
Abstract—
Advances in the research of molecular factors involved in the onset and progression of Alzheimer’s disease, have led to the creation of several pathogenesis concepts of the most common neurodegenerative disease in the world, and amyloid, cholinergic, and neuroinflammatory hypotheses became leading. Over past twenty years, based on these hypotheses, hundreds of drug prototypes were developed, but none of them were able to stop the development of Alzheimer’s disease. In this review, based on the latest experimental data on structure-function properties of chemically modified amyloid-beta isoforms, the concept of the origin and the mechanism of action of amyloid-beta with isomerized Asp7 residue, as a molecular agent of Alzheimer’s disease pathogenesis, is presented. This concept makes it possible not only to combine the most important aspects of existing hypotheses but also indicates ways of creating agents for fighting Alzheimer’s disease with a principally new mechanism of action, “disease-modifying drugs.”
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Alzheimer’s disease (AD) is today’s most widespread neurodegenerative proteinopathy [1], which affects more than 44 million people [2]. Clinical manifestations of AD are expressed in inevitable cognitive decline and accompanied by organic brain destruction, which, in the end, results in patient death via respiratory arrest. Hereditary variants make less than 1% of total AD cases and are associated with mutations [3, 4], resulting in constitutional excess above the physiologically normal level of amyloid-beta (Aβ), a short polypeptide of 39‒43 amino-acid residues length with a heterogeneous С-terminal site [5]. Causes of the onset of sporadic variants, which make over 95% of all AD cases, remain unknown, but they are closely associated with abnormal aggregation of endogenous Aβ. There exist three main neuromorphological signs, which unambiguously confirm (postmortem) the diagnosis of all AD cases: (1) the presence, in certain brain regions, of typical extracellular aggregates (amyloid plagues), whose main components are various Aβ isoforms and biometal ions (zinc, as well as iron and copper); (2) the formation of intracellular neurofibrillary tangles (whose main component is hyperphosphorylated tau protein); and (3) neuronal degeneration [6].
Extensive experimental data support a contemporary version of the amyloid cascade hypothesis of AD pathogenesis [7]. This hypothesis states that slow steady accumulation of Aβ aggregates in the brain (cerebral amyloidogenesis, CA) initiates AD development by triggering a complex signaling cascade, which accelerates the pathological transformation of tau and results in neurodegeneration and clinical dementia. Such factors as familial mutations, traumatic brain injuries, specifics of lifestyle and environment, inflammation processes, and cardiovascular diseases increase the risk of the disease or influence pathological processes [8].
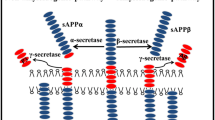
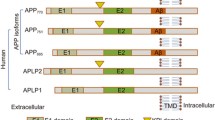
Endogenous Aβ is formed as a result of proteolysis of the amyloid protein precursor [9]; it is present in brain tissues and in peripheral organs over a lifetime. The Aβ level in blood and cerebrospinal fluid varies depending on age and individual specifics of patients, hence it cannot serve as an AD biomarker [10]. The physiological role of Aβ can be the regulation of a synaptic function, protection from infections, repairing damaged sites of the blood-brain barrier, and compensation of trauma consequences [11]. Aβ concentration in cerebrospinal fluid is 5 to 15 times higher than in plasma. It is not clear how Aβ is removed from blood, however it is established that about 60% Aβ goes from the brain into the peripheral circulatory system [12]. At the same time, Aβ molecules circulating in blood, which are mainly generated by platelets, can get into the brain at system circulation disturbance and significantly accelerate AD development [10].
According to the neuroinflammation hypothesis, disturbance in the regulation of the central nervous system immune response is considered a key factor of AD etiology, and neuroinflammation is regarded as an integral mechanism of AD pathogenesis [13]. The persistent anti-inflammatory response in the brain of AD patients was earlier considered a reaction to numerous neuronal loss. However, at present, the persistent immune response in the brain is not only linked to neurodegeneration but also considered a driving force of pathological processes involving Aβ and tau [14]. It is shown that peripheral inflammation and abnormalities of peripheral immune response in AD are associated with oxidative stress increase in the peripheral circulatory system and in the brain [15]. An increase in oxidative stress is described in peripheral blood lymphocytes obtained from AD patients. Neuroinflammatory processes in the brain are mediated mainly via the innate immune system, which includes astrocytes and microglial cells, as well as cytokines, chemokines, and growth factors. At the same time, the central nervous system is accessible for lymphocytes and monocytes, circulating in blood, which indicates an intensive interaction between immune and central nervous systems, aimed at maintaining homeostasis [16]. Aβ molecules, generated by platelets, may act as mediators of this interaction, and, at Aβ metabolism disturbance in the brain, may be a source for the formation and distribution of amyloid plaques in AD [17].
Besides Aβ and its chemically modified isoforms, amyloid plaques contain many other components, including various proteoglycans and glycosaminoglycanes, carbohydrate-binding proteins of the innate immunity system (in amyloid plaques, serum amyloid protein P (SAP) is represented the most), nucleic acids, biometal ions, lipids, and transport proteins. It is accepted that these cofactors may seriously affect Aβ aggregating processes [18]. However, by now, only for zinc ions, the absolute necessity for amyloid plaque formation in model animals is experimentally proven [19]. Zinc, released into glutamatergic synapse due to ZnT3 transporter activity, is vital for the maintenance of memory and cognitive functions [20]. ZnT3 is expressed only in damaged via amyloid pathology grey matter areas. Zinc, released into glutamatergic synapse through ZnT3, causes amyloid pathology in transgenic mice [21, 22], which explains the abnormally high content of zinc ions in amyloid plaques [23, 24]. Very low in physiological conditions, extracellular concentration of zinc ions (<1 μM) in neuronal activity can run up to 100‒300 μM in synaptic clefts of glutamatergic synapses [25]. It is important to notice that zinc ions also promote the formation of highly toxical zinc-bound Aβ oligomers [26].
