
Abstract—In the cell, protein folding begins during protein synthesis/translation and thus is a co-translational process. Co-translational protein folding is tightly linked to translation elongation, which is not a uniform process. While there are many reasons for translation non-uniformity, it is generally believed that non-uniform synonymous codon usage is one of the key factors modulating translation elongation rates. Frequent/optimal codons as a rule are translated more rapidly than infrequently used ones and vice versa. Over 30 years ago, it was hypothesized that changes in synonymous codon usage affecting translation elongation rates could impinge on co-translation protein folding and that many synonymous codons are strategically placed within mRNA to ensure a particular translation kinetics facilitating productive step-by-step co-translational folding of proteins. It was suggested that this particular translation kinetics (and, specifically, translation pause sites) may define the window of opportunity for the protein parts to fold locally, particularly at the critical points where folding is far from equilibrium. It was thus hypothesized that synonymous codons may provide a secondary code for protein folding in the cell. Although, mostly accepted now, this hypothesis appeared to be difficult to prove and many convincing results were obtained only relatively recently. Here, I review the progress in the field and explain, why this simple idea appeared to be so challenging to prove.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Protein folding is one of the most fundamental mechanisms in the cell. For any protein to function properly, the polypeptide chain produced by the ribosome, the protein-synthesizing factory of the cell, has to fold into the correct three-dimensional structure. Misfolding and aggregation are implicated in a number of diseases, including many neurodegenerative proteinopathies, such as Alzheimer’s disease, prion diseases and Parkinson’s disease to name a few [1–3]. Therefore, knowledge of how proteins acquire their spatial structure (folds) is extremely important.
For decades the mechanism by which the polypeptide chain acquires its native structure has been investigated mainly by in vitro denaturation/renaturation experiments [4, 5]. Seminal research performed by Christian Anfinsen and his colleagues on the reversible denaturation of ribonuclease (in the 1950–60’s) prompted their suggestion that a protein’s amino acid sequence contains all the information necessary to specify its unique three-dimensional structure [6]. These original experiments were followed by many in depth in vitro unfolding/refolding as well as in silico studies and provided a wealth of information suggesting that protein folding obeys a sequential model which postulates a unique pathway with defined intermediates [4, 5, 7, 8]. Subsequently, the concept of the folding funnel has been developed further suggesting that there could be multiple pathways which guide protein folding to a native conformation with the lowest free energy minimum [7, 9]. The majority of these studies employed relatively small proteins that can be successfully refolded in aqueous solutions, and these observations supported the Anfinsen’s principle [4, 5, 7–9]. Yet, many attempts to achieve in vitro 100% refolding of isolated denatured proteins were only partially successful [4]. In addition, in most cases reconstitution in a test tube was found to be exceedingly slow and not comparable with times required/expected for a protein to acquire its native structure in the cell [10].
It thus became evident that comprehensive understanding of the mechanism of protein folding requires elucidation of the folding pathway under native conditions, such as those that exist in vivo in the cell [11–14]. These conditions are quite different from those in a test tube. Firstly, folding in vivo takes place in a crowded cellular environment and thus is thought to be affected by a number of factors, such as concentration and activity/affinity of surrounding macromolecules and the presence of folding catalysts and accessory proteins [15, 16]. Systematic investigations of these effects revealed an important role played by folding catalysts and accessory proteins in acceleration of the rate of in vivo protein folding and in the prevention of protein misfolding and aggregation [15, 16]. However, it has been also suggested that both chaperones and folding catalysts are mainly involved in kinetic partitioning between proper folding and aggregation; thus, they thought to affect the yield (of the correctly folded protein) rather than the folding mechanism. Secondly, and most importantly, it became recognized that in vivo, protein folding begins co-translationally as nascent peptide chains emerge from the ribosome [17–25]. In the early 1960s and 1970s, the first observations were made [26–32] suggesting that in vivo protein folding starts while the growing peptide chain is still bound to the ribosome and that it is a vectorial process; i.e. the polypeptide chain is synthesized and is being folded predominantly from the N-terminal to the C-terminal end. Co-translational folding of a nascent polypeptide was thus suggested to result in a sequential structuring of distinct regions of the polypeptide emerging from the ribosome at different points in time and hierarchical condensation has therefore been considered to be the most likely mechanism that governs assembly of the nascent polypeptide into the native protein during its synthesis in vivo. Earlier studies on the co-translational protein folding were followed by many modern experiments and are continuing to date, revealing many interesting features of the process ([17–25] and ref. therein).
It became also clear, that co-translational folding starts almost immediately after the first amino acid residues begin to polymerize at the ribosomal peptidyl transferase center ([17–25] and ref. therein), with alpha-helices forming inside the ribosome tunnel and some elements of the supersecondary and tertiary structure possibly forming already in the so-called vestibule (lower/wider) region of the exit tunnel [33–36]. Once the polypeptide chain emerges from the exit tunnel into the cytosol, the folding continues governed by the thermodynamics and kinetics of polypeptide chain, leading to the formation of subsequent co-translational folding intermediates and, finally, the native structure, which is usually being completely formed after the chain’s release [17–25].
It should be noted that the ribosome reads the mRNA codons one-by-one and translates them into the sequence of amino acids of the protein. However, it became clear that not all codons are read with the same speed: periods of rapid translation are separated by translation pauses [17, 37, 38]. Therefore, it can’t be excluded that variations in local translation rates may affect/facilitate protein folding by allowing ordered, sequential structuring of the discrete nascent polypeptide chain portions synthesized by the ribosome and that kinetics of protein synthesis may thus influence/ fine-tune the co-translational protein folding.
In the late 1980’s Alistair Brown’s group in the Institute of Genetic at Glasgow University, UK [39] and our group in the Department of Molecular Biology at Moscow State University, Russia [40–42] suggested that sequential folding events, which can take place during co-translational folding of proteins, might be separated by translational pauses and that such regions of slowed translation might serve as interpunctuations during co-translational protein folding. This hypothesis was put forward based on observations that revealed a certain correlation between the locations of rare (slowly translated) codons in mRNA with either the domain boundaries in the encoded proteins [39, 41], or with the boundaries of the smaller structural units such as secondary or supersecondary structure elements [40, 42].
This hypothesis relied on several assumptions. First, it was based on the presumed assumption that non-uniformity in synonymous codon usage along mRNA would lead to a particular translation kinetics, resulting in ribosome pausing (at rare codon clusters) or ribosome acceleration (at frequent codon clusters), respectively ([17] for a review, [39–42]). Second, it assumed that synonymous codons are placed in mRNA non-randomly and strategically (thus facilitating ordered co-translational protein folding) and that changes in synonymous codon usage would lead to a different translation kinetics that in turn may alter protein folding ([17, 39–42]). Third, it also assumed that altered kinetics of translation will affect the conformation of the ribosome-bound nascent chains on the first place, subsequently potentially also changing the final conformation of the released protein and/or altering the equilibrium between different protein conformers (native and near-native and/or non-native), which in turn could lead to e.g. enhanced protein aggregation and/or degradation (co- or post-translational), or a change of the protein’s specific activity [17].
