
Abstract—
Polyglutamine diseases are rare, inherited neurodegenerative pathologies that arise as a result of expansion of trinucleotide CAG repeats in the coding segment of certain genes. This expansion leads to the appearance of mRNA with abnormally long repetitive CAG triplets (mCAG-RNA) and proteins with polyglutamine (PolyQ) tracts in the cells, which is why these pathologies are commonly termed polyglutamine diseases, or PolyQ diseases. To date, nine PolyQ diseases have been described: Huntington’s disease, dentatorubral pallidoluysian atrophy (DRPLA), spinal and bulbar muscular atrophy (SBMA), and six different types of spinocerebellar ataxia (SCA 1, 2, 3, 6, 7, and 17). PolyQ diseases lead to serious, constantly progressing dysfunctions of the nervous and/or muscular systems, and there currently exists no efficacious therapy for any of them. Recent studies have convincingly shown that mCAG-RNA can actively participate in the pathological process during the development of PolyQ diseases. Mutant RNA is involved in a wide range of molecular mechanisms, ultimately leading to disruption of the functions of transcription, splicing, translation, cytosol structure, RNA transport from the nucleus to the cytoplasm, and, finally, to neurodegeneration. This review discusses the involvement of mutant mCAG-RNA in neurodegenerative processes in PolyQ diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Polyglutamine (PolyQ) diseases exhibit a number of common properties, such as neuron death, inverse correlation between the number of CAG repeats and the onset age, instability of the CAG repeat number in generations, and the tendency of protein products to form large intracellular aggregates. All PolyQ diseases are autosomal dominant disorders, except for X-linked spinal and bulbar muscular atrophy (SBMA), which affects only adult men. We note that, although PolyQ diseases have similar properties, their neurological presentation is different, since each of them causes predominant degradation of different neuron types (Table 1).
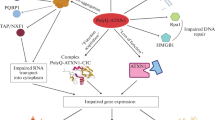
The molecular mechanisms that underlie the pathogenesis of PolyQ diseases are still insufficiently understood. We note the toxic effects of PolyQ proteins, especially their intracellular aggregates, have attracted the most attention. For instance, it is known that, in Huntington’s disease, mutant HTT protein (mHTT) impairs autophagy, vesicular transport, neurotransmission, and the functioning of mitochondria. At the same time, it has recently become clear that the pathological process may also actively involve mCAG-RNA (Fig. 1). Secondary RNA structures formed by excessive sequence repeats affect transcription and splicing, inducing global changes in the cell transcriptome. They can interfere with translation, which gives rise to nonfunctional or toxic protein products. Secondary structures and long repeats also trigger the formation on intranuclear foci accumulating abnormal RNA.

Molecular mechanisms of potential mutant RNA involvement in the pathogenesis of polyglutamine diseases.
In this review, we consider the issues related to the role of mCAG-RNA in neurodegenerative processes in PolyQ diseases. The interest to mCAG-RNA is due not only to its potential involvement in the pathogenesis but also to the fact that one of the most promising approaches to PolyQ diseases therapy is the use of antisense oligonucleotides (ASO) complementary to mCAG-RNA [1‒3]. Huntington’s disease, as the most comprehensively studied PolyQ disease, will be discussed in more detail.
HTT GENE AND ITS TRANSCRIPTION IN HUNTINGTON’S DISEASE
Huntington’s disease is caused by expansion of trinucleotide CAG repeats in HTT exon 1. This gene located in the short arm of chromosome 4 (4p16.3) comprises 67 exons and encodes a protein of approximately 350 kDa. HTT is expressed in all cell types at similar levels [4], although brain cells contain slightly more HTT. Expansion of CAG triplets in HTT exon 1 leads to degeneration of the corpus striatum and manifests as an autosomal dominant gain-of-function mutation. If the number of CAG repeats in HTT exceeds 40, the mutation is 100% penetrant. The longer the CAG repeat stretch, the earlier the disease manifests. If the number of repeats slightly exceeds 40, first clinical signs are usually detected after the age of 40‒50 years. Repeat numbers of over 70 lead to juvenile Huntington’s disease where first signs are diagnosed during childhood [5]. Complete HTT deletion results in embryo death at an early stage of development [6]; hemizygosity does not result in an anomalous phenotype, and the presentation of heterozygous and homozygous CAG triplet expansion is identical [7, 8].
In the terminal form of Huntington’s disease, the abundance of transcripts generated by the mutant HTT allele in specimens of the frontal lobe brain tissue was ~25% lower the level of normal transcripts. In juvenile Huntington’s disease, fibroblasts also contained lower levels of mutant transcripts [9]. No difference in the expression of normal and mutant HTT alleles with a moderate number of CAG repeats was detected in lymphocyte cultures or fibroblasts [10].
