 cells predominantly [30, 65]. Surprisingly, when CRISPR/Cas9-induced damage is repaired, HRR factors are localized in caps even in G1 cells, where sister chromatids have still not form. Intact rDNA repeats presumably serve as a template for homologous recombination in cis in this case. Thus, the choice of HRR for rDNA repair may be determined by the lesion characteristics rather than by the cell cycle phase. I-PpoI and AsiSI introduce simple breaks, which are repaired quickly, and rDNA with a relatively small amount of such breaks can be repaired immediately within the nucleolus by NHEJ before transcriptional repression arises and caps form. Breaks induced with CRISPR/Cas9 are more complex and display relatively slow repair kinetics (up to 10 h) [90]. NHEJ is not efficient enough in this case, and rDNA moves to nucleolar caps, where repair proceeds via the HRR mechanism [87]. Thus, the type and complexity of the lesion can primarily determine the choice between the rDNA repair pathways.
CONCLUSIONS
The nucleolus is the largest subnuclear compartment and its main function consists in ribosome biogenesis, which is one of the most energy-consuming metabolic processes. Timely transcriptional regulation in the nucleolus serves, first, to maintain the rDNA stability upon rDNA damage and, second, to redistribute the energy resources that ensure cell homeostasis in stress. Data obtained by our and other teams confirm that the nucleolus and pol I act as multifunctional sensors of cell stress. The level of gene transcription with pol I is far higher than that with RNA polymerase II, and the nucleolus is consequently far more sensitive to various stress factors. The nucleolus-specific response to DNA damage can trigger various pathways of the cell stress response. Activation of ATR and ATM kinases can lead to activation of p53 and cell cycle checkpoint kinases, thus arresting cell proliferation or triggering the cell death. It is of interest that rDNA transcription arrest itself is capable of activating p53. An arrest of nucleolar transcription leads to a release of ribosomal proteins, which suppress MDM2, and MDM2 suppression leads to p53 activation. The structural changes that occur in the nucleolus upon pol I inhibition can induce a redistribution of the nucleolar proteins (B23, fibrillarin, and nucleolin) that are involved in the cell stress response. However, in spite of the long history of studies of the nucleolus, its functional role as a cell stress sensor is still far from fully understood.
REFERENCES
Pfeiffer P., Goedecke W., Obe G. 2000. Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis. 15, 289–302.
García-Muse T., Aguilera A. 2016. Transcription-replication conflicts: How they occur and how they are resolved. Nat. Rev. Mol. Cell. Biol. 17, 553–563.
Ganem N.J., Pellman D. 2012. Linking abnormal mitosis to the acquisition of DNA damage. J. Cell. Biol. 199, 871–881.
Lindahl T. 1993. Instability and decay of the primary structure of DNA. Nature. 362, 709–715.
Sonoda E., Morrison C., Yamashita Y.M., Takata M., Takeda S. 2001. Reverse genetic studies of homologous DNA recombination using the chicken B-lymphocyte line, DT40. Philos. Trans. R Soc. Lond. B. 356, 111–117.
Vilenchik M.M., Knudson A.G. 2003. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. U. S. A. 100, 12871–12876.
Jackson S.P., Bartek J. 2009. The DNA-damage response in human biology and disease. Nature. 461, 1071–1078.
Bartkova J., Horejsí Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J.M., Lukas C., Ørntoft T., Lukas J., Bartek J. 2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 434, 864–870.
Harper J.W., Elledge S.J. 2007. The DNA damage response: ten years after. Mol. Cell. 28, 739–745.
Ceccaldi R., Rondinelli B., D’Andrea A.D. 2016. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 26, 52–64.
Mladenov E., Magin S., Soni A., Iliakis G. 2016. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 37–38, 51–64.
Clouaire T., Legube G. 2015. DNA double strand break repair pathway choice: A chromatin based decision? Nucleus. 6, 107–113.
Bzymek M., Lovett S.T. 2001. Instability of repetitive DNA sequences: The role of replication in multiple mechanisms. Proc. Natl. Acad. Sci. U. S. A. 98, 8319–8325.
Durkin S.G., Glover T.W. 2007. Chromosome fragile sites. Annu. Rev. Genet. 41, 169–192.