Cholinergic synapses are spread everywhere in the human central nervous system, and the studying of synaptic neurotransmission in norm and AD led to the creation of the cholinergic hypothesis, which associates the progressive loss of limbic and neocortical cholinergic innervation with cognitive decline and neurodegeneration [27]. The following facts underlay the cholinergic hypothesis: (1) the detection of presynaptic cholinergic marker depletion in the brain cortex in AD; (2) the discovery that nucleus basalis of Meynert in the basal forebrain is a source of cortical cholinergic innervation, which is subjected to grave neurodegeneration in AD; (3) cholinergic antagonists impair memory, while agonists exert the opposite effect. The cholinergic hypothesis was practically implied in the development and successful application of cholinesterase inhibitors as a means of symptomatic treatment for AD patients. Five anti-AD drugs, approved by the US Food and Drug Administration, are used worldwide and include four cholinesterase inhibitors (Tacrine, Donepezil, Rivastigmine, Galantamine) [28].
It is suggested that amyloid plaques cause the degeneration of cholinergic fiber endings [29]; at the same time, at early stages of AD, first of all occurs the impairment of nicotine cholinoceptors (nAChR) of α4β2 subtype (α4β2-nAChR) [30], with which Aβ forms specific complexes [31, 32]. α4β2-nAChR receptors are widespread in the brain cortex. The loss of cognitive functions via middle-stage AD patients is accompanied by a substantial reduction in availability (according to data of positron emission tomography) of precisely α4β2-nAChR [33]. The 11-Glu-Val-His-His-14 (11EVHH14) site of Aβ is critical for interactions between Aβ and nAChR [34]. With the help of bioinformatic approaches, it is established that the extracellular N-terminal domain of the α4β2-nAChR α4 subunit includes 35HAEE38—the 35-His-Ala-Glu-Glu-38 fragment (conservative in human, mouse, and chicken), which is ionic-complementary to the 11EVHH14 Aβ site. An experimental test using an optical biosensor on a surface plasmon resonance effect showed that a synthetic [Acetyl]-His-Ala-Glu-Glu-[Amide] (Ac-HAEE-NH2) analogue of the 35HAEE38 site is specifically bound to the 11EVHH14 site of the immobilized metal-binding 1‒16 Aβ domain [35]. Thus, we suggest that the interaction interface of Aβ and α4β2-nAChR is formed via 11EVHH14 and 35HAEE38 sites of these molecules, correspondingly.
The ability of Aβ aggregates, extracted postmortem from AD patient brains, inducing AD development was for the first time demonstrated on primates, to whom the proper autopsy material was administered intracerebrally [36, 37]. Further, in a series of studies on animal AD models [38‒41], it was established that a molecular agent causing the formation of pathological amyloid plaques in brain tissues constitutes a conformational or chemically modified variant of Aβ [42‒44]. It was also shown that, in CA induction, amyloid plaques are formed in model animals in a day, then, after one to two days, the activation of microglial cells and their migration to the plaques are observed, which is accompanied by local alterations in axons and dendrites of adjacent neurons [45]. The spreading of amyloid plaques through brain structures occurs extracellularly [46]. As a result of incubation in hippocampus splice culture, synthetic Aβ becomes a pathological agent causing CA via seeding mechanism (at presence of a minor amount of brain homogenate from AD patients) [47]. The seeding CA initiation mechanism in AD suggests a significant alteration of the native structure of endogenous Aβ under the influence of interaction with pathological Aβ molecules [48], present in amyloid plaques. This mechanism is confirmed by the fact that minor amounts of pathologically changed Aβ molecules, contained in material from AD patients, caused the transition of native Aβ molecules into a pathological state [49, 50].
The tendency of full-size Aβs to spontaneous aggregation prevents determination of their spatial structures with modern biophysical methods [51]. However, the N-terminal 1‒16 (Aβ16) fragment, which is also present in the human organism as an independent factor [52], is an autonomous metal-binding Aβ domain, stable in vitro [53]. The structure of this domain is successfully determined in a free state and in complex with zinc ion in physiologically relevant conditions [54]. It turned out that rigid spatial organization of the main polypeptide chain of the 11EVHH14 site remains practically unchanged in intact Aβ16 and in the Aβ16 complex with zinc ion [54, 55]. The secondary structure of the 11EVHH14 site constitutes a left-handed helix of the polyprolin II type [56], possessing high predisposition to participate in protein-protein interactions [57].
The molecular mechanism of interaction between zinc ions and Aβ16 is established. First, zinc ion is chelated by Glu11, His13, and His14 side chains, then takes place the folding of N-terminal 1‒8 Aβ16 site with the following formation of additional coordination linkage between zinc ion and His6 side chain, resulting in the appearance of a well-ordered compact structure of the whole 1‒16 domain [58]. It is also shown that the 11EVHH14 site of amyloid-beta acts not only as primary center of zinc ion recognition but also controls zinc-induced Aβ dimerization processes [59, 60].