It should be noted that while overall hypothesis (broadly stating that synonymous codon usage along mRNA may serve as a kinetic guide for co-translational protein folding in the cell) has now been generally accepted by the scientific community, many of its postulates remain subjects of intense debate. Nevertheless, advances in modern techniques such as Next Generation Sequencing (NGS), single-molecule and time-resolved fluorescent approaches are transforming our ability to study co-translational protein folding and allowing to obtain key evidence in support of this hypothesis.
Below, I’ll review some of this key evidence in support of each subsequent postulate of the hypothesis.
TRANSLATION IS A NON-UNIFORM PROCESS GOVERNED BY SYNONYMOUS CODON USAGE
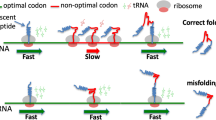
The genetic code is degenerate [43]. With the exception of two amino acids (Met and Trp), all other amino acid residues are each encoded by multiple, so-called synonymous codons [37]. Synonymous codons are however not present at equal frequencies in individual mRNAs as well as entire genomes [37, 44–46]. This pattern of non-uniform codon use is known as codon usage bias [37, 44–46] (Fig. 1a). Codon usage bias varies between organisms and represents a unique feature of an organism [47–50] (Fig. 1a). This suggests that codon choice might have functional implications beyond amino acid coding [37, 44–46]. Organism-specific codon choice is related to organism-specific differences in populations of cognate tRNAs [44]. It was generally found that in both unicellular and multicellular organisms there exists a strong positive correlation between codon usage and cellular tRNA content [47, 51, 52], meaning that codon bias would likely have a direct impact on translation elongation rates. Indeed, frequently used codons were, as a rule, found to be translated more rapidly than infrequently used ones due to the more ready availability (during translation) of corresponding frequent cognate tRNAs and vise versa (see [22, 37, 53–55] for reviews) (Fig. 1b). Several additional lines of evidence supported this notion (i) highly expressed genes were found to harbor more preferred/frequent codons compared to lowly expressed genes, which were found to be enriched in synonymous un-preferred codons [56–59]; (ii) substitution of synonymous frequently used codons by infrequently used codons (or vice versa) affected protein expression levels (see [22, 37, 53–55] for reviews) (iii) alterations in the level of expression/abundance of particular tRNAs also altered protein expression levels [60–64].

Genetic code redundancy and non-uniform/non-random codon utilization shape codon usage bias and govern non-uniform translation. (a) Codon usage bias in Escherichia coli (EC), Saccharomyces cerevisiae (SC) and Homo sapiens (HS) (https://www.kazusa.or.jp/codon/). These examples reveal substantial differences in usage of codons between the three species. Frequency per thousand codons is shown. (b) Preferentially used (frequent) codons are translated faster than infrequently used (rare) codons due to the more ready availability (during translation) of the corresponding frequent cognate tRNAs. Rare codons, as a rule, lead to a substantial increase in ribosome residence time due to an increased waiting period of the ribosome for a cognate tRNA. A simplified scheme is shown (omitting the elongation factor 1A, which promotes the GTP-dependent binding of aminoacyl-tRNA to the A-site of ribosomes during protein biosynthesis). Colors of tRNAs/codons correspond to the differential frequency of their usage.
While it was generally accepted that frequent codons would accelerate translation and rare codons would cause a translation pause, what appeared to be extremely challenging is to prove that a particular rare codon (or a cluster of rare codons) would determine the appearance of the corresponding ribosome-mediated translational pause at a particular place in mRNA.
Earlier experiments attempted to verify this notion by analyzing the sizes of nascent chains attached to the ribosome [65–68]. As discontinuous elongation rates were presumed to increase the residence time of a ribosome at particular positions along mRNA (enriched in rare codons), it was expected that this would lead to an increase in the amount of nascent peptides of the corresponding sizes present on polyribosomes. At first, the accumulation of nascent peptides of discrete sizes has been monitored by the use of gel-filtration [65, 66] and/or gel electrophoresis [67, 68]. Although, it was generally observed that enrichment in rare codons leads to enhanced pausing [65–68], precise determination and localization of translation pause sites appeared to be quite challenging due to low resolution of these methods. The development of a micrococcal nuclease protection assay [69, 70] and, subsequently, the ribosome profiling (which combined ribosome footprinting with deep sequencing of mRNA fragments protected by ribosomes during translation) have allowed the direct analysis of ribosome distribution along mRNA at codon resolution [71–73]. However, ribosome profiling experiments, at first, yielded puzzling results. While local variations in translation rates have been clearly detected, no reliable correlation between the position of ribosomes and rare codons has been originally observed [71]. It appeared however that detecting pause sites and their relation to rare codons in the original ribosome profiling data have been challenging, because the methods used to arrest translation, involved antibiotics (like cycloheximide), which skewed the position of ribosomes on messages and obscured the enrichment of ribosome density at non-optimal codons [71, 74]. A systematically-revised ribosome profiling method revealed pause sites at rare codons at single-codon resolution [75]. The new data also revealed a clear negative correlation between ribosome density and codon adaptation index, consistent with the expectation that rare codons will be decoded by lower-abundance tRNAs more slowly than more abundant codons [75].
SYNONYMOUS CODONS ARE PLACED IN mRNA NON-RANDOMLY AND STRATEGICALLY
The neutral theory of molecular evolution suggests that synonymous codons (encoding the same amino acid) will be largely unaffected by the selective pressure and thus they should be distributed in mRNA and substituted during evolution randomly [76]. It appeared however that less then half of all synonymous substitutions are under neutral expectation and that synonymous mutations are subjects to constraints [77, 78]. Close examination of mRNA sequences revealed biases in the distribution of codons within mRNA open reading frames (ORFs), a phenomenon originally recognized as codon context [79–81]. In addition to biases in synonymous codon usage relative to neighboring codons in an mRNA (codon context [79–81] and the so-called codon pair bias [82–86]), it has also been established that codon choice (particularly for rare codons) is biased according to a codon’s specific conserved location in an mRNA [87–94]. Rare/infrequent codons, specifically, have been shown to occur in clusters, enriched at a number of specific locations in mRNAs. These include clusters of rare codons located at 5' and 3' ORF termini [88–92], the so-called “+70” rare codon cluster (located ~35–40 codons downstream of the signal sequences (or transmembrane segments) in secreted proteins [87, 93], and many other internal clusters located at specific positions (88). Although earlier studies (focused on investigation of codon choice relative to codon placement at specific positions in mRNA [39–42, 94–97]) have not been comprehensive enough (partially owing to a lack of sufficient structure and sequence information), it was nevertheless concluded that the locations of rare codon clusters along mRNAs are highly conserved throughout evolution, as for example, evidenced by their similarity across homologous protein families from different organisms [17, 41, 42, 88, 94, 98–100]. This observation supported the assumption that such placement of these rare codon clusters may be linked to protein structure [17, 88]. Strategically placed rare codon clusters were specifically observed to occur, for example, at regions encoding (or close to) domain/subdomain linkers (Fig. 2) and such location of these clusters were suggested to allow temporal separation of domain and/or sub-domain folding on the ribosome [17, 88, 94].