Under normal conditions, HTT is transcribed producing two alternative transcripts (10.3 and 13.7 kb long) that differ in the length of the 3'-untranslated region (3'-UTR). Along with two full-size transcripts, expression of mutant HTT can give rise to a truncated polyadenylated transcript that comprises exon 1 and a part of intron 1 [11]. This transcript of approximately 7.9 kb can be translated to produce a highly toxic truncated PolyQ protein. The aberrant transcript is produced as a result of polyadenyation using a weak polyA signal in intron 1, while the splicing of exons 1 and 2 does not occur. This signal works, if exon 1 contains an elongated CAG repeat stretch, and there is a positive correlation between the length of this fragment and the level of abnormal polyadenylation. Presumably, suppression of splicing between exons 1 and 2 is caused by absorption of splicing factors, in particular SRSF6, on the CAG repeats [12]. In addition, the weak polyA signal in intron 1 may become more available for polyadenylation, because CAG repeats decelerate RNA polymerase II during transcription [13].
HTT transcripts differ in their stability and polyA-tail length, as well as in their location in the neuron body or projections. They have different sites for RNA-binding proteins and microRNAs [14, 15]. The short transcript carries a polyA tail ~50 b long, whereas the medium and the long HTT mRNAs have polyA sequences of only 5 and 10 b. The short transcript has the longest half-life period, and the long HTT-mRNA is the least stable. The medium and the long transcripts have more miRNA binding sites than the short, and miRNA-221 is selective for medium and long mRNAs. Differences in cellular localization of medium and long HTT-mRNAs, as well as in effectiveness of their translation have been described [16]. Although the relative effectiveness of translation of short HTT-mRNA has not been determined, it can be expected that short mRNA, due to its higher stability and lower number of regulation sites, would be translated more effectively than normal full-size transcripts [13].
In some hereditary diseases caused via expansion of microsatellite repeats, antisense transcripts including the expansion fragment may be functional or have an additional pathogenic effect [17, 18]. HTT occupies a long fragment of chromosome 4, therefore it was not a surprising finding to detect mRNAs transcribed from its antisense strand. Some of them, in particular, HTT-AS alternative transcripts, include the expansion region; accordingly, these transcripts contain a CUG repeat fragment. Investigation of these transcripts showed that nearly all cells types express them at low levels. In brain tissue specimens of patients with Huntington’s disease, the HTT-AS promoter was significantly less active than the standard HTT promoter, and the length of triplet repeat fragment showed inverse correlation with the level of antisense transcript expression [19]. Thus, it seem unlikely that HTT antisense transcripts contribute significantly to the pathogenesis of Huntington’s disease. However, it cannot be ruled out that antisense transcripts of the corresponding genes are somehow involved in other PolyQ diseases. In particular, antisense transcripts with CUG repeats were detected in SCA2. Expression of the ATXN2-AS gene, which encodes these transcripts, was detected in brain tissues of patients with SCA2, as well as in different types of cells from these patients, including induced pluripotent stem cells (IPSC). Toxicity of mRNA transcribed from ATXN2-AS with an elevated number of CTG repeats was demonstrated in cell models [20].
Thus, when studying potential pathogenic effects of mRNAs in PolyQ diseases, it is important to characterize all alternative transcripts of the genes with CAG repeat expansion, including illegitimate transcripts from hidden promoters and polyadenylation sites. Furthermore, it is also necessary to assess the possible contribution of transcription from the antisense strand.
NUCLEAR FOCI OF mRNAs WITH LONG TRINUCLEOTIDE REPEAT REGIONS
The CAG repeat region in HTT mRNAs transcribed from a wild-type or mutant gene can form hairpins, the stability of which is maintained due to adjacent short fragments of CCG triplets. The only difference between mCAG-RNA and normal HTT mRNA is a longer hairpin stem composed of CAG repeats only. Nevertheless, this suffices to cause mCAG-RNA accumulation in the nucleus with formation of foci that can be visualized using fluorescent in situ hybridization (RNA-FISH) [21, 22]. In Huntington’s disease, these RNA foci were detected in different types of cells, including fibroblasts, lymphocytes, and neuronal precursors [23].
We note that the phenomenon of nuclear RNA foci has been studied best in type 1 myotonic dystrophy (DM1), a disease that is also caused by trinucleotide (CTG) repeat expansion in 3'-UTR of DMPK gene. The first description of nuclear foci in cells and tissues of patients with DM1 dates back to 1995 [24]. Many studies of nuclear RNA foci in Huntington’s disease were inspired by earlier research in DM; therefore, we will discuss its principal results.
Similarly to PolyQ diseases, DM1 is characterized with a pathogenicity threshold of CTG repeat number (>50) and exhibits an inverse correlation between the number of triplets and the age of disease onset [25]. In contrast to PolyQ diseases, in which CAG expansion leads to production of abnormal protein and abnormal mRNA, pathogenesis of DM1 does not involve the protein encoded by mutant DMPK. It is assumed that DM1 is induced by toxic mRNA containing a large number of CUG repeats (mCUG-RNA). CUG repeats form a hairpin structure where GC pairs are opposed by complementary GC pairs (GC‒CG) in antiparallel RNA chains, whereas uracyls oppose uracyls (U‒U). The resulting CUG hairpin represents an RNA helix with a conformation that is very close to the A form. In the minor groove of this RNA helix, alternating positive and negative charges form a characteristic pattern recognized by certain RNA-binding proteins [26]. The toxic effect of mCUG-RNA results from the fact that the CUG repeat hairpin promotes formation of intranuclear mCUG-RNA aggregates (foci) that capture RNA-binding proteins, in particular, MBNL1 and other splicing-regulating proteins of the MBNL family.