Complexes of the zinc ion with a metal-binding Aβ domain may constitute either compactly folded monomers, where zinc ion is coordinated via four chelator (His6, Glu11, His13, and His14) of one Aβ16 molecule, or structurally unfolded dimers, where one zinc ion and Glu11 and His14 amino acid residues of interacting subunits form an intermolecular interface. In the presence of zinc ions, the metal-binding domain within full-size forms of Aβ is mainly unfolded and is involved in intermolecular bonds that stabilize polymorphic oligomers and aggregates of Aβ [61]. The molecular mechanism of zinc-dependent Aβ16 oligomerization includes the formation of unfolded dimer, then in each subunit of the dimer occurs the regrouping of side chains His6 and His13, which lets these residues from two adjacent Aβ16 molecules form an additional molecular interface with one zinc ion. As a result, each Aβ16 subunit within oligomers has two intermolecular zinc-dependent interfaces [62]. Thus, the Aβ16-zinc-Aβ16 dimer is a seed of zinc-dependent oligomerization of Aβ16 molecules via “chain” mechanism. In forming oligomers, the conformation of the main chain of the 11EVHH14 fragment of each domain remains the same in free and in bound with zinc ion Aβ16 molecules [62]. By the total of all properties, 11EVHH14 site constitutes a structure-function determinant not only for Aβ16 in zinc-bound oligomers but, perhaps, also for full-size Aβ molecules within amyloid plaques. Therefore, a new antiamyloid strategy for AD therapy seems to be prospective, where substances, specifically binding to the 11EVHH14 site, will be used as medicinal agents preventing pathological Aβ aggregation and the destruction of already formed amyloid plaques in AD [63].
The isomerization of Asp7 residue in Aβ metal-binding domain plays an especially significant role in the interaction of Aβ with zinc ions [64]. This chemical modification significantly increases the ability of Aβ16 to undergo zinc-dependent oligomerization due to intramolecular conformational changes, sterically facilitating the 11EVHH14 site ability to bind with other Aβ molecules via zinc-dependent interfaces [65, 66]. In amyloid plaques, Aβ with isomerized Asp7 residue (isoAβ) is present in abnormally high amounts (about 50% of total Aβ content) [67, 68]. IsoAβ is formed spontaneously as a result of “protein aging” [69] both of circulating and aggregated Aβ molecules [51]. It is shown that isoAβ accumulation in brain tissues is a common process for elderly people; however, the isoAβ level is significantly higher in AD patients [70].
We made a suggestion that, under the action of inner or outer stress factors in the peripheral circulatory system (which seem to be more susceptible to different kinds of stress than the central nervous system), isoAβ is formed. Further, isoAβ, in interaction with the 35HAEE38 site of the extracellular N-terminal domain of the α4 subunit of α4β2-nAChR, forms an abnormally long-lived complex, or “amyloid matrix,” while an intact Aβ forms a realtively short-lived complex with the same site of the receptor, and, in such a complex, the normal function of Aβ in the organism is fulfilled. On the contrary, the “amyloid matrix,” on the synaptic burst of zinc ion concentration, initiates zinc-dependent oligomerization of endogenous Aβ molecules, as a consequence of which, in a short time, an amyloid plaque is formed on “amyloid matrix,” whose growth can be restricted by a microglial response. This extracellular plaque can exist for decades and does no direct harm to the neuron, since Aβ molecules are not transported from it in significant amounts (because of their condensed state) into the cell and, correspondingly, no signal cascades are activated. Under favorable circumstances, amyloid plaques would be practically inert Aβ deposits, at first insignificantly aggravating the life of neurons, but eventually causing their apoptosis (most likely, through the induction of intraneuronal tau protein phosphorylation [71, 72]), and, as a consequence, a cognitive decline in the patient in the long term. However, if, for some reason, for instance, as a result of neuroinflammation, the necrosis of at least one neuron starts, then different cytosolic proteins will get into extracellular space, in particular, one of the most widespread cell proteins—glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH). GAPDH is involved in AD pathogenesis [73], presumably, as a result of the formation of neurotoxic aggregates with Aβ [74]. Upon reaching a certain critical number of amyloid plaques in brain tissues, the initial focus of neuronal necrosis may trigger a chain reaction of mass neuron loss, typical for late stages of AD. Thus, isoAβ, which is considered an end product of the pathological transformation of physiologically normal Aβ undergoing various kinds of stress, can be a molecular agent of initiation of AD pathogenesis.
We tested this hypothesis on the animal model of AD. In 2013, it was for the first time shown that intravenous injection of synthetic isoAβ to transgenic mice of the B6C3-Tg(APPswe, PSEN1-dE9)85Dbo/j line results in abrupt acceleration of CA [75]. At the same time, there was no trace of amyloid plaques in wild-type control mice, which confirms the role of isoAβ as a seed of human endogenic Aβ aggregation. Five years later, the possibility of the transition of a molecular agent, causing AD, from the peripheral system into the brain, was confirmed in independent studies using the parabiosis of the APPswe/PS1dE9 line transgenic mice and wild-type mice [76], and on transgenic mice of the APPswe/PS1dE9 line, to which were intravenously administered preparations of AD patients brain homogenates [77]. With the help of 5XFAD line transgenic mice, we established that the precisely metal-binding domain of isoAβ (isoAβ16) is necessary and sufficient for exogenous induction of CA in animals [78]. Thus, using synthetic peptides isoAβ and isoAβ16, we can arbitrarily turn on the molecular mechanism of CA in animal models of AD, where human Aβ is constitutively expressed.