Crosetto N., Mitra A., Silva M.J., Bienko M., Dojer N., Wang Q., Karaca E., Chiarle R., Skrzypczak M., Ginalski K., Pasero P., Rowicka M., Dikic I. 2013. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods. 10, 361–365.
Lensing S.V., Marsico G., Hänsel-Hertsch R., Lam E.Y., Tannahill D., Balasubramanian S. 2016. DSB capture: in situ capture and sequencing of DNA breaks. Nat. Methods. 13, 855–857.
Mourad R., Ginalski K., Legube G., Cuvier O. 2018. Predicting double-strand DNA breaks using epigenome marks or DNA at kilobase resolution. Genome Biol. 19, 34.
Tchurikov N.A., Yudkin D.V., Gorbacheva M.A., Kulemzina A.I., Grischenko I.V., Fedoseeva D.M., Sosin D.V., Kravatsky Y.V., Kretova O.V. 2016. Hot spots of DNA double-strand breaks in human rDNA units are produced in vivo. Sci. Rep. 6, 25866.
Lindström M.S., Jurada D., Bursac S., Orsolic I., Bartek J., Volarevic S. 2018. Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene. 37, 2351–2366.
Santos-Pereira J.M., Aguilera A. 2015. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 16, 583–597.
Aguilera A., García-Muse T. 2012. R loops: From transcription byproducts to threats to genome stability. Mol. Cell. 46, 115–124.
Sollier J., Cimprich K.A. 2015. Breaking bad: R-loops and genome integrity. Trends Cell. Biol. 25, 514–522.
Chastain M., Zhou Q., Shiva O., Fadri-Moskwik M., Whitmore L., Jia P., Dai X., Huang C., Ye P., Chai W. 2016. Human CST facilitates genome-wide RAD51 recruitment to GC-rich repetitive sequences in response to replication stress. Cell Rep. 16, 1300–1314.
Dehé P.-M., Gaillard P.-H.L. 2017. Control of structure-specific endonucleases to maintain genome stability. Nat. Rev. Mol. Cell. Biol. 18, 315–330.
Rhodes D., Lipps H.J. 2015. G-Quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 43, 8627–8637.
Stults D.M., Killen M.W., Williamson E.P., Hourigan J.S., Vargas H.D., Arnold S.M., Moscow J.A., Pierce A.J. 2009. Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 69, 9096–9104.
Hallgren J., Pietrzak M., Rempala G., Nelson P.T., Hetman M. 2014. Neurodegeneration-associated instability of ribosomal DNA. Biochim. Biophys. Acta. 1842, 860–868.
Diesch J., Hannan R.D., Sanij E. 2014. Perturbations at the ribosomal genes loci are at the centre of cellular dysfunction and human disease. Cell. Biosci. 4, 43.
Korsholm L.M., Gál Z., Lin L., Quevedo O., Ahmad D.A., Dulina E., Luo Y., Bartek J., Larsen D.H. 2019. Double-strand breaks in ribosomal RNA genes activate a distinct signaling and chromatin response to facilitate nucleolar restructuring and repair. Nucleic Acids Res. 47, 8019–8035.
Mooser C., Symeonidou I.E., Leimbacher P.A., Ribeiro A., Shorrocks A.K., Jungmichel S., Larsen S.C., Knechtle K., Jasrotia A., Zurbriggen D., Jeanrenaud A., Leikauf C., Fink D., Nielsen M.L., Blackford A.N., Stucki M. 2020. Treacle controls the nucleolar response to rDNA breaks via TOPBP1 recruitment and ATR activation. Nat. Commun. 11, 123.
Velichko A.K., Petrova N.V., Luzhin A.V., Strelkova O.S., Ovsyannikova N., Kireev I.I., Petrova N.V., Razin S.V., Kantidze O.L. 2019. Hypoosmotic stress induces R loop formation in nucleoli and ATR/ATM-dependent silencing of nucleolar transcription. Nucleic Acids Res. 47, 6811–6825.
Larsen D.H., Hari F., Clapperton J.A., Gwerder M., Gutsche K., Altmeyer M., Jungmichel S., Toledo L.I., Fink D., Rask M.B., Grøfte M., Lukas C., Nielsen M.L., Smerdon S.J., Lukas J., Stucki M. 2014. The NBS1-Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat. Cell. Biol. 16, 792–803.