It is known that MBNL1 forms a ring-shaped structure that binds to the RNA helix composed of CUG repeats. More specifically, the N-terminal fragment of MBNL1 binds to RNA, while its C-end is involved in homotypic interactions, which can stabilize contacts within or between the rings [27]. The capturing of MBNL in nuclear RNA foci and the resulting MBNL deficit impairs splicing, which leads to deficiency of essential transcripts and simultaneously to the appearance of alternative fetal transcripts that do not correspond to the state of terminal muscle cell differentiation [28]. Among the targets of MBNL-mediated splicing, there are transcripts such as those of troponin, insulin receptor, and one of the chloride channel proteins that are important for muscle cells [29, 30]. Thus, the leading pathogenic factor in DM1 is mutant RNA, and RNA-binding proteins act as intermediate agents.
Dynamical observations showed that nuclear foci of mCUG-RNA in DM1 are unstable structures that constantly disaggregate and stochastically reassemble again. Nuclear mCUG-RNA foci can fall apart, recombine, and fuse with neighboring foci, exhibiting dynamics that is rather that of a viscous liquid than of a solid body [31]. The number of nuclear mCUG-RNA foci in the cells of DM1 patients can be diminished by decreasing MBNL1 levels, which implicates this protein as a key factor of RNA foci formation [32]. Formation of toxic RNA foci can be suppressed by decreasing mCUG-RNA abundance using RNA interference [33] or ASO [34]. A possible alternative approach to suppressing the formation of RNA foci is interaction of small molecules with the secondary structure of mCUG-RNA formed by the CUG expansion region [35, 36]. Suppression of mCUG-RNA foci formation in the nucleus normalizes the cellular transcriptome, decreasing the frequency of abnormal splicing events.
As it has already been mentioned, CAG repeats also form an RNA hairpin. Moreover, MBNL1 has nearly the same affinity to CUG and CAG hairpins, because it interacts mainly with the GC element of the repeat sequence [37]. However, there is also a number of fine molecular differences between CUG and CAG repeats. For instance, parameters of the dsRNA helix formed by a CAG hairpin are intermediate between A and B conformations [38]. The thermodynamic stability of a hairpin formed by 20 CAG triplet repeats is approximately 1.5 times lower than that of a similar CUG oligonucleotide. In contrast to CUG structures, CAG oligonucleotides are highly sensitive to RNases, since they contain a large number of CpA phosphodiester bonds [39]. In addition to minor molecular differences in the secondary structures of RNA containing CUG and CAG repeats, it is necessary to mention a significant difference in repeat numbers. In Huntington’s disease, the number of repeats rarely exceeds 100, whereas patients with the classical DM1 form usually inherit alleles with several hundreds CTG repeats. In DM1, expansion of CTG repeats continues in somatic cells, reaching several thousands in skeletal and cardiac muscle tissue. Nuclear RNA foci formed by mCAG-RNA are significantly more difficult to visualize through FISH [23]. CAG foci also have some morphological differences from RNA foci in DM1: they are relatively loose and occupy a larger area [40]. Interestingly, nontranslated mCUG-RNA exogenously expressed in HeLa cells formed more foci than mCAG-RNA with the same number of repeats [41].
It was found that the number and morphology of RNA foci in fibroblasts of patients with different PolyQ diseases (e.g., HD, DRPA, SCA1, SCA3, SCA7) are similar, although the cells of DRPA patients had the largest RNA foci with the greatest total area. The number of RNA foci depended on the length of CAG repeats, however not on the level of mCAG-RNA expression. In PolyQ fibroblasts, there were no focal aggregates of mRNA transcribed from CTG repeats of the antisense strand. In contrast to RNA foci in DM1, RNA foci in the nuclei of fibroblasts from patients with PolyQ diseases exhibited only partial colocalization with MBNL1. At the same time, in all analyzed PolyQ fibroblasts, RNA foci colocalized with SC35, a splicing speckle marker SC35 [39]. This is another difference between PolyQ diseases and DM1, where the pattern of colocalization between RNA foci and SC35 speckle marker was random [24].
Thus, there exists a number of differences between RNA foci observed in DM1 and in PolyQ diseases; that is, the role of focal nuclear accumulation of mCAG-RNA in the pathogenesis of PolyQ diseases is still to be determined. Moreover, we note that the presence of numerous and pronounced RNA foci in the nucleus does not necessarily lead to cell death. At any rate, this is suggested by the results of a study of RNA toxicity in amyotrophic lateral sclerosis with frontal-temporal dementia, in which pathological changes are caused by expansion of GGGGCC repeats in C9orf72 intron 1. In particular, it was shown that the ability of RNA containing GGGGCC repeats to aggregate into nuclear foci in neural cells did not correlate with enhanced neurodegeneration [42].