It is important to note that, according to this mechanism, amyloid plaques can be formed only on condition that endogenous Aβ molecules with an amino acid sequence, identical to the human Aβ sequence, are present in the organism. Three amino acid replacements (Arg5Gly, Tyr10Phe, and His13Arg) in the metal-binding domain Aβ of rats and mice distinguish them from other mammals and are associated with resistance to Alzheimer type neurodegeneration in these rodents [79]. According to our hypothesis, precisely the His13Arg mutation completely prevents zinc-dependent oligomerization in these animals. Aβ of the naked mole rat (Heterocephalus glaber) contains only this replacement, and this animal never has Alzheimer type neurodegeneration [80]. Earlier it was reported that, in the Aβ of degu (Octodon degu), a single His13Arg replacement is also present, but that degu are allegedly susceptible to spontaneous Alzheimer type neurodegeneration [81], however, later independent studies refute these data [82, 83].
Based on the critical role of “amyloid matrices,” i.e., complexes between isoAβ and α4β2-nAChR, in the initiation of zinc-dependent formation of amyloid plaques in AD, we suggested that compounds, capable either to inhibit the “amyloid matrix” formation or hinder zinc-dependent Aβ oligomerization on this matrix, can block CA and destroy amyloid plaques. A synthetic peptide [Acetyl]-His-Ala-Glu-Glu-[Amide] (Ac-HAEE-NH2)—N-acetylated and C-amidated analog the 35-His-Ala-Glu-Glu-38 (35HAEE38) site of the subunit α4 of nicotinic acetylcholine receptor α4β2-nAChR—was tested as a substance for destroying amyloid matrices. Intravenous injection of Ac-HAEE-NH2 to transgenic mice of the B6C3-Tg(APPswe, PSEN1-dE9)85Dbo/j line resulted in an abrupt decrease in the number of amyloid plaques in animal brains [84]. We have tested the ability of synthetic Aβ and isoAβ analogs, phosphorylated at Ser8 residue, to inhibit zinc-dependent Aβ oligomerization. The choice of these molecules is based on the influence of Ser8 residue phosphorylation on the blockage of zinc-dependent oligomerization of metal-binding Aβ domain established earlier by us [60]. Intravenous injections of phosphorylated Aβ and isoAβ resulted in significant retardation of CA in model animals [85, 86].
The following integral scenario of the CA molecular mechanism in AD is suggested: (1) because of unknown causes (most likely stress and aging), isoAβ appears in the brain; (2) the 11EVHH14 site of this chemically modified Aβ isoform interacts with the 35HAEE38 site of the α4 subunit of α4β2-nAChR, resulting in the “amyloid matrix”—a long-lived complex of isoAβ and α4β2-nAChR—forming on the external surface of the neuron; (3) upon zinc ion concentration burst in the synaptic cleft, the intact Aβ molecule “sticks” to the “amyloid matrix” via a zinc-dependent mechanism, herewith the 11EVHH14 fragment participates in the interface on behalf of this Aβ molecule, while, on behalf of the “amyloid matrix,” the His6 and His13 residues of isoAβ molecules do so—and an already insoluble aggregate appears, at which on the external side an endogenous Aβ molecule is located, however, already in the pathological conformation (as a result of interaction with isoAβ from “amyloid matrix”); (4) further, by the same pattern, endogenous Aβ molecules, newly arrived from the extracellular space, “stick” on Aβ molecules, aggregated on the initial “amyloid matrix,” and the growth of amyloid plaque goes on (to a certain canonic volume).
From this mechanism of amyloid plaque formation, it follows that the most effective and safe way to destroy this plaque would be to use structure analogs (peptide or peptidomimetics) of the 35HAEE38 site of the α4 subunit of α4β2-nAChR, for example, an Ac-HAEE-NH2 substance, which has a potential therapeutic effect already confirmed on an animal AD model [84]. When interacting with an amyloid plaque, these analogs will progressively destroy intermolecular interfaces involving Aβ fragments 11EVHH14 due to the specific binding with 11EVHH14 sites of aggregated Aβ molecules. Herewith, the “amyloid matrix,” underlying this pathological pyramid eventually also will be destroyed [63], and the neuron that for many long years or even decades was burdened with amyloid plaques, will return to normal life.
The “amyloid matrices” concept, introduced in this work, is based on the convergence of leading hypotheses of AD pathogenesis and makes it possible not only to suggest an integral molecular mechanism of CA in this disease, but indicates the possible role of amyloid plaques in neurodegeneration in AD, and also the perspectives of developing disease modifying drugs, capable of effectively stopping the clinical course of the disease.
REFERENCES
Shelkovnikova T.A., Kulikova A.A., Tsvetkov F.O., Peters O., Bachurin S.O., Bukhman V.L., Ninkina N.N. 2012. Proteinopathies, neurodegenerative disorders with protein aggregation-based pathology. Mol. Biol. (Moscow). 46 (3), 362–374.
Alzheimer'sassociation (2014. 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 10 (2), e47‒e92.
Rogaev E.I., Sherrington R., Rogaeva E.A., Levesque G., Ikeda M., Liang Y., Chi H., Lin C., Holman K., Tsuda T., Mar L., Sorbi S., Nacmias B., Piacentini S., Amaducci L., et al. 1995. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 376, 775‒778.
Sherrington R., Rogaev E.I., Liang Y., Rogaeva E.A., Levesque G., Ikeda M., Chi H., Lin C., Li G., Holman K., Tsuda T., Mar L., Foncin J.F., Bruni A.C., Montesi M.P., et al. 1995. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 375, 754‒760.
Querfurth H.W., Laferla F.M. 2010. Alzheimer’s disease. N. Engl. J. Med. 362, 329‒344.