Sakai K., Ohta T., Minoshima S., Kudoh J., Wang Y., de Jong P.J., Shimizu N. 1995. Human ribosomal RNA gene cluster: identification of the proximal end containing a novel tandem repeat sequence. Genomics. 26, 521–526.
Gonzalez I.L., Sylvester J.E. 1995. Complete sequence of the 43-kb human ribosomal DNA repeat: Analysis of the intergenic spacer. Genomics. 27, 320–328.
Stults D.M., Killen M.W., Pierce H.H., Pierce A.J. 2008. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 18, 13–18.
Gibbons J.G., Branco A.T., Godinho S.A., Yu S., Lemos B. 2015. Concerted copy number variation balances ribosomal DNA dosage in human and mouse genomes. Proc. Natl. Acad. Sci. U. S. A. 112, 2485–2490.
McStay B., Grummt I. 2008. The epigenetics of rRNA genes: From molecular to chromosome biology. Annu. Rev. Cell. Dev. Biol. 24, 131–157.
Grummt I., Pikaard C.S. 2003. Epigenetic silencing of RNA polymerase I transcription. Nat. Rev. Mol. Cell. Biol. 4, 641–649.
Russell J., Zomerdijk J.C. 2005. RNA-polymerase-I-directed rDNA transcription, life and works. Trends Biochem. Sci. 30, 87–96.
Miller O.L., Jr., Beatty B.R. 1969. Visualization of nucleolar genes. Science. 164, 955–957.
Olson M.O., Dundr M. 2005. The moving parts of the nucleolus. Histochem. Cell. Biol. 123, 203–216.
Maiser A., Dillinger S., Längst G., Schermelleh L., Leonhardt H., Németh A. 2020. Super-resolution in situ analysis of active ribosomal DNA chromatin organization in the nucleolus. Sci. Rep. 10, 7462.
Yao R.W., Xu G., Wang Y., Shan L., Luan P.F., Wang Y., Wu M., Yang L.Z., Xing Y.H., Yang L., Chen L.L. 2019. Nascent pre-rRNA sorting via phase separation drives the assembly of dense fibrillar components in the human nucleolus. Mol. Cell. 76, 767–783.
Raska I., Shaw P.J., Cmarko D. 2006. Structure and function of the nucleolus in the spotlight. Curr. Opin. Cell. Biol. 18, 325–334.
Banani S.F., Lee H.O., Hyman A.A., Rosen M.K. 2017. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell. Biol. 18, 285–298.
Razin S.V., Gavrilov A.A. 2020. The role of liquid–liquid phase separation in the compartmentalization of cell nucleus and spatial genome organization. Biochemistry (Moscow). 85 (6), 643–650.
Mangan H., Gailín M., McStay B. 2017. Integrating the genomic architecture of human nucleolar organizer regions with the biophysical properties of nucleoli. FEBS J. 284, 3977–3985.
Brangwynne C.P., Mitchison T.J., Hyman A.A. 2011. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. U. S. A. 108, 4334–4339.
Feric M., Vaidya N., Harmon T.S., Mitrea D.M., Zhu L., Richardson T.M., Kriwacki R.W., Pappu R.V., Brangwynne C.P. 2016. Coexisting liquid phases underlie nucleolar subcompartments. Cell. 165, 1686–1697.
Reynolds R.C., Montgomery P.O., Hughes B. 1964. Nucleolar “caps” produced by actynomycin D. Cancer Res. 24, 1269–1277.
Zhang Y., Lu H. 2009. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 16, 369–377.
Velichko A.K., Petrova N.V., Razin S.V., Kantidze O.L. 2015. Mechanism of heat stress-induced cellular senescence elucidates the exclusive vulnerability of early S-phase cells to mild genotoxic stress. Nucleic Acids Res. 43, 6309–6320.
Zhao Z., Dammert M.A., Hoppe S., Bierhoff H., Grummt I. 2016. Heat shock represses rRNA synthesis by inactivation of TIF-IA and lncRNA-dependent changes in nucleosome positioning. Nucleic Acids Res. 44, 8144–8152.