Rare codon clusters are often non-randomly and strategically placed within mRNAs. Top—codon usage profile for Bovine (Bos taurus) βB2 crystallin revealing an extended cluster of rare codons partially encoding the domain linker and the adjacent downstream region of the protein structure. Bottom—backbone/cartoon structure of the βB2 crystallin (PDB 2BB2). The N‑terminal domain is in blue, the C-terminal domain is in yellow and a portion of the linker connecting the two domains is shown in gray. Positions of Pro80 and Lys89 at the beginning and the end of the linker peptide connecting the domains are indicated; Asn95 marks the end of the first β-structure in the βB2 C-terminal domain.
In general, genome-wide analyses of ORFeomes from prokaryotic and eukaryotic organisms revealed that rare codon clustering (i) is not limited to a particular set of genes or genotype, (ii) does not depend on and is not related to the overall GC content of the organism’s genome, and (iii) is significantly more abundant than would be expected based on random selection [88, 98]. While the precise functional roles of many conserved codon clusters has yet to be determined, it is becoming evident that codon choice has functional implications beyond amino acid coding and support of the predefined translation levels of a protein and that mRNA (and thus a genetic code) might indeed contain a secondary information linked to protein structure/folding.
CHANGES IN SYNONYMOUS CODON USAGE AFFECT LOCAL TRANSLATION ELONGATION RATES
It is widely believed that the major influence of codon usage is on global translation rate. Approaches involving substitution of the majority (or a subset) of infrequently used codons with synonymous frequently used ones, have been widely used for optimization of protein expression (54, 101–103) and ref. therein). Use of gene sequences optimized through this strategy often yielded large amounts of recombinant proteins, indicating that protein synthesis/translation elongation rates of the engineered proteins have been substantially accelerated [54, 101–103].
It must be noted however that in addition to the effects of codon usage on translation, synonymous codon choice can also impact the turnover/stability of mRNA template itself [55, 104–106]. mRNA turnover is a critical determinant of gene expression, and mRNAs with longer half-lives would typically produce more protein. It was recently found that stable/long-lived mRNAs harbor mainly preferred/optimal codons while many unstable/short-lived mRNAs have a higher frequency of un-preferred/non-optimal/rare codons within their ORFs [104–106]. Substitution of preferred codons with synonymous, un-preferred codons resulted in dramatic destabilization of the mRNA and vice versa [104–106].
These and other experiments brought about awareness of the scientific community to the impact of synonymous codon usage and codon adaptation index (CAI) (as a measure of synonymous codon usage bias [107]) on the efficiency of translation and protein expression ([101–107] and ref. therein). However, despite this general acceptance of the idea that substitution of synonymous codons in a gene can dramatically affect the rate/efficiency of synthesis of the encoded protein and increase or decrease its yield, only limited studies have attempted to investigate, how local substitutions of synonymous codons would affect the elongation rate(s) at the particular region(s) of mRNA, where these changes were introduced. Nonetheless, such evidence has been obtained.
Earlier studies attempted to monitor increase and/or decrease in the residence time of a ribosome at particular positions along mRNA after changing synonymous codons via monitoring the increase/decrease of the amount of nascent peptides of the corresponding sizes [68, 108]. Renewed attempts have been made recently to estimate the ribosome residence time at different synonymous codons using ribosome profiling and time-resolved single-molecule fluorescence methods ([38, 75, 109, 110] and ref. therein). Ensemble real-time fluorescence approaches have been also used to determine how bulk substitutions of synonymous codons would affect the speed of ribosome movement [36, 111]. These experiments combined with the experiments that measured the influence of tRNA availability, clearly indicated that there is a direct link between synonymous codons usage and the local translation elongation rates and that changes in synonymous codon usage do affect translational kinetics.
FOLDING in vivo IS A CO-TRANSLATIONAL PROCESS
A direct demonstration of the influence of synonymous codon usage on co-translational protein folding required two pieces of evidence to be obtained: (i) indicating that codon usage may affect the final conformation of a protein and (ii) indicating that these conformational changes may originate within the nascent chains bound to the ribosome i.e. co-translationally. As has been mentioned above, the original hypothesis was put forward in the late 1980s [39–42]. However, at that time, even the basic idea that folding in vivo is a co-translational process was not widely accepted yet.
So, at first, it was necessary to obtain solid evidence in support of co-translational protein folding. The majority of the earlier experiments in support of co-translational folding involved isolation/fractionation of ribosome-bound nascent chain complexes through a sucrose density gradient, followed by assessment of the structural properties of the nascent chains through measurement of (i) their specific enzymatic activities, (ii) their recognition by specific/conformational antibodies, or (iii) formation of correct disulfide cross-bridges within and/or between nascent chains ([23, 24] and ref. therein). Subsequently, other methods have been introduced ([23, 24] and ref. therein), such as those involving e.g., measurement of the resistance of ribosome attached nascent chains to proteolytic digestion and/or the ability of co-factors and ligands (such as heme) to bind the growing polypeptide chain (as an indication that a binding-competent conformation has been achieved).
Our group together with Alexander S. Spirin’s laboratory at the institute of Protein Research in Pushchino used heme binding to probe co-translational folding of the α-globin chains [112, 113]. Using in vitro translation reactions performed in the presence of [3H]hemin and [35S]methionine together with sucrose gradient centrifugation and puromycin treatment, we showed that ribosome-bound α-globin chains are capable of efficient heme binding [113]. In addition, we found that incomplete α-globin nascent chains attached to the ribosome are capable of co-translational heme binding, indicating that a structure that allows for heme binding in the nascent chain is achieved prior to the completion of α-globin synthesis. These results provided strong support for co-translational folding of the α-globin molecule [113].
However, back at a time, one of the most solid supports of co-translational protein folding came from the work done by Kolb, Makeyev and Spirin, who pioneered the study of co-translational protein folding using real-time measurements [114, 115]. These authors developed a technique allowing to continuously monitor enzymatic activity of newly synthesized firefly luciferase in a cell-free system in a luminometer cuvette and showed that luciferase activity (indicative of folding of the protein) can be detected as soon as the full-length molecule was formed in the translation reaction [114]. Importantly, such rapid acquisition of the enzyme’s activity was incompatible with a post-translational folding scenario [114]. Furthermore, subsequently, the same authors demonstrated that ribosome-bound luciferase can be enzymatically active and concluded that folding of the firefly luciferase protein occurs during the course of translation [115].
More recently, a plethora of modern technologies, such as NMR spectroscopy, cryo-electron microscopy (cryo-EM) and fluorescent techniques (Fluorescence Resonance Energy Transfer (FRET) and fluorescence anisotropy/dynamic fluorescence depolarization) as well as some other approaches have been introduced to study co-translational folding ([19, 23, 24] and ref. therein). These and other experiments provided overwhelming evidence in support of co-translational protein folding and the idea of co-translational protein folding has now become widely accepted [17–25]. What remained unclear however, is to what extent the co-translational folding pathway is unique and whether it can be indeed influenced by the kinetics of translation?