EFFECTS OF mCAG-RNA ON THE TRANSCRIPTOME
Early stages of Huntington’s disease are characterized with abnormal expression of numerous genes in those brain areas that are most susceptible to degeneration [43]. Some researchers suppose that these abnormalities are related to the ability of mCAG-RNA to induce illegitimate RNA interference that affects a number of intracellular transcripts. It was shown that mCAG-RNA that contains long CAG hairpins is a target of Dicer ribonuclease, the principal component of RNA interference [44]. Basically, all sufficiently long CNG hairpins (>17 b) can be Dicer substrates. Dicer interacts with the stem of the RNA-hairpin, because this double-stranded structure resembles a perfect RNA duplex, in spite of incomplete strand complementarity. In vitro experiments showed that Dicer cleaves a CAG hairpin producing short RNA duplexes (sCAG) 21 bp long. Most probably, this process can also occur in vivo, because sCAG fragments of similar length were detected in post mortem brain specimens of patients with Huntington’s disease. Presumably, Dicer ribonuclease activity may be responsible for the already mentioned fact that the levels of mutant HTT transcripts are decreased in comparison to normal HTT-mRNA [44]. After a CAG hairpin is cleaved, one sCAG fragment binds to the RISC complex, which cleaves target RNA due to nuclease activity of Argonaute protein (Ago1). Potential mRNA targets of the RISC complex are transcripts containing CTG repeats in the coding region or in 3'-UTR. However, we note that in vitro experiments performed in SH-SY5Y neuroblastoma cells detected only a slight (~10%) decrease in the expression of these transcripts after the cells were transfected with mHTT-RNA containing 80 CAG repeats [45]. Similarly, sCAG were not found in a Drosophila line that serves as a model of PolyQ diseases [46]. In other words, the sCAG phenomenon appears controversial, and the contribution of sCAG in the pathogenesis of PolyQ diseases requires further investigation.
Interesting results unrelated to sCAG involvement in neurodegeneration were reported recently by Murmann et al. [47], who claim that sCAG is highly toxic for tumor cells in vitro and in vivo, however not for nontransformed cells. According to the authors, this may explain the established fact of decreased incidence of malignancies in patients with Huntington’s disease [48].
mCAG-RNA EFFECTS ON SPLICING
As it has already been mentioned, mCAG-RNA binds the splicing factor MBNL1. As a consequence, the splicing patterns of known mRNA targets of MBNL1 are altered in Huntington’s disease. This was also confirmed in cell models of Huntington’s disease and SCA3 [41].
The role of mutant RNA in damaging splicing mechanisms was studied in detail by J. Schilling et al. [49]. Mass-spectrometry of proteins that predominantly bind mutant HTT-RNA showed that these were mainly spliceosome proteins, in particular, PRPF8, SF3B2, SNRNP40, and SON. PRPF8 and SNRNP40 are components of small spliceosome subunit U5, and SF3B3 makes part of U2, another small spliceosome subunit. Apparently, SON facilitates the interaction between spliceosome protein SRSF2 and RNA polymerase II. This interaction is required for the assembly of the pre-early ATP-dependent splicing complex and for interaction of U1 and U2 spliceosome subunits with-mRNA.
It was found that expression of mCAG-RNA caused abnormal splicing in a cell model based on SHSY5Y neuroblastoma cells, while overexpression of Prp8, a homolog of PRPF8 splicing factor, alleviated the Huntington’s disease phenotype in Drosophila melanogaster [49]. Finally, to find out whether abnormal splicing occurred in brain cells in Huntington’s disease, quantitative RT‒PCR was used to compare CREB1 transcripts in brain tissue specimens from patients and control subjects. It was shown that, in mRNA specimens from the patients, CREB1 intron was retained at significantly higher levels [49].
We noted that involvement of mCAG-RNA in splicing abnormalities and the binding of splicing proteins was demonstrated only in Huntington’s disease and SCA3, however, to date, has not been observed in other PolyQ diseases.
NUCLEOLAR DYSFUNCTION IN PolyQ DISEASES
In neurons of model mice and in cells of patients with Huntington’s disease, a repeated finding is a significant decrease in rRNA expression, which may lead to nucleolar stress with subsequent apoptosis [50]. A research group from the Chinese University of Hong Kong was able to unfold the chain of events linking mCAG-RNA to apoptosis induced by nucleolar stress [51]. It was shown that mCAG-RNA is associated with nucleolin: a multifunctional protein that plays a critical role in the transcription and processing of pre-rRNA and in pre-ribosome assembly. Interaction with mCAG-RNA prevents nucleolin from binding to a regulatory element of rRNA. This, in turn, causes hypermethylation of the regulatory element and downregulation of rRNA transcription. Due to a decrease in rRNA levels, there appear free ribosomal proteins (RpL5, RpL11, and RpL23), which associate with E3 ubiquitin ligase MDM2 involved in proteasomal degradation of p53. Interaction of ribosomal proteins with MDM2 impairs p53 degradation; as a result, the level of p53 in mCAG-RNA-expressing cells increases, the protein is transported to mitochondria and activates apoptosis [51].
Subsequently, the same research group designed a peptide termed BIND (beta-structured inhibitor for neurodegenerative diseases) that can block nucleolin binding to mCAG-RNA [52]. BIND was fused to the translocation peptide TAT to enable its intracellular delivery, and it was shown that TAT-BIND significantly decreased the death of HEK293 cells expressing exogenous mCAG-RNA. Moreover, TAT-BIND diminished structural abnormalities of the nucleoli typical for cells under nucleolar stress. The decreased level of 45S rRNA expression, another characteristic sign of nucleolar stress, was also reversed to normal. Experiments in mice and Drosophila confirmed the results from cell models. Furthermore, they demonstrated specificity of the corrective effect of TAT-BIND in PolyQ diseases.