Cummings J.L. 2004. Alzheimer’s disease. N. Engl. J. Med.351, 56‒67.
Karran E., Mercken M., De Strooper B. 2011. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698‒712.
Golde T.E., Dekosky S.T., Galasko D. 2018. Alzheimer’s disease: The right drug, the right time. Science. 362, 1250‒1251.
Müller U.C., Zheng H. 2012. Physiological functions of APP family proteins. Cold Spring Harb. Perspect. Med. 2 (2), a006288.
Roher A.E., Esh C.L., Kokjohn T.A., Castano E.M., Van Vickle G.D., Kalback W.M., Patton R.L., Luehrs D.C., Daugs I.D., Kuo Y.M., Emmerling M.R., Soares H., Quinn J.F., Kaye J., Connor D.J., et al. 2009. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimers Dement. 5, 18‒29.
Brothers H.M., Gosztyla M.L., Robinson S.R. 2018. The physiological roles of amyloid-β peptide hint at new ways to treat Alzheimer’s disease. Front. Aging Neurosci. 10, 118.
Wang J., Gu B.J., Masters C.L., Wang Y.J. 2017. A systemic view of Alzheimer disease: Insights from amyloid-beta metabolism beyond the brain. Nat. Rev. Neurol. 13, 612‒623.
Cao W., Zheng H. 2018. Peripheral immune system in aging and Alzheimer’s disease. Mol. Neurodegener. 13, 51.
Kinney J.W., Bemiller S.M., Murtishaw A.S., Leisgang A.M., Salazar A.M., Lamb B.T. 2018. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. (NY). 4, 575‒590. https://doi.org/10.1016/j.trci.2018.06.014
Morris G., Berk M., Maes M., Puri B.K. 2019. Could Alzheimer’s disease originate in the periphery and if so how so? Mol. Neurobiol. 56, 406‒434.
Esteras N., Alquezar C., De La Encarnacion A., Martin-Requero A. 2016. Lymphocytes in Alzheimer’s disease pathology: Altered signaling pathways. Curr. Alzheimer Res. 13, 439‒449.
Inyushin M.Y., Sanabria P., Rojas L., Kucheryavykh Y., Kucheryavykh L. 2017. Aβ: Peptide originated from platelets promises new strategy in anti-Alzheimer’s drug development. BioMed. Res. Int.2017, 10.
Stewart K.L., Radford S.E. 2017. Amyloid plaques beyond Abeta: A survey of the diverse modulators of amyloid aggregation. Biophys. Rev. 9, 405‒419.
Bush A.I. 2013. The metal theory of Alzheimer’s disease. J. Alzheimer’s Dis. 33, S277‒S281.
Adlard P.A., Parncutt J.M., Finkelstein D.I., Bush A.I. 2010. Cognitive loss in zinc transporter-3 knock-out mice: A phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 30, 1631‒1636.
Friedlich A.L., Lee J.-Y., Van Groen T., Cherny R.A., Volitakis I., Cole T.B., Palmiter R.D., Koh J.-Y., Bush A.I. 2004. Neuronal zinc exchange with the blood vessel wall promotes cerebral amyloid angiopathy in an animal model of Alzheimer’s disease. J. Neurosci. 24, 3453‒3459.
Lee J.Y., Cole T.B., Palmiter R.D., Suh S.W., Koh J.Y. 2002. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. U. S. A.99, 7705‒7710.
Lovell M.A., Robertson J.D., Teesdale W.J., Campbell J.L., Markesbery W.R. 1998. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 158, 47‒52.
Miller L.M., Wang Q., Telivala T.P., Smith R.J., Lanzirotti A., Miklossy J. 2006. Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 155, 30‒37.
Xie X.M., Smart T.G. 1991. A physiological role for endogenous zinc in rat hippocampal synaptic neurotransmission. Nature. 349, 521‒524.
Lee M.C., Yu W.C., Shih Y.H., Chen C.Y., Guo Z.H., Huang S.J., Chan J.C.C., Chen Y.R. 2018. Zinc ion rapidly induces toxic, off-pathway amyloid-beta oligomers distinct from amyloid-beta derived diffusible ligands in Alzheimer’s disease. Sci. Rep. 8, 4772.
Hampel H., Mesulam M.M., Cuello A.C., Farlow M.R., Giacobini E., Grossberg G.T., Khachaturian A.S., Vergallo A., Cavedo E., Snyder P.J., Khachaturian Z.S. 2018. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain. 141, 1917‒1933.
Cummings J., Morstorf T., Zhong K. 2014. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 6, 37.
Auld D.S., Kornecook T.J., Bastianetto S., Quirion R. 2002. Alzheimer’s disease and the basal forebrain cholinergic system: relations to beta-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 68, 209‒245.
Perry E.K., Morris C.M., Court J.A., Cheng A., Fairbairn A.F., Mckeith I.G., Irving D., Brown A., Perry R.H. 1995. Alteration in nicotine binding sites in Parkinson’s disease, Lewy body dementia and Alzheimer’s disease: Possible index of early neuropathology. Neuroscience. 64, 385‒395.
Paterson D., Nordberg A. 2000. Neuronal nicotinic receptors in the human brain. Prog. Neurobiol. 61, 75‒111.
Wu J., Kuo Y.-P., George A.A., Xu L., Hu J., and Lukas R.J. 2004. β-Amyloid directly inhibits human α4β2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J. Biol. Chem. 279, 37842‒37851.