Therefore, many researchers in the field turned their attention to the analysis of the influence of kinetics of translation on co-translational protein folding. These experiments appeared to be extremely challenging, because of the numerous quality control mechanisms existing in a cell, by which a cell monitors proteins to ensure that they are appropriately folded and if not—guides them to degradation [12–16]. It was thus possible that changes in co-translational folding caused by synonymous codon-driven alterations in elongation kinetics could not be substantial enough to overcome the effects of cellular quality control and chaperone network machineries and become detectable/visible.
SYNONYMOUS CODON USAGE INFLUENCES CO-TRANSLATIONAL PROTEIN FOLDING
The choice of cell-free translation system(s), where the effects of the cellar quality control mechanisms could be less pronounced, or could be deliberately controlled by omitting members of the quality control machineries seemed to be therefore logical at first and is one of the preferred approaches for the analysis of the influence of synonymous codon usage on co-translational protein folding at present time. The in vitro translational systems and, especially, the fully reconstituted in vitro systems also allow easy control of many other factors/components necessary for protein synthesis and folding, therefore allowing answering many key questions in the field.
In 1999, we have provided one the first observations showing that synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation of a model protein, chloramphenicol acetyltransferase (CAT) [108]. In this study, sixteen consecutive rare codons in the CAT gene have been replaced by frequent ones and this led to the acceleration of the ribosome traffic through the mutated region and at the same time affected the specific activity of the enzyme (in comparison with the wild-type protein) [108]. Since specific activity of a given protein could be considered as a measure of its proper folding, we have concluded that CAT folding was affected. We have further suggested that accelerated rates of translation of a selected CAT region potentially allowed the particular part of the polypeptide chain to appear earlier in time during translation and this might have led to an affected interaction of the extruded polypeptide region with the preceding one, which (we speculated) was not yet properly folded due to a lack of time [108]. This was one of the first experimental confirmations of the hypothesis stating that kinetics of protein translation can influence the in vivo protein folding pathway. However, the idea was yet not well accepted then.
The next breakthrough came 8 years later. In 2007, Chava Kimchi-Sarfaty, Michael Gottesman and their colleagues showed that substrate specificity of P-glycoprotein, the product of the multidrug resistance 1 (MDR1) gene, is altered by synonymous single nucleotide polymorphisms (SNPs) presumed to be silent [116]. The authors carefully investigated ex vivo (in transfected cells) the effect of naturally occurring polymorphic MDR1 variants on P-gp mRNA translation and protein expression, activity of the protein and its substrate specificity. They have concluded that a single synonymous mutation (C3435T; Ile-ATC>ATT, changing a relatively frequent ATC codon into a more rare ATT and, therefore, presumably affecting translation elongation rates in this region, although this was not demonstrated directly) can alter P-gp conformation and protein activity/substrate specificity [116]. This study was of immense importance as it for the first time demonstrated that naturally occurring synonymous/silent SNPs can lead to the synthesis of the protein product with the same amino acid sequence but different structural and functional properties. It also explained differences (observed previously in several clinical studies) in P-gp pharmacokinetics in individuals carrying this silent SNP [116].
The significance of synonymous codon usage for protein folding were highlighted by a number of subsequent studies showing that synonymous codon substitutions can affect proteins’ sensitivity to limited proteolysis [117, 118], phosphorylation profiles [118], spectroscopic properties [119], aggregation propensity [119–121] and specific activity [122], which ultimately can cause diseases [123–128]. Synonymous codon choice has been also suggested to affect efficient interaction of nascent polypeptides with the signal recognition particle [93], thus affecting protein secretion.
Of special interest is the study performed by Patricia Clark and colleagues [119], who took advantage of the so-called bimolecular fluorescence complementation (BiFC) assay [129, 130] to design a fluorescent protein consisting of three half-domains, where the N-(yellow) and C-terminal (cyan) half-domains compete each other to interact with the central half-domain [119]. The outcome of this competition determined the fluorescence properties of the resulting folded structure. Using a cellular expression system and monitoring FRET between the YFP/CFP labels, they demonstrated that the rate at which a nascent protein emerges from the ribosome (which was affected by synonymous codon substitutions at the inter-half-domain linker) can specify the final folded conformation of a protein [119].
The studies above, had however a number of drawbacks. As a rule, they investigated just only one aspect of the hypothesis and predominantly looked at the properties of the polypeptide chains released from the ribosome. Analyses of the released polypeptide chains in all the above experiments were done using indirect (e.g. specific activity, limited proteolysis), rather than direct (NMR, X-ray) structure probing approaches. Also, few studies attempted to investigate the structure of ribosome bound nascent chains produced from the silently mutated mRNA in comparison with the wild-type. Changes in the kinetics of protein synthesis as a result of synonymous mutations were not usually simultaneously monitored in these studies and potential effects of miscoding (that could potentially arise from synonymous codon changes) were frequently neglected. Finally, none of the studies employed real-time measurements to demonstrate that synonymous mutations may not only affect kinetics of protein synthesis, but they, at the same time, may also affect the real-time kinetics of co-translational protein folding.
Recently, we have filled in the gaps above and in collaboration with Harald Schwalbe from the Johann Wolfgang Goethe-Universität in Frankfurt, Germany and Marina V. Rodnina from the Max Planck Institute for Biophysical Chemistry in Goettingen, Germany executed a study, which addressed all these outstanding questions [111].
To investigate how differential usage of synonymous codons affects translation kinetics, co- and post-translational folding, and protein conformation and stability, we analyzed in vivo expression of the recombinant bovine eye lens protein gamma-B crystallin in Escherichia coli cells and in vitro in a completely reconstituted high-performance translation system from E. coli. We have chosen gamma-B-crystallin, because previously we have shown that translation of this two-domain protein is a non-uniform process [68]. We also suggested that the codon usage and translation rates in gamma-B-crystallin are optimized to tune the synthesis and folding of this protein in the cell [68]; however the direct experimental evidence in support of this suggestion was lacking. We therefore designed two variants of the mRNA coding for gamma-B crystallin, one with the codon usage that would be optimal for protein translation in E. coli (with an mRNA codon distribution (codon usage profile) similar to that found in B. taurus, which was expected to result in more natural translation kinetics) and the other with unaltered codon composition un-optimal for translation in E. coli [111].
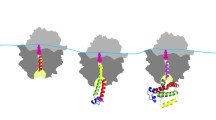
Our analysis of the effects of synonymous codon choice on the translation of gamma-B crystallin mRNAs showed that codon choice alters local and global translation rates and results in the formation of alternative conformations of the protein [111]. We showed using real time measurements, which employed fluorescence and FRET, that kinetics of synthesis and co-translational folding of gamma-B crystallin is indeed altered by synonymous codon substitutions. Moreover, for the first time, we detected considerable structural heterogeneity of the purified, mature synonymous gamma-B crystallin protein variants by using direct structure elucidation approach (2D NMR). We concluded that the synonymous polymorphisms altered the distribution of populations within the landscape of accessible protein conformations both on the ribosome and after chain’s release [111]. Importantly, we have carefully verified by using Mass Spectrometry and Microsequencing that synonymous gamma-B crystallin variants led to the synthesis of the polypeptide chains with identical amino acid sequence. Together our results provided a strong support to the hypothesis and showed that synonymous codons may indeed serve as a secondary code for protein folding in the cell [111]. We thus concluded that codon usage specifies a unique translation kinetics that affects the partitioning of the folding intermediates both on the ribosome and after chain’s release and that non-natural codon usage and translation kinetics can result in a kinetically trapped folding intermediates. These intermediates can be converted, with (or without) the help of molecular chaperones (co- or post-translationally), to the native protein state through reshuffling reactions. However, such kinetically trapped intermediates could also remain stable and drive the overall folding into a non-native and/or aggregation-prone state. Non-productive, trapped species could be also degraded (co- or post-translationally) (Fig. 3).