It is currently unclear, what place nucleolar stress has in the chain of pathogenetic events in PolyQ diseases. Nevertheless, it has been established that nucleolar abnormalities caused by interaction with mutant mRNA are promising targets for development of therapeutic agents.
ABNORMAL mCAG-RNA TRANSLATION IN PolyQ DISEASES
mCAG-RNA can interfere not only with those processes that take place in the nucleus, but also with extranuclear, and, first of all, translation. One abnormality is atypical repeat-associated non-ATG translation (RAN), which produces nonfunctional peptides and proteins, sometimes with toxic properties. RAN translation occurs because the hairpin formed by the CAG expansion region in mRNA can initiate translation without a canonical ATG triplet. In this case, RAN translation can begin from a noncanonical initiation site, or from the repeat region. In 2015, it was first shown that brain tissues of patients with Huntington’s disease accumulate four protein homopolymers: polyalanine, polyserine, polyleucine, and polycysteine [53]. This suggests that Huntington’s disease involves RAN translation from the sense and the antisense HTT strand. The abundance of these proteins was the highest in the brain areas that exhibited neuron loss, microglial activation, and apoptosis, in particular, the caudate nucleus, the white matter, and the cerebellum (in the juvenile form). Homotypic proteins generated by RAN translation were also detected in the mouse model of Huntington’s disease [53]. Thus, in Huntington’s disease, in addition to mHTT, cells synthesize nonfunctional and potentially toxic proteins, which is caused by abnormal secondary structure of mCAG-RNA. In more detail, the phenomenon of RAN translation in PolyQ diseases was discussed in [54].
It has repeatedly been noticed mCAG-RNA is translated more efficiently than normal mRNA. One of the translation-enhancing mechanisms is triggered by association of the expanded CAG repeat to RNA-binding protein MID1, which, in turn, recruits protein phosphatase 2A (PP2A) and 40S ribosome kinase S6K. MID1 is an E3 ubiquitin ligase, S6K is a translational regulator, and PP2A phosphatase suppresses phosphate-dependent activity of S6K. Upon binding to PP2A, MID1 catalyzes its ubiquitin-dependent degradation, and acts as a negative regulator of its phosphatase activity. Without phosphatase activity of PP2A, S6K, which is recruited by MID1 to mCAG-RNA, stimulates enhanced translation of mutant RNA [55].
Subsequently, it was shown that the binding of MID1 to mCAG-RNAs encoded by ATXN2, ATXN3, and ATXN7 upregulated translation of mutant proteins in cultures of transgenic mouse neurons, as well as in fibroblasts of patients with the corresponding PolyQ diseases [56]. Thus, it has been established that MID1 is a promising target for development of PolyQ disease therapy aimed at decreasing the amount of mutant proteins in neurons.
WHY DO POLYQ DISEASES DEVELOP AFTER A THRESHOLD REPEAT NUMBER?
In all currently known diseases associated with expansion of short nucleotide repeats, their symptoms manifest only if the number of repeats exceeds a certain critical limit; that is, RNA acquires pathological properties after reaching a specific threshold length [57, 58]. Several hypotheses have been proposed to explain this phenomenon [59]. In our opinion, the most convincing one holds that the triggering of the pathological process depends on the ability of mutant mRNA to form additional or more stable secondary structures (mainly hairpins), in contrast to mRNA containing a normal number of repeats. These secondary structures can engage in illegitimate interactions with RNA-binding proteins, such as splicing proteins, Dicer ribonuclease, or other proteins of RNA metabolism, thus impairing normal functions.
Another hypothesis concerning the threshold repeat number effect assumes that the pathological condition develops because of structural abnormalities caused by the presence of mutant RNA. These abnormalities may be because extended repeat regions in mRNA are capable of intermolecular self-association, which leads to phase separation, in other words, to intracellular structural segregation of molecules. For instance, intramolecular interactions among mutant mRNAs can produce complex branched supramolecular RNA complexes, which may grow to form colloidal particles capable of fusing into hydrogel. Subsequently, these amorphous particles can acquire a thermodynamically optimal ordered solid structure. The structural hypothesis is supported by the results of a study published in 2017, which showed that (CNG)n-RNA solutions undergo phase transition in vitro [60], as observed by droplet aggregation of (CNG)n-RNA. This process depends on the number of repeats in the RNA and on their sequence. For instance, clusterization of RNA containing (CAG)n and (CUG)n repeats was recorded at n ≥ 31, while RNA with (CCGGG)n clusterized at n > 5. The number of repeats determines the ability of RNA to intermolecular association via complementary and/or Hoogsteen interactions of heterocycles. The formation of micron-sized RNA gel particles is facilitated by known factors of polynucleotide compaction, such as the presence of bivalent metal ions, RNA concentration, and molecular crowding.
We note that RNA may facilitate phase separation not only due to RNA‒RNA interactions but also via RNA‒protein contacts. Some researchers think that absorption of specific RNA-binding proteins on RNA-repeats may promote phase separation by generating nuclear RNA foci and contributing to nucleoplasm structuring [61].