Kruhlak M., Crouch E.E., Orlov M., Montaño C., Gorski S.A., Nussenzweig A., Misteli T., Phair R.D., Casellas R. 2007. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature. 447, 730–734.
Iarovaia O.V., Minina E.P., Sheval E.V., Onichtchouk D., Dokudovskaya S., Razin S.V., Vassetzky Y.S. 2019. Nucleolus: A central hub for nuclear functions. Trends Cell. Biol. 29, 647–659.
Boulon S., Westman B.J., Hutten S., Boisvert F.M., Lamond A.I. 2010. The nucleolus under stress. Mol. Cell. 40, 216–227.
Moore H.M., Bai B., Boisvert F.M., Latonen L., Rantanen V., Simpson J.C., Pepperkok R., Lamond A.I., Laiho M. 2011. Quantitative proteomics and dynamic imaging of the nucleolus reveal distinct responses to UV and ionizing radiation. Mol. Cell. Proteomics. 10, M111 009241.
Siebenwirth C., Greubel C., Drexler G.A., Reindl J., Walsh D.W.M., Schwarz B., Sammer M., Baur I., Pospiech H., Schmid T.E., Dollinger G., Friedl A.A. 2019. Local inhibition of rRNA transcription without nucleolar segregation after targeted ion irradiation of the nucleolus. J. Cell Sci. 132, jcs232181.
Harding S.M., Boiarsky J.A., Greenberg R.A. 2015. ATM dependent silencing links nucleolar chromatin reorganization to DNA damage recognition. Cell. Rep. 13, 251–259.
van Sluis M., McStay B. 2015. A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev. 29, 1151–1163.
Warmerdam D.O., van den Berg J., Medema R.H. 2016. Breaks in the 45S rDNA lead to recombination-mediated loss of repeats. Cell Rep. 14, 2519–2527.
Ellison E.L., Vogt V.M. 1993. Interaction of the intron-encoded mobility endonuclease I-PpoI with its target site. Mol. Cell. Biol. 13, 7531–7539.
Monnat R.J., Jr., Hackmann A.F., Cantrell M.A. 1999. Generation of highly site-specific DNA double-strand breaks in human cells by the homing endonucleases I-PpoI and I-CreI. Biochem. Biophys. Res. Commun. 255, 88–93.
Berkovich E., Monnat R.J., Jr., Kastan M.B. 2007. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell. Biol. 9, 683–690.
Marnef A., Finoux A.L., Arnould C., Guillou E., Daburon V., Rocher V., Mangeat T., Mangeot P.E., Ricci E.P., Legube G. 2019. A cohesin/HUSH- and LINC-dependent pathway controls ribosomal DNA double-strand break repair. Genes Dev. 33, 1175–1190.
Clouaire T., Rocher V., Lashgari A., Arnould C., Aguirrebengoa M., Biernacka A., Skrzypczak M., Aymard F., Fongang B., Dojer N., Iacovoni J.S., Rowicka M., Ginalski K., Côté J., Legube G. 2018. Comprehensive mapping of histone modifications at DNA double-strand breaks deciphers repair pathway chromatin signatures. Mol. Cell. 72, 250–262.
Kantidze O.L., Velichko A.K., Luzhin A.V., Petrova N.V., Razin S.V. 2018. Synthetically lethal interactions of ATM, ATR, and DNA-PKcs. Trends Cancer. 4, 755–768.
Shiloh Y., Ziv Y. 2013. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell. Biol. 14, 197–210.
Kuhn A., Gottlieb T.M., Jackson S.P., Grummt I. 1995. DNA-dependent protein kinase: A potent inhibitor of transcription by RNA polymerase I. Genes Dev. 9, 193–203.
Calkins A.S., Iglehart J.D., Lazaro J.-B. 2013. DNA damage-induced inhibition of rRNA synthesis by DNA-PK and PARP-1. Nucleic Acids Res. 41, 7378–7386.
Matsuoka S., Ballif B.A., Smogorzewska A., McDonald E.R., 3rd, Hurov K.E., Luo J., Bakalarski C.E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S.P., Elledge S.J. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 316, 1160–1166.
Larsen D.H., Stucki M. 2016. Nucleolar responses to DNA double-strand breaks. Nucleic Acids Res. 44, 538–544.