Synonymous codon usage alters kinetics of protein translation and can direct co-translational folding towards different protein conformation(s). A model illustrating the influence of codon usage/translation kinetics on the final conformation of the synthesized protein. (a) Natural (native) kinetics of translation leads to the efficient formation of the native structure through the number of productive co-translational intermediates. (b) Altered codon usage/translation kinetics might create kinetically trapped intermediates. These intermediates might then be converted, with (or without) the help of molecular chaperones (co- or post-translationally), to the native protein through reshuffling reactions. However, such kinetically trapped intermediates could also remain stable and drive the overall folding into a non-native and/or aggregation-prone state. Nonproductive, trapped species could be also degraded (co- or post-translationally). Various end-points of non-productive folding arising due to altered synonymous codon usage can lead to a disease.
Importantly, non-productive folding arising due to altered synonymous codon usage can also lead to a disease [123–127]. In collaboration with Chava Kimchi-Sarfaty, we have recently demonstrated that a synonymous mutation, c.459G>A (GTG>GTA, p.Val153Val or Val107Val (Val107 is the amino acid number after the prepro-petide cleavage on secretion)) that has been previously identified in F9 gene (encoding blood coagulation factor FIX) in patients with mild haemophilia B [128], alters FIX synthesis and affects its conformation resulting in decreased extracellular protein level [127]. As such, we were able to determine the pathogenic basis for a single synonymous mutation in the F9 gene associated with haemophilia B [127]. This case remained a mystery for about 10 years, since its discovery in 2008 [128], as back at a time it couldn’t be explained by altered mRNA properties (mRNA levels, splicing/exon skipping or retention of introns, or stability), a common cause of many diseases associated with synonymous mutations [125, 126].
CONCLUSIONS AND FUTURE PERSPECTIVES
The work on the effects of synonymous codon usage undoubtedly deepens our understanding of protein folding in the cell. Importantly, it, indeed, helps to explain cases of genetic diseases linked to synonymous mutations; cases which couldn’t be explained by previously known effects of synonymous mutations on mRNA splicing and/or mRNA stability [125, 126]. These studies further brought about increased awareness of the scientific community to the impact of the synonymous mutations on gene function and phenotype linked to protein folding and stability [123–127]. Finally, this work gave a novel tool to upscale the production of functionally active recombinant proteins and provided explanation for a long-standing paradigm, revealing that approaches involving substitution of the majority of infrequently used codons with synonymous frequently used ones (targeted to inflate CAI), which have been widely used for optimization of heterologous and homologous protein production, frequently yielded biologically inactive insoluble protein aggregates [54]. Our work made it clear that maximizing the speed and output of translation may put conflicting demands on the protein synthesis machinery, resulting in improper protein folding.
However, much work has yet to be done. There is yet a limited understanding, of how exactly the structure of co-translational folding intermediates is affected by the synonymous mutations. Novel methods, such as single molecule FRET and/or time-resolved cryo-EM [131] may potentially allow improved analysis of the effects of synonymous mutations on the appearance and conformation of co-translational folding intermediates. However, application of time-resolved cryo-EM for analysis of nascent chain dynamics remains challenging at the present time and FRET per se will not allow direct visualization of nascent chain structures.
Nevertheless, our improved understanding of the impact of synonymous codon usage on protein folding strongly supports the view that synonymous codon usage serves as a guide for co-translational protein folding in the cell (Fig. 3).
REFERENCES
Hartl F.U. 2017. Protein misfolding diseases. Annu. Rev. Biochem.86, 21‒26.
Chiti F., Dobson C.M. 2017. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem.86, 27‒68.
Soto C., Pritzkow S. 2018. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci.21, 1332‒1340.
Fersht A.R. 2008. From the first protein structures to our current knowledge of protein folding: Delights and skepticisms. Nat. Rev. Mol. Cell Biol.9, 650‒654.
Ferina J., Daggett V. 2019. Visualizing protein folding and unfolding. J. Mol. Biol.431, 1540–1564.
Anfinsen C.B. 1973. Principles that govern the folding of protein chains. Science.181, 223‒230.
Finkelstein A.V. 2018. 50+ years of protein folding. Biochemistry (Moscow). 83 (Suppl. 1), S3‒S18.
Bartlett A.I., Radford S.E. 2009. An expanding arsenal of experimental methods yields an explosion of insights into protein folding mechanisms. Nat. Struct. Mol. Biol.16, 582‒588.
Abaskharon R.M., Gai F. 2016. Meandering down the energy landscape of protein folding: Are we there yet? Biophys. J.110, 1924‒1932.
Jaenicke R. 1991. Protein folding: local structures, domains, subunits, and assemblies. Biochemistry.30, 3147‒3161.
Hartl F.U., Hayer-Hartl M. 2009. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol.16, 574‒581.
Hingorani K.S. Gierasch L.M. 2014. Comparing protein folding in vitro and in vivo: Foldability meets the fitness challenge. Curr. Opin. Struct. Biol.24, 81‒90.
Balchin D., Hayer-Hartl M., Hartl F.U. 2016. In vivo aspects of protein folding and quality control. Science. 353, aac4354.
Gruebele M., Dave K., Sukenik S. 2016. Globular protein folding in vitro and in vivo. Annu. Rev. Biophys.45, 233‒251.
Dahiya V., Buchner J. 2019. Functional principles and regulation of molecular chaperones. Adv. Protein Chem. Struct. Biol.114, 1‒60.
Jayaraj G.G., Hipp M.S., Hartl F.U. 2019. Functional modules of the proteostasis network. Cold Spring Harb. Perspect. Biol. Mar. 4, pii: a033951. https://doi.org/10.1101/cshperspect.a033951
Komar A.A. 2009. A pause for thought along the co-translational folding pathway. Trends Biochem. Sci.34, 16‒24.
Kramer G., Boehringer D., Ban N., Bukau B. 2009. The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat. Struct. Mol. Biol.16, 589‒597.
Cabrita L.D., Dobson C.M., Christodoulou J. 2010. Protein folding on the ribosome. Curr. Opin. Struct. Biol.20, 33‒45.
Pechmann S., Willmund F., Frydman J. 2013. The ribosome as a hub for protein quality control. Mol. Cell.49, 411‒421.
Gloge F. Becker A.H. Kramer G., Bukau B. 2014. Co-translational mechanisms of protein maturation. Curr. Opin. Struct. Biol.24, 24‒33.