Phase separation is involved in formation of membraneless organelles, such as the nucleolus, Cajal bodies, stress granules, etc. [62]. Furthermore, it has recently been shown that intracellular structural segregation via phase separation also occurs during interactions of molecular complexes on superenhancers [63], heterochromatin formation [64], DNA repair [65], and response to oxidative stress [66]. Cell structures and processes of this type may be disrupted in the presence of mCAG-RNA, for instance, because of competition for compaction factors.
Thus, the pathological effects caused by mutant mRNA where the number of CAG repeats exceeds the threshold limit may be due to their interference with intracellular processes and structures. Obviously, structural and functional abnormalities are not mutually exclusive phenomena, however their quantitative and qualitative contribution to the development of the pathological phenotype in PolyQ diseases are still to be determined.
MUTANT mRNA AS A THERAPEUTIC TARGET
The fact that mCAG-RNA is a promising target for therapeutic intervention was demonstrated as long ago as in 2005, when a decrease in mCAG-RNA levels achieved using RNA interference was shown to alleviate pathological signs in HD-N171-82Q mice that serve as a model of Huntington’s disease [67]. A construct based on the adeno-associated virus that expressed a short hairpin RNA was injected into different brain regions, and this approach was proven safe.
Currently, several companies are conducting clinical trials of drugs designed to inhibit the expression of mCAG-RNA in Huntington’s disease.
Voyager Therapeutics and uniQure are developing a therapeutic approach based on miRNA. They have constructed modified virus vectors that are delivered directly into the brain. More specifically, the AMT-130 vector designed by uniQure based on the adeno-associated virus (AAV5) constantly expresses RNA that selectively blocks mCAG-RNA. The miRNA-expressing АMT-130 is administered by injection directly into affected brain tissues, causing selective knockdown of the mutant gene. In April 2018, uniQure presented a review of preclinical data that demonstrated the efficacy of the AMT-130 concept. The results of preclinical animal trials suggest that a single-time AMT-130 injection led to a consistent dose-dependent decrease in mHTT content in the cortex and in deep brain structures. A study in primates showed that, six months after AMT-130 administration, mHTT amounts decreased by 68% in the striatum and by 47% in the frontal cortex. Preclinical trials in rodents showed that a decrease in mHTT content resulted in a significant improvement of the neuronal function of the striatum and motor coordination, diminished body weight loss, and increased median survival by 24% in comparison to the control group. In April 2019, the FDA approved phase I/II clinical trials of AMT-130 by uniQure [68].
Another approach is being developed by Ionis Pharmaceuticals: mCAG-RNA expression should be suppressed by means of ASO delivered intrathecally. Compound RG6042 proposed by this company is a synthetic oligonucleotide with a phosphate backbone that contains phosphorothioate bonds instead of phosphodiester. The end nucleotides of this ASO are modified with 2'-O-methoxyethyl groups. This combination of modifications affects ASO distribution in brain tissues, its half-life, cellular assimilation, and RNase activity [69]. It was supposed that RG6042 might decrease not only mHTT levels but also the amount of wild-type protein. In vitro experiments performed in fibroblasts from patients with Huntington’s disease showed that the therapeutic ASO decreased the levels of mutant and normal HTT mRNA by 83 and 43%.
Large-scale preclininical studies conducted in YAC128 and BACHD mice that model slowly progressing forms of Huntington’s disease, as well as in R6/2 mice as a model of rapidly progressing disease, demonstrated the safety and efficacy of RG6042. The drug has already completed several stages of clinical trials. A phase I/II clinical trial of RG6042 safety and tolerability (NCT02519036) included 46 adult patients with early-stage Huntington’s disease and was conducted in 2015–2017 [70]. It was found that intrathecal administration of RG6042 repeated four times with four-week intervals decreased mHTT levels in the cerebrospinal fluid by up to 60%. This corresponded to a decrease in mHTT levels in the brain cortex and the spinal cord by 55‒85% and 20‒50%. The drug was distributed in all brain tissues: neurons and the glia. The results using cognitive tests and encephalographic evaluation were also positive.
Phase III of multicenter clinical trials on the safety and efficacy of RO7234292 (RG6042) administration began in December 2018. This study is conducted by Ionis Pharmaceuticals in collaboration with the pharmaceutical giant Roche; it will involve 46 medical centers and 660 patients with manifest chorea. The study should be completed in 2022. Two treatment regimens are to be tested: the drug will be administered every four or eight weeks intrathecally (either by subarachnoidal or by epidural intralumbar injection on the level of lumbar vertebrae L4‒L5). The principal goal of the study is to demonstrate that the drug can efficaciously alter the dynamics of motor dysfunction. The efficacy will also be evaluated using 17 neurological parameters (motor, cognitive, and psychomotor functions), as well as biomarkers that characterize the effects of the drug, in particular, the concentration of the mutant protein.
Along with Ionis Pharmaceuticals, an ASO-based approach to therapy is being developed by Wave Life Sciences. Currently, this company is testing two oligonucleotide preparations that block mCAG-RNA expression, WVE-120101 and WVE-120102. The mutant allele is identified by the presence of a linked single-nucleotide polymorphism (SNP). WVE-120101 and WVE-120102 are designed for patients with two different SNP types. Currently, Wave Life Sciences are conducting phase I/II clinical trials (NCT03225833 and NCT03225846) to evaluate safety and tolerability of these agents in patients with early stage of the disease. The drug is designed to be administered into the spinal canal.