Sabri O., Meyer P.M., Graf S., Hesse S., Wilke S., Becker G.A., Rullmann M., Patt M., Luthardt J., Wagenknecht G., Hoepping A., Smits R., Franke A., Sattler B., Tiepolt S., et al. 2018. Cognitive correlates of alpha4beta2 nicotinic acetylcholine receptors in mild Alzheimer’s dementia. Brain. 141, 1840‒1854.
Lawrence J.L.M., Tong M., Alfulaij N., Sherrin T., Contarino M., White M.M., Bellinger F.P., Todorovic C., Nichols R.A. 2014. Regulation of presynaptic Ca2+, synaptic plasticity and contextual fear conditioning by a N-terminal β-amyloid fragment. J. Neurosci. 34, 14210‒14218.
Mediannikov O., Morozov A. 2014). Peptide compound useful for inhibiting amyloid plaque formation. France Patent 2,966,827, filed October 10, 2010; issued August 22, 2014.
Baker H.F., Ridley R.M., Duchen L.W., Crow T.J., Bruton C.J. 1994. Induction of beta (A4)-amyloid in primates by injection of Alzheimer’s disease brain homogenate. Comparison with transmission of spongiform encephalopathy. Mol. Neurobiol. 8, 25‒39.
Ridley R.M., Baker H.F., Windle C.P., Cummings R.M. 2006. Very long term studies of the seeding of beta-amyloidosis in primates. J. Neural. Transm. 113, 1243‒1251.
Langer F., Eisele Y.S., Fritschi S.K., Staufenbiel M., Walker L.C., Jucker M. 2011. Soluble Abeta seeds are potent inducers of cerebral beta-amyloid deposition. J. Neurosci. 31, 14488‒14495.
Morales R., Duran-Aniotz C., Castilla J., Estrada L.D., Soto C. 2012. De novo induction of amyloid-β deposition in vivo.Mol. Psychiatry. 17, 1347‒1353.
Rosen R.F., Fritz J.J., Dooyema J., Cintron A.F., Hamaguchi T., Lah J.J., Levine H., 3rd, Jucker M., Walker L.C. 2012. Exogenous seeding of cerebral beta-amyloid deposition in betaAPP-transgenic rats. J. Neurochem. 120, 660‒666.
Watts J.C., Giles K., Grillo S.K., Lemus A., Dearmond S.J., Prusiner S.B. 2011. Bioluminescence imaging of Aβ deposition in bigenic mouse models of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A.108, 2528‒2533.
Eisele Y.S., Bolmont T., Heikenwalder M., Langer F., Jacobson L.H., Yan Z.X., Roth K., Aguzzi A., Staufenbiel M., Walker L.C., Jucker M. 2009. Induction of cerebral beta-amyloidosis: Intracerebral versus systemic Abeta inoculation. Proc. Natl. Acad. Sci. U. S. A.106, 12926‒12931.
Eisele Y.S., Obermuller U., Heilbronner G., Baumann F., Kaeser S.A., Wolburg H., Walker L.C., Staufenbiel M., Heikenwalder M., Jucker M. 2010. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 330, 980‒982.
Meyer-Luehmann M., Coomaraswamy J., Bolmont T., Kaeser S., Schaefer C., Kilger E., Neuenschwander A., Abramowski D., Frey P., Jaton A.L., Vigouret J.M., Paganetti P., Walsh D.M., Mathews P.M., Ghiso J., et al. 2006. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 313, 1781‒1784.
Meyer-Luehmann M., Spires-Jones T., Prada C., Garcia-Alloza M., De Calignon A., Rozkalne A., Koenigsknecht-Talboo J., Holtzman D., Bacskai B., Hyman B. 2008. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 451, 720‒724.
Mezias C., Raj A. 2017. Analysis of amyloid-β pathology spread in mouse models suggests spread is driven by spatial proximity, not connectivity. Front. Neurol. 8, 653.
Novotny R., Langer F., Mahler J., Skodras A., Vlachos A., Wegenast-Braun B.M., Kaeser S.A., Neher J.J., Eisele Y.S., Pietrowski M.J., Nilsson K.P., Deller T., Staufenbiel M., Heimrich B., Jucker M. 2016. Conversion of synthetic Abeta to in vivo active seeds and amyloid plaque formation in a hippocampal slice culture model. J. Neurosci. 36, 5084‒5093.
Jucker M., Walker L.C. 2018. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 21, 1341‒1349.
Jaunmuktane Z., Mead S., Ellis M., Wadsworth J.D., Nicoll A.J., Kenny J., Launchbury F., Linehan J., Richard-Loendt A., Walker A.S., Rudge P., Collinge J., Brandner S. 2015. Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature. 525, 247‒250.
Purro S.A., Farrow M.A., Linehan J., Nazari T., Thomas D.X., Chen Z., Mengel D., Saito T., Saido T., Rudge P., Brandner S., Walsh D.M., Collinge J. 2018. Transmission of amyloid-beta protein pathology from cadaveric pituitary growth hormone. Nature. 564, 415‒419.
Masters C.L., Selkoe D.J. 2012. Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harbor Persp. Med. 2, a006262.
Portelius E., Price E., Brinkmalm G., Stiteler M., Olsson M., Persson R., Westman-Brinkmalm A., Zetterberg H., Simon A.J., Blennow K. 2011. A novel pathway for amyloid precursor protein processing. Neurobiol. Aging. 32, 1090‒1098.
Kozin S.A., Zirah S., Rebuffat S., Hui Bon Hoa G., Debey P. 2001. Zinc binding to Alzheimer’s Aβ(1‒16) peptide results in stable soluble complex. Biochem. Biophys. Res. Commun. 285, 959‒964.