Chaney J.L., Clark P.L. 2015. Roles for synonymous codon usage in protein biogenesis. Annu. Rev. Biophys.44, 143‒166.
Thommen M., Holtkamp W., Rodnina M.V. 2017. Co-translational protein folding: Progress and methods. Curr. Opin. Struct. Biol.42, 83‒89.
Komar A.A. 2018. Unraveling co-translational protein folding: Concepts and methods. Methods.137, 71‒81.
Williams N.K, Dichtl B. 2018. Co-translational control of protein complex formation: A fundamental pathway of cellular organization? Biochem. Soc. Trans.46, 197‒206.
Cowie D.B., Spiegelman S., Roberts R.B., Duerksen J.D. 1961. Ribosome-bound beta-galactosidase. Proc. Natl. Acad. Sci. U. S. A.47, 114‒122.
Zipser D., Perrin D. 1963. Complementation on ribosomes. Cold Spring Harbor. Symp. Quant. Biol.28, 533‒537.
Kiho Y., Rich A. 1964. Induced enzyme formed on bacterial polyribosomes. Proc. Natl. Acad. Sci. U. S. A.51, 111‒118.
Hamlin J., Zabin I. 1972. β-Galactosidase: Immunological activity of ribosome-bound, growing polypeptide chains. Proc. Natl. Acad. Sci. U. S. A.69, 412‒416.
Bergman L.W., Kuehl W.M. 1979. Formation of intermolecular disulfide bonds on nascent immunoglobulin polypeptides. J. Biol. Chem.254, 5690‒5694.
Bergman L.W., Kuehl W.M. 1979. Formation of an intrachain disulfide bond on nascent immunoglobulin light chains. J. Biol. Chem.254, 8869‒8876.
Bergman L.W., Kuehl W.M. 1979. Co-translational modification of nascent immunoglobulin heavy and light chains. J. Supramol. Struct.11, 9‒24.
Gilbert R.J., Fucini P., Connell S., Fuller S.D., Nierhaus K.H., Robinson C.V., Dobson C.M., Stuart D.I. 2004. Three-dimensional structures of translating ribosomes by Cryo-EM. Mol. Cell.14, 57‒66.
Kosolapov A., Deutsch C. 2009. Tertiary interactions within the ribosomal exit tunnel. Nat. Struct. Mol. Biol.16, 405‒411.
Tu L., Khanna P., Deutsch C. 2014. Transmembrane segments form tertiary hairpins in the folding vestibule of the ribosome. J. Mol. Biol.426, 185‒198.
Holtkamp W., Kokic G., Jäger M., Mittelstaet J., Komar A.A., Rodnina M.V. 2015. Cotranslational protein folding on the ribosome monitored in real time. Science. 350, 1104‒1107.
Komar A.A. 2018. The Yin and Yang of codon usage. Hum. Mol. Genet.25 (R2), R77–R85.
Schuller A.P., Green R. 2018. Roadblocks and resolutions in eukaryotic translation. Nat. Rev. Mol. Cell Biol.19, 526‒541.
Purvis I.J., Bettany A.J., Santiago T.C., Coggins J.R., Duncan K., Eason R., Brown A.J. 1987. The efficiency of folding of some proteins is increased by controlled rates of translation in vivo. A hypothesis. J. Mol. Biol.193, 413‒417.
Krasheninnikov I.A., Komar A.A., Adzhubeĭ I.A. 1988. Role of the rare codon clusters in defining the boundaries of polypeptide chain regions with identical secondary structures in the process of co-translational folding of proteins. Dokl. Akad. Nauk SSSR.303, 995‒999.
Krasheninnikov I.A., Komar A.A., Adzhubeĭ I.A. 1989. Frequency of using codons in mRNA and coding of the domain structure of proteins. Dokl. Akad. Nauk SSSR.305, 1006‒1012.
Krasheninnikov I.A., Komar A.A., Adzhubeĭ I.A. 1989. Role of the code redundancy determining cotranslational protein folding. Biokhimiya.5, 187‒200.
Crick F.H., Barnett L., Brenner S., Watts-Tobin R.J. 1961. General nature of the genetic code for proteins. Nature. 192, 1227‒1232.
Hershberg R., Petrov D.A. 2008. Selection on codon bias. Annu. Rev. Genet.42, 287‒299.
Sharp P.M., Emery L.R. Zeng K. 2010. Forces that influence the evolution of codon bias. Philos. Trans. R. Soc. Lond. B.365, 1203‒1212.
Behura S.K.m Severson D.W. 2013. Codon usage bias: Causative factors, quantification methods and genome-wide patterns, with emphasis on insect genomes. Biol. Rev. Camb. Philos. Soc.88, 49‒61.
Ikemura T. 1985. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol.2, 13‒34.
Sharp P.M., Cowe E., Higgins D.G., Shields D.C., Wolfe K.H., Wright F. 1988. Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens: A review of the considerable within-species diversity. Nucleic Acids Res.16, 8207‒8211.
Andersson S.G., Kurland C.G. 1990. Codon preferences in free-living microorganisms. Microbiol. Rev.,54, 198‒210.
Nakamura Y., Gojobori T., Ikemura T. 2000. Codon usage tabulated from the international DNA sequence databases: Status for the year 2000. Nucleic Acids Res.28, 292.
Ikemura T. 1981. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J. Mol. Biol.146, 1‒21.
Ikemura T. 1982. Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. Differences in synonymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer RNAs. J. Mol. Biol.158, 573‒597.
Quax T.E., Claassens N.J., Söll D., van der Oost J. 2015. Codon bias as a means to fine-tune gene expression. Mol. Cell.59, 149‒161.
Komar A.A. 2016. The art of gene redesign and recombinant protein production: approaches and perspectives. In: Protein Therapeutics. Eds Zuben E. Sauna, Chava Kimchi-Sarfaty. Springer, pp 161‒177. doi.org/https://doi.org/10.1007/978-3-319-41818-6
Hanson G., Coller J. 2018. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol.19, 20‒30.
Sharp P.M., Tuohy T.M., Mosurski K.R. 1986. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res.14, 5125‒5143.
Shields D.C., Sharp P.M. 1987. Synonymous codon usage in Bacillus subtilis reflects both translational selection and mutational biases. Nucleic Acids Res.15, 8023‒8040.
Sharp P.M., Devine K.M. 1989. Codon usage and gene expression level in Dictyostelium discoideum: Highly expressed genes do “prefer” optimal codons. Nucleic Acids Res.17, 5029‒5039.
Karlin S., Mrázek J., Campbell A.M. 1998. Codon usages in different gene classes of the Escherichia coli genome. Mol. Microbiol.29, 1341‒1355.
Brinkmann U., Mattes R.E., Buckel P. 1989. High-level expression of recombinant genes in Escherichia coli is dependent on the availability of the dnaY gene product. Gene.85, 109‒114.
Chen K.S., Peters T.C., Walker J.R. 1990. A minor arginine tRNA mutant limits translation preferentially of a protein dependent on the cognate codon. J. Bacteriol.172, 2504‒2510.
Chen G.T., Inouye M. 1994. Role of the AGA/AGG codons, the rarest codons in global gene expression in Escherichia coli.Genes Dev.8, 2641‒2652.