There also exist other approaches to decreasing mHTT levels by affecting the mRNA target. For instance, studies on the possibility to employ small molecules with high bioavailability for oral administration have been in progress since 2014 [71, 72].
CONCLUSIONS
Investigation of mechanisms that underlie chronic neurodegenerative diseases and the search for new ways of compensating the damage and protecting neurons in these patients is a topical issue of modern medical science. It is especially important to elucidate the causes of neurodegeneration and to study changes that occur during the development of PolyQ diseases, because of their severity and because there are no efficacious therapeutic agents that could be used to treat these nosological forms. The functions of mutant and normal genes in most PolyQ diseases remain insufficiently studied; it has not been determined whether most toxicity is induced by mutant protein or mutant RNA. It is unknown why PolyQ diseases selectively affect the nervous system, although mutant genes and the proteins they encode are widely present in all tissues. It is also unclear what cellular organelles and signaling pathways are most susceptible to toxic damage, which triggering mechanisms are common for all diseases of this groups, and which are specific for each mutant gene.
Recently, the role of mutant RNA in the pathogenesis of PolyQ diseases has become a subject of extensive discussion. A number of studies have convincingly demonstrated that, in Huntington’s disease, mCAG-RNA is involved in abnormalities of transcription, splicing, translation, cytosol structure, and RNA transportation from the nucleus into cytosol. Nevertheless, it is possible that the contribution of mechanisms associated with PolyQ proteins or mCAG-RNA is different for each PolyQ disease. For instance, a recent study showed that the complex of mutant ataxin 1 with its protein partner CIC has a critical toxic effect in SCA1. Mice with a mutation that prevented the binding of ataxin 1 to CIC did not exhibit degeneration of Purkinje cells, although they expressed mCAG- mRNA with a long stretch of CAG repeats [73].
Until recently, investigation of PolyQ diseases was limited to post-mortem specimens of human neurons, transformed cell lines, and transgenic animals. Thanks to a relatively novel technique of obtaining [74] and differentiating IPSC into neural cells, the range of neurodegenerative disease models can be extended. The use of IPSC-based models is now recognized as a promising approach to investigation of RNA involvement in the pathogenesis of neurodegenerative diseases [75]. It is also of interest to perform comparative studies of PolyQ diseases to reveal their general and specific mechanisms.
REFERENCES
Aronin N., DiFiglia M. 2014. Huntingtin-lowering strategies in Huntington’s disease: Antisense oligonucleotides, small RNAs, and gene editing. Movement Disorders.29, 1455‒1461.
Hu J., Matsui M., Gagnon K.T., Schwartz J.C., Gabillet S., Arar K., Wu J., Bezprozvanny I., Corey D.R. 2009. Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat. Biotechnol.27, 478.
Hu J., Liu J., Corey D.R. 2010. Allele-selective inhibition of huntingtin expression by switching to an miRNA-like RNAi mechanism. Chem. Biol.17, 1183‒1188.
Sharp A.H., Loev S.J., Schilling G., Li S.H., Li X.J., Bao J., Wagster M.V., Kotzuk J.A., Steiner J.P., Lo A., Hedreen J. 1995. Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron. 14, 1065‒1074.
Illarioshkin S.N., Klyushnikov S.A., Seliverstov Yu.A. 2018. Bolezn’ Gentingtona (Huntington’s Disease). Moscow: Atmosfera.
Nasir J., Floresco S.B., O’Kusky J.R., Diewert V.M., Richman J.M., Zeisler J., Borowski A., Marth J.D., Phillips A.G., Hayden M.R. 1995. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 81, 811‒823.
Wexler N.S., Young A.B., Tanzi R.E., Travers H., Starosta-Rubinstein S., Penney J.B., Snodgrass S.R., Shoulson I., Gomez F., Ramos Arroyo M.A., Penchaszadeh G.K. 1987. Homozygotes for Huntington’s disease. Nature. 326, 194.
Myers R.H., Leavitt J.L., Farrer L.A., Jagadeesh J., McFarlane H., Mastromauro C.A., Mark R.J., Gusella J.F. 1989. Homozygote for Huntington disease. Am. J. Hum. Genet.45, 615.
Evers M.M., Schut M.H., Pepers B.A., Atalar M., van Belzen M.J., Faull R.L., Roos R.A., van Roon-Mom W.M. 2015. Making (anti-)sense out of huntingtin levels in Huntington disease. Mol. Neurodegener.10, 21.
Shin A., Shin B., Shin J.W., Kim K.H., Atwal R.S., Hope J.M., Gillis T., Leszyk J.D., Shaffer S.A., Lee R., Kwak S., MacDonald M.E., Gusella J.F., Seong I.S., Lee J.M. 2017. Novel allele-specific quantification methods reveal no effects of adult onset CAG repeats on HTT mRNA and protein levels. Hum. Mol. Genet.26, 1258‒1267.
Neueder A., Landles C., Ghosh R., Howland D., Myers R.H., Faull R.L., Tabrizi S.J., Bates G.P. 2017. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci. Rept.7, 1307.