Zirah S., Kozin S.A., Mazur A.K., Blond A., Cheminant M., Segalas-Milazzo I., Debey P., Rebuffat S. 2006. Structural changes of region 1–16 of the Alzheimer disease amyloid β-peptide upon zinc binding and in vitro aging. J. Biol. Chem. 281, 2151‒2161.
Nisbet R.M., Nuttall S.D., Robert R., Caine J.M., Dolezal O., Hattarki M., Pearce L.A., Davydova N., Masters C.L., Varghese J.N., Streltsov V.A. 2013. Structural studies of the tethered N-terminus of the Alzheimer’s disease amyloid-β peptide. Proteins: Struct. Funct. Bioinformatics. 81, 1748‒1758.
Adzhubei A.A., Anashkina A.A., Makarov A.A. 2017. Left-handed polyproline-II helix revisited: Proteins causing proteopathies. J. Biomol. Struct. Dyn. 35, 2701‒2713.
Adzhubei A.A., Sternberg M.J.E., Makarov A.A. 2013. Polyproline-II helix in proteins: Structure and function. J. Mol. Biol. 425, 2100‒2132.
Tsvetkov P.O., Kulikova A.A., Golovin A.V., Tkachev Y.V., Archakov A.I., Kozin S.A., Makarov A.A. 2010. Minimal Zn2+ binding site of amyloid-β. Biophys. J.99, L84‒L86.
Kozin S.A., Mezentsev Y.V., Kulikova A.A., Indeykina M.I., Golovin A.V., Ivanov A.S., Tsvetkov P.O., Makarov A.A. 2011. Zinc-induced dimerization of the amyloid-β metal-binding domain 1–16 is mediated by residues 11–14. Mol. BioSystems. 7, 1053‒1055.
Kulikova A.A., Tsvetkov P.O., Indeykina M.I., Popov I.A., Zhokhov S.S., Golovin A.V., Polshakov V.I., Kozin S.A., Nudler E., Makarov A.A. 2014. Phosphorylation of Ser8 promotes zinc-induced dimerization of the amyloid-beta metal-binding domain. Mol. Biosyst. 10, 2590‒2596.
Miller Y., Ma B., Nussinov R. 2010. Zinc ions promote Alzheimer Abeta aggregation via population shift of polymorphic states. Proc. Natl. Acad. Sci. U. S. A.107, 9490‒9495.
Istrate A.N., Kozin S.A., Zhokhov S.S., Mantsyzov A.B., Kechko O.I., Pastore A., Makarov A.A., Polshakov V.I. 2016. Interplay of histidine residues of the Alzheimer’s disease Aβ peptide governs its Zn-induced oligomerization. Sci. Rep. 6, 21734.
Kozin S.A., Barykin E.P., Mitkevich V.A., Makarov A.A. 2018. Antiamyloid therapy of Alzheimer’s disease: Current state and prospects. Biochemistry (Moscow). 83 (9), 1057‒1067.
Kulikova A.A., Makarov A.A., Kozin S.A. 2015. Roles of zinc ions and structural polymorphism of β-amyloid in the development of Alzheimer’s disease. Mol. Biol. (Moscow). 49 (2), 217–230.
Mezentsev Y.V., Medvedev A.E., Kechko O.I., Makarov A.A., Ivanov A.S., Mantsyzov A.B., Kozin S.A. 2016. Zinc-induced heterodimer formation between metal-binding domains of intact and naturally modified amyloid-beta species: implication to amyloid seeding in Alzheimer’s disease? J. Biomol. Struct. Dyn. 34, 2317‒2326.
Tsvetkov P.O., Popov I.A., Nikolaev E.N., Archakov A.I., Makarov A.A., Kozin S.A. 2008. Isomerization of the Asp7 residue results in zinc-induced oligomerization of Alzheimer’s disease amyloid β(1‒16) peptide. Chembiochem. 9, 1564‒1567.
Hosoda R., Saido T.C., Otvos L.J., Arai T., Mann D.M.A., Lee V.M.-Y., Trojanowski J.Q., Iwatsubo T. 1998. Quantification of modified amyloid β peptides in Alzheimer disease and Down Syndrome brains. J. Neuropathol. Exp. Neurol. 57, 1089‒1095.
Roher A.E., Lowenson J.D., Clarke S., Wolkow C., Wang R., Cotter R.J., Reardon I.M., Zurcher-Neely H.A., Heinrikson R.L., Ball M.J., Greenberg B.D. 1993. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 268, 3072‒3083.
Orpiszewski J., Schormann N., Kluve-Beckerman B., Liepnieks J.J., Benson M.D. 2000. Protein aging hypothesis of Alzheimer disease. FASEB J.14, 1255‒1263.
Moro M.L., Phillips A.S., Gaimster K., Paul C., Mudher A., Nicoll J.A.R., Boche D. 2018. Pyroglutamate and isoaspartate modified amyloid-beta in ageing and Alzheimer’s disease. Acta Neuropathol. Commun.6, 3.
Mitkevich V.A., Petrushanko I.Y., Yegorov Y.E., Simonenko O.V., Vishnyakova K.S., Kulikova A.A., Tsvetkov P.O., Makarov A.A., Kozin S.A. 2013. Isomerization of Asp7 leads to increased toxic effect of amyloid-β42 on human neuronal cells. Cell Death Dis. 4, e939.