Zahn K., Landy A. 1996. Modulation of lambda integrase synthesis by rare arginine tRNA. Mol. Microbiol.21, 69‒76.
Del Tito B.J., Jr., Ward J.M., Hodgson J., Gershater C.J., Edwards H., Wysocki L.A., Watson F.A., Sathe G., Kane J.F. 1995. Effects of a minor isoleucyl tRNA on heterologous protein translation in Escherichia coli.J. Bacteriol.177, 7086‒7091.
Protzel A., Morris A.J. 1974. Gel chromatographic analysis of nascent globin chains. Evidence of nonuniform size distribution. J. Biol. Chem.249, 4594‒4600.
Chaney W.G., Morris A.J. 1978. Nonuniform size distribution of nascent peptides: The role of messenger RNA. Arch. Biochem. Biophys.191, 734‒741.
Krasheninnikov I.A. Komar A.A., Adzhubeĭ I.A. 1991. Nonuniform size distribution of nascent globin peptides, evidence for pause localization sites, and a cotranslational protein-folding model. J. Protein Chem.10, 445–454.
Komar A.A., Jaenicke R. 1995. Kinetics of translation of gamma B crystallin and its circularly permutated variant in an in vitro cell-free system: Possible relations to codon distribution and protein folding. FEBS Lett.376, 195‒198.
Wolin S.L., Walter P. 1988. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J.7, 3559‒3569.
Hollingsworth M.J., Kim J.K., Stollar N.E. 1998. Heelprinting analysis of in vivo ribosome pause sites. Methods Mol. Biol.77, 153‒165.
Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science.324, 218‒223.
McGlincy N.J., Ingolia N.T. 2017. Transcriptome-wide measurement of translation by ribosome profiling. Methods.126, 112‒129.
Ingolia NT, Hussmann JA, Weissman JS. 2018. Ribosome profiling: Global views of translation. Cold Spring Harb. Perspect. Biol.11 (5), pii: a032698. https://doi.org/10.1101/cshperspect.a032698
Hussmann J.A., Patchett S., Johnson A., Sawyer S., Press W.H. 2015. Understanding biases in ribosome profiling experiments reveals signatures of translation dynamics in yeast. PLoS Genet.11, e1005732.
Mohammad F., Green R., Buskirk A.R. 2019. A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. eLife.8, e42591.
Kimura M. 1984. The Neutral Theory of Molecular Evolution Cambridge. Cambridge, UK: Cambridge Univ. Press.
Zhao Z., Fu Y.X., Hewett-Emmett D., Boerwinkle E. 2003. Investigating single nucleotide polymorphism (SNP) density in the human genome and its implications for molecular evolution. Gene.312, 207‒213.
Chamary J.V., Parmley J.L., Hurst L.D. 2006. Hearing silence: Non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet.7, 98‒108.
Yarus M., Folley L.S. 1985. Sense codons are found in specific contexts. J. Mol. Biol.182, 529‒540.
Buckingham R.H. 1990. Codon context. Experientia. 46, 1126‒1133.
Buckingham, R.H. 1994. Codon context and protein synthesis: enhancements of the genetic code. Biochimie. 76, 351‒354.
Gutman G.A., Hatfield G.W. 1989. Nonrandom utilization of codon pairs in Escherichia coli.Proc. Natl. Acad. Sci. U. S. A.86, 3699‒3703.
Tats A., Tenson T., Remm M. 2008. Preferred and avoided codon pairs in three domains of life. BMC Genomics. 9, 463.
Diambra L.A. 2017. Differential bicodon usage in lowly and highly abundant proteins. PeerJ. 5, e3081.
Brule C.E., Grayhack E.J. 2017). Synonymous codons: Choose wisely for expression. Trends Genet.33, 283‒297.
Alexaki A., Kames J.M., Holcomb D.D., Athey J., Santana-Quintero L.V., Lam P.V., Hamasaki-Katagiri N., Osipova E., Simonyan V., Bar H., Komar A.A., Kimchi-Sarfaty C. 2019. Codon and Codon-Pair Usage Tables (CoCoPUTs): Facilitating genetic variation analyses and recombinant gene design. J. Mol. Biol.431 (13), 2434‒2441. https://doi.org/10.1016/j.jmb.2019.04.021
Képès F. 1996. The “+70 pause”: Hypothesis of a translational control of membrane protein assembly. J. Mol. Biol.262, 77‒86.
Clarke T.F., 4th, Clark P.L. 2008. Rare codons cluster. PLoS One.3, e3412.
Kudla G., Murray A.W., Tollervey D., Plotkin J.B. 2009. Coding-sequence determinants of gene expression in Escherichia coli.Science.324, 255‒258.
Clarke T.F., 4th, Clark P.L. 2010. Increased incidence of rare codon clusters at 5' and 3' gene termini: Implications for function. BMC Genomics.11, 118.
Goodman D.B., Church G.M., Kosuri S. 2013. Causes and effects of N-terminal codon bias in bacterial genes. Science.342, 475‒479.
Bentele K., Saffert P., Rauscher R., Ignatova Z., Bluthgen N. 2013. Efficient translation initiation dictates codon usage at gene start. Mol. Syst. Biol.9, 675.
Pechmann S., Chartron J.W., Frydman J. 2014. Local slowdown of translation by nonoptimal codons promotes nascent-chain recognition by SRP in vivo.Nat. Struct. Mol. Biol.21, 1100‒1105.
Thanaraj T.A., Argos P. 1996. Ribosome-mediated translational pause and protein domain organization. Protein Sci.5, 1594‒1612.
Thanaraj T.A., Argos P. 1996. Protein secondary structural types are differentially coded on messenger RNA. Protein Sci.5, 1973‒1983.
Adzhubei A.A., Adzhubei I.A., Krasheninnikov I.A., Neidle S. 1996. Non-random usage of 'degenerate' codons is related to protein three-dimensional structure. FEBS Lett.399, 78‒82.
Oresic M., Shalloway D. 1998. Specific correlations between relative synonymous codon usage and protein secondary structure. J. Mol. Biol.281, 31‒48.
Chartier M., Gaudreault F., Najmanovich R. 2012. Large-scale analysis of conserved rare codon clusters suggests an involvement in co-translational molecular recognition events. Bioinformatics. 28, 1438‒1445.
Widmann M., Clairo M., Dippon J., Pleiss J. 2008. Analysis of the distribution of f unctionally relevant rare codons. BMC Genomics.9, 207.
McKownvR.L., Raab R.W., Kachelries P., Caldwell S., Laurie G.W. 2013. Conserved regional 3' grouping of rare codons in the coding sequence of ocular prosecretory mitogen lacritin. Invest. Ophthalmol. Vis. Sci.54, 1979‒1987.
Gustafsson C., Govindarajan S., Minshull J. 2004. Codon bias and heterologous protein expression. Trends Biotechnol.22, 346–353.
Wu G., Zheng Y., Qureshi I., Zin H.T., Beck T., Bulka B., Freeland S.J. 2007. SGDB: A database of synthetic genes re-designed for optimizing protein over-expression. Nucleic Acids Res.35, D76–D79.