Sathasivam K., Neueder A., Gipson T.A., Landles C., Benjamin A.C., Bondulich M.K., Smith D.L., Faull R.L., Roos R.A., Howland D., Detloff P.J., Housman D.E., Bates G.P. 2013. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U. S. A.110, 2366‒2370.
Neueder A., Dumas A.A., Benjamin A.C., Bates G.P. 2018. Regulatory mechanisms of incomplete huntingtin mRNA splicing. Nat. Commun.9, 3955.
Romo L., Ashar-Patel A., Pfister E., Aronin N. 2017. Alterations in mRNA 3′ UTR isoform abundance accompany gene expression changes in human Huntington’s disease brains. Cell Rept.20, 3057‒3070.
Romo L., Mohn E.S., Aronin N. 2018. A fresh look at huntingtin mRNA processing in Huntington’s disease. J. Huntington’s Dis.7, 101‒108.
Xu H., An J.J., Xu B. 2017. Distinct cellular toxicity of two mutant huntingtin mRNA variants due to translation regulation. PLoS One.12, e0177610.
Khalil A.M., Faghihi M.A., Modarresi F., Brothers S.P., Wahlestedt C. 2008. A novel RNA transcript with antiapoptotic function is silenced in fragile X syndrome. PLoS One.3, e1486.
Daughters R.S., Tuttle D.L., Gao W., Ikeda Y., Moseley M.L., Ebner T.J., Swanson M.S., Ranum L.P. 2009. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genetics. 5, e1000600.
Chung D.W., Rudnicki D.D., Yu L., Margolis R.L. 2011. A natural antisense transcript at the Huntington’s disease repeat locus regulates HTT expression. Hum. Mol. Genet.20, 3467‒3477.
Li P.P., Sun X., Xia G., Arbez N., Paul S., Zhu S., Peng H.B., Ross C.A., Koeppen A.H., Margolis R.L., Pulst S.M., Ashizawa T., Rudnicki D.D. 2016. ATXN2-AS, a gene antisense to ATXN2, is associated with spinocerebellar ataxia type 2 and amyotrophic lateral sclerosis. Ann. Neurol.80, 600‒615.
de Mezer M., Wojciechowska M., Napierala M., Sobczak K., Krzyzosiak W.J. 2011. Mutant CAG repeats of huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res.39, 3852‒3863.
Wojciechowska M., Krzyzosiak W.J. 2011. Cellular toxicity of expanded RNA repeats: Focus on RNA foci. Hum. Mol. Genet.20, 3811‒3821.
Urbanek M.O., Krzyzosiak W.J. 2016. RNA FISH for detecting expanded repeats in human diseases. Methods.98, 115‒123.
Taneja K.L., McCurrach M., Schalling M., Housman D., Singer R.H. 1995. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell. Biol.128, 995‒1002.
Wheeler T.M., Thornton C.A. 2007. Myotonic dystrophy: RNA-mediated muscle disease. Curr. Opin. Neurol.20, 572‒576.
Mooers B.H., Logue J.S., Berglund J.A. 2005. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc. Natl. Acad. Sci. U. S. A.102, 16626‒16631.
Yuan Y., Compton S.A., Sobczak K., Stenberg M.G., Thornton C.A., Griffith J.D., Swanson M.S. 2007. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res.35, 5474‒5486.
Nakamori M., Sobczak K., Puwanant A., Welle S., Eichinger K., Pandya S., Dekdebrun J., Heatwole C.R., McDermott M.P., Chen T., Cline M., Tawil R., Osborne R.J., Wheeler T.M., Swanson M.S., et al. 2013. Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol.74, 862‒872.
Botta A., Vallo L., Rinaldi F., Bonifazi E., Amati F., Biancolella M., Gambardella S., Mancinelli E., Angelini C., Meola G., Novelli G. 2006. Gene expression analysis in myotonic dystrophy: Indications for a common molecular pathogenic pathway in DM1 and DM2. Gene expression, J. Liver Res.13, 339‒351.
Salvatori S., Furlan S., Fanin M., Picard A., Pastorello E., Romeo V., Trevisan C.P., Angelini C. 2009. Comparative transcriptional and biochemical studies in muscle of myotonic dystrophies (DM1 and DM2). Neurol. Sci.30, 185‒192.
Querido E., Gallardo F., Beaudoin M., Ménard C., Chartrand P. 2011. Stochastic and reversible aggregation of mRNA with expanded CUG-triplet repeats. J. Cell. Sci.124, 1703‒1714.
Konieczny P., Stepniak-Konieczna E., Sobczak K. 2014. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res.42, 10873‒10887.
Sobczak K., Wheeler T.M., Wang W., Thornton C.A. 2013. RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol. Therapy.21, 380‒387.
Jauvin D., Chrétien J., Pandey S. K., Martineau L., Revillod L., Bassez G., Thornton C.A. 2017. Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol. Therapy–Nucl. Acids.7, 465‒474.
Rzuczek S.G., Colgan L.A., Nakai Y., Cameron M.D., Furling D., Yasuda R., Disney M.D. 2017. Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol.13, 188.
Angelbello A.J., Rzuczek S.G., Mckee K.K., Chen J.L., Olafson H., Cameron M.D., Moss W.