Trainor P.A., Dixon J., Dixon M.J. 2009. Treacher Collins syndrome: Etiology, pathogenesis and prevention. Eur. J. Hum. Genet. 17, 275–283.
Ciccia A., Huang J.-W., Izhar L., Sowa M.E., Harper J.W., Elledge S.J. 2014. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc. Natl. Acad. Sci. U. S. A. 111, 18631–18636.
Sokka M., Rilla K., Miinalainen I., Pospiech H., Syväoja J.E. 2015. High levels of TopBP1 induce ATR-dependent shut-down of rRNA transcription and nucleolar segregation. Nucleic Acids Res. 43, 4975–4989.
Pefani D.E., Tognoli M.L., Pirincci Ercan D., Gorgoulis V., O’Neill E. 2018. MST2 kinase suppresses rDNA transcription in response to DNA damage by phosphorylating nucleolar histone H2B. EMBO J. 37, e98760.
Iannelli F., Galbiati A., Capozzo I., Nguyen Q., Magnuson B., Michelini F., D’Alessandro G., Cabrini M., Roncador M., Francia S., Crosetto N., Ljungman M., Carninci P., d’Adda di Fagagna F. 2017. A damaged genome’s transcriptional landscape through multilayered expression profiling around in situ-mapped DNA double-strand breaks. Nat. Commun. 8, 15656.
Pankotai T., Bonhomme C., Chen D., Soutoglou E. 2012. DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat. Struct. Mol. Biol. 19, 276–282.
Ayrapetov M.K., Gursoy-Yuzugullu O., Xu C., Xu Y., Price B.D. 2014. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. U. S. A. 111, 9169–9174.
Gong F., Clouaire T., Aguirrebengoa M., Legube G., Miller K.M. 2017. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J. Cell. Biol. 216, 1959–1974.
Kruhlak M.J., Celeste A., Dellaire G., Fernandez-Capetillo O., Müller W.G., McNally J.G., Bazett-Jones D.P., Nussenzweig A. 2006. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell. Biol. 172, 823–834.
Verkuijl S.A., Rots M.G. 2019. The influence of eukaryotic chromatin state on CRISPR-Cas9 editing efficiencies. Curr. Opin. Biotechnol. 55, 68–73.
Falahati H., Pelham-Webb B., Blythe S., Wieschaus E. 2016. Nucleation by rRNA dictates the precision of nucleolus assembly. Curr. Biol. 26, 277–285.
Aymard F., Bugler B., Schmidt C.K., Guillou E., Caron P., Briois S., Iacovoni J.S., Daburon V., Miller K.M., Jackson S.P., Legube G. 2014. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 21, 366–374.
Caridi C.P., D’Agostino C., Ryu T., Zapotoczny G., Delabaere L., Li X., Khodaverdian V.Y., Amaral N., Lin E., Rau A.R., Chiolo I. 2018. Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature. 559, 54–60.
Lottersberger F., Karssemeijer R.A., Dimitrova N., de Lange T. 2015. 53BP1 and the LIN–C complex promote microtubule-dependent DSB mobility and DNA repair. Cell. 163, 880–893.
van Sluis M., McStay B. 2019. Nucleolar DNA double-strand break responses underpinning rDNA genomic stability. Trends Genet. 35, 743–753.
Blackford A.N., Jackson S.P. 2017. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell. 66, 801–817.
Britton S., Coates J., Jackson S.P. 2013. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell. Biol. 202, 579–595.
Brinkman E.K., Chen T., de Haas M., Holland H.A., Akhtar W., van Steensel B. 2018. Kinetics and fidelity of the repair of Cas9-induced double-strand DNA breaks. Mol. Cell. 70, 801–813.
Funding
This work was supported by the Russian Foundation for Basic Research (project no. 17-00-00098) and the Russian Science Foundation (project no. 19-74-10009).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest. This work does not contain any studies involving animals or human subjects performed by any of the authors.
Additional information
Translated by T. Tkacheva
Rights and permissions
About this article

Cite this article
Velichko, A.K., Razin, S.V. & Kantidze, O.L. DNA Damage Response in Nucleoli. Mol Biol 55, 182–192 (2021). https://doi.org/10.1134/S002689332102014X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S002689332102014X