Quax T.E., Claassens N.J., Soll D., van der Oost J. 2015. Codon bias as a means to fine-tune gene expression. Mol. Cell. 59, 149–161.
Presnyak V., Alhusaini N., Chen Y.H., Martin S., Morris N., Kline N., Olson S., Weinberg, D., Baker K.E., Graveley B.R., Coller J. 2015. Codon optimality is a major determinant of mRNA stability. Cell.160, 1111‒1124.
Boël G., Letso R., Neely H., Price W.N., Wong K.H., Su M., Luff J.D., Valecha M., Everett J.K., Acton T.B., Xiao R., Montelione G.T., Aalberts D.P., Hunt J.F. 2016. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature.529, 358‒363.
Mishima Y., Tomari Y. 2016. Codon usage and 3' UTR length determine maternal mRNA stability in zebrafish. Mol. Cell.61, 874‒885.
Sharp P.M., Li W.H. 1987. The Codon Adaptation Index: A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281‒1295.
Komar A.A., Lesnik T., Reiss C. 1999. Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett.462, 387‒391.
Uemura S., Aitken C.E., Korlach J., Flusberg B.A., Turner S.W., Puglisi J.D. 2010. Real-time tRNA transit on single translating ribosomes at codon resolution. Nature.464, 1012‒1017.
Prabhakar A., Puglisi E.V., Puglisi J.D. 2019. Single-molecule fluorescence applied to translation. Cold Spring Harb. Perspect. Biol.11, pii: a032714.
Buhr F., Jha S., Thommen M., Mittelstaet J., Kutz F., Schwalbe H., Rodnina M.V., Komar A.A. 2016. Synonymous codons direct cotranslational folding toward different protein conformations. Mol. Cell.61, 341‒351.
Komar A.A., Kommer A., Krasheninnikov I.A., Spirin A.S. 1993. Cotranslational heme binding to nascent globin chains. FEBS Lett.326, 261‒263.
Komar A.A., Kommer A., Krasheninnikov I.A., Spirin A.S. 1997. Cotranslational folding of globin. J. Biol. Chem.272, 10646‒10651.
Kolb V.A., Makeyev E.V., Spirin A.S. (1994). Folding of firefly luciferase during translation in a cell-free system. EMBO J.13, 3631‒3637.
Makeyev E.V., Kolb V.A., Spirin A.S. 1996. Enzymatic activity of the ribosome-bound nascent polypeptide. FEBS Lett.378, 166‒170.
Kimchi-Sarfaty C., Oh J.M., Kim I.W., Sauna Z.E., Calcagno A.M., Ambudkar S.V., Gottesman M.M. 2007. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science.315, 525‒528.
Zhang G., Hubalewska M., Ignatova Z. 2009. Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol.16, 274‒280.
Zhou M., Guo J., Cha J., Chae M., Chen S., Barral J.M., Sachs M.S., Liu Y. 2013. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature.495, 111‒115.
Sander I.M., Chaney J.L., Clark P.L. 2014. Expanding Anfinsen’s principle: Contributions of synonymous codon selection to rational protein design, J. Am. Chem. Soc.136, 858‒861.
Hu S., Wang M., Cai G., He M. 2013. Genetic code-guided protein synthesis and folding in Escherichia coli.J. Biol. Chem.288, 30855‒30861.
Kim S.J., Yoon J.S., Shishido H., Yang Z., Rooney L.A., Barral J.M., Skach W.R. 2015. Protein folding. Translational tuning optimizes nascent protein folding in cells. Science.348, 444‒448.
Yu C.H., Dang Y., Zhou Z., Wu C., Zhao F., Sachs M.S., Liu Y. 2015. Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol. Cell.59, 744‒754.
Komar A.A. 2007. SNPs, silent but not invisible. Science.315, 466‒467.
Komar A.A. 2007. Silent SNPs: Impact on gene function and phenotype. Pharmacogenomics.8, 1075–1080.
Sauna. Z.E., Kimchi-Sarfaty C. 2011. Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet.12, 683‒691.
Hunt R.C., Simhadri V.L., Iandoli M., Sauna Z.E., Kimchi-Sarfaty C. 2014. Exposing synonymous mutations. Trends Genet.30, 308‒321.
Simhadri V.L., Hamasaki-Katagiri N., Lin B.C., Hunt R., Jha S., Tseng S.C., Wu A., Bentley A.A., Zichel R., Lu Q., Zhu L., Freedberg D.I., Monroe D.M., Sauna Z.E., Peters R., Komar A.A., Kimchi-Sarfaty C. 2017. Single synonymous mutation in factor IX alters protein properties and underlies haemophilia B. J. Med. Genet.54, 338‒345.
Knobe K.E., Sjorin E., Ljung R.C. 2008. Why does the mutation G17736A/Val107Val (silent) in the F9 gene cause mild haemophilia B in five Swedish families? Haemophilia. 14, 723–728.
Shyu Y.J., Liu H., Deng X., Hu C.D. 2006. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques.40, 61‒66.
Shyu Y.J., Hu C.D. 2008. Fluorescence complementation: An emerging tool for biological research. Trends Biotechnol.26, 622‒630.
Chen B., Kaledhonkar S., Sun M., Shen B., Lu Z., Barnard D., Lu T.M., Gonzalez R.L., Jr., Frank J. 2015. Structural dynamics of ribosome subunit association studied by mixing-spraying time-resolved cryogenic electron microscopy. Structure. 23, 1097‒1105.
ACKNOWLEDGMENTS
This work would not be possible without the original contribution of Ivan A. Adzhubei and Igor A. Krasheninnikov, further support from Slava Kolb, Aigar Kommer, Lev P. Ovchinnikov and Alexander S. Spirin, followed by collaborations with Rainer Jaenicke, Claude Reiss and more recently with Chava Kimchi-Sarfaty, Harald Schwalbe and Marina V. Rodnina.
I am indebted to all my colleagues and collaborators for their extremely generous and inspiring discussions and invaluable contributions.
I also apologize to those whose work or original publications could not be cited in this short review article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
FUNDING
In recent years, this work in my laboratory was supported by grants from the Human Frontier Science Program Organization [HFSP grant #RGP0024/2010], the American Heart Association [AHA grant 13GRNT17070025], the National Institutes of Health [NIH grant HL121779], the Center for Gene Regulation in Health and Disease (GRHD) at CSU, and the biotechnology company, DAPCEL, Inc, that for more that 10 years has been successfully utilizing ideas developed during this study for synonymous gene optimization aimed at production (in any desired host organism) of correctly folded, soluble proteins.
COMPLIANCE WITH ETHICAL STANDARDS
The author declares that he has no conflict of interest. This article does not contain any studies involving animals or human participants performed by the author.
ADDITIONAL INFORMATION
The text was submitted by the author(s) in English.
Rights and permissions
About this article

Cite this article
Komar, A.A. Synonymous Codon Usage—a Guide for Co-Translational Protein Folding in the Cell. Mol Biol 53, 777–790 (2019). https://doi.org/10.1134/S0026893319060098
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026893319060098