Zatsepina O.G., Kechko O.I., Mitkevich V.A., Kozin S.A., Yurinskaya M.M., Vinokurov M.G., Serebryakova M.V., Rezvykh A.P., Evgen’ev M.B., Makarov A.A. 2018. Amyloid-β with isomerized Asp7 cytotoxicity is coupled to protein phosphorylation. Sci. Rep. 8, 3518.
Li Y., Nowotny P., Holmans P., Smemo S., Kauwe J.S., Hinrichs A.L., Tacey K., Doil L., Van Luchene R., Garcia V., Rowland C., Schrodi S., Leong D., Gogic G., Chan J., et al. 2004. Association of late-onset Alzheimer’s disease with genetic variation in multiple members of the GAPD gene family. Proc. Natl. Acad. Sci. U. S. A.101, 15688‒15693.
Itakura M., Nakajima H., Kubo T., Semi Y., Kume S., Higashida S., Kaneshige A., Kuwamura M., Harada N., Kita A., Azuma Y.-T., Yamaji R., Inui T., Takeuchi T. 2015. Glyceraldehyde-3-phosphate dehydrogenase aggregates accelerate amyloid-β amyloidogenesis in Alzheimer disease. J. Biol. Chem. 290, 26072‒26087.
Kozin S.A., Cheglakov I.B., Ovsepyan A.A., Telegin G.B., Tsvetkov P.O., Lisitsa A.V., Makarov A.A. 2013. Peripherally applied synthetic peptide isoAsp7-Aβ(1‒42) triggers cerebral β-amyloidosis. Neurotox. Res.24, 370‒376.
Bu X.L., Xiang Y., Jin W.S., Wang J., Shen L.L., Huang Z.L., Zhang K., Liu Y.H., Zeng F., Liu J.H., Sun H.L., Zhuang Z.Q., Chen S.H., Yao X.Q., Giunta B., et al. 2018. Blood-derived amyloid-β protein induces Alzheimer’s disease pathologies. Mol. Psychiatry. 23, 1948‒1956.
Burwinkel M., Lutzenberger M., Heppner F.L., Schulz-Schaeffer W., Baier M. 2018. Intravenous injection of beta-amyloid seeds promotes cerebral amyloid angiopathy (CAA). Acta Neuropathol. Commun. 6, 23.
Kulikova A.A., Cheglakov I.B., Kukharsky M.S., Ovchinnikov R.K., Kozin S.A., Makarov A.A. 2016. Intracerebral Injection of metal-binding domain of Abeta comprising the isomerized Asp7 increases the amyloid burden in transgenic mice. Neurotox. Res. 29, 551‒557.
Istrate A.N., Tsvetkov P.O., Mantsyzov A.B., Kulikova A.A., Kozin S.A., Makarov A.A., Polshakov V.I. 2012. NMR solution structure of rat Aβ(1‒16): Toward understanding the mechanism of rats’ resistance to Alzheimer’s disease. Biophys. J.102, 136‒143.
Edrey Y.H., Medina D.X., Gaczynska M., Osmulski P.A., Oddo S., Caccamo A., Buffenstein R. 2013. Amyloid beta and the longest-lived rodent: The naked mole-rat as a model for natural protection from Alzheimer’s disease. Neurobiol. Aging. 34, 2352‒2360.
Ardiles A.O., Tapia-Rojas C.C., Mandal M., Alexandre F., Kirkwood A., Inestrosa N.C., Palacios A.G. 2012. Postsynaptic dysfunction is associated with spatial and object recognition memory loss in a natural model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A.109, 13835‒13840.
Bourdenx M., Dovero S., Thiolat M.-L., Bezard E., Dehay B. 2017. Lack of spontaneous age-related brain pathology in Octodon degus: A reappraisal of the model. Sci. Repts. 7, 45831.
Steffen J., Krohn M., Paarmann K., Schwitlick C., Brüning T., Marreiros R., Müller-Schiffmann A., Korth C., Braun K., Pahnke J. 2016. Revisiting rodent models: Octodon degus as Alzheimer’s disease model? Acta Neuropathol. Commun.4, 91.
Tsvetkov P.O., Cheglakov I.B., Ovsepyan A.A., Mediannikov O.Y., Morozov A.O., Telegin G.B., Kozin S.A. 2015. Peripherally applied synthetic tetrapeptides HAEE and RADD slow down the development of cerebral beta-amyloidosis in AbetaPP/PS1 transgenic mice. J. Alzheimers Dis. 46, 849‒853.
Barykin E.P., Petrushanko I.Y., Kozin S.A., Telegin G.B., Chernov A.S., Lopina O.D., Radko S.P., Mitkevich V.A., Makarov A.A. 2018. Phosphorylation of the amyloid-beta peptide inhibits zinc-dependent aggregation, prevents Na,K-ATPase inhibition, and reduces cerebral plaque deposition. Front. Mol. Neurosci. 11, 302.
Kozin S.A., Barykin E.P., Telegin G.B., Chernov A.S., Adzhubei A.A., Radko S.P., Mitkevich V.A., Makarov A.A. 2018. Intravenously injected amyloid-β peptide with isomerized Asp7 and phosphorylated Ser8 residues inhibits cerebral β-amyloidosis in AβPP/PS1 transgenic mice model of Alzheimer’s disease. Front. Neurosci.12, 518.
Funding
The work is supported by Russian Science Foundation (grant no. 19-74-30007).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Translated by A. Khaitin
Rights and permissions
About this article

Cite this article
Kozin, S.A., Makarov, A.A. The Convergence of Alzheimer’s Disease Pathogenesis Concepts. Mol Biol 53, 896–903 (2019). https://doi.org/10.1134/S0026893319060104
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026893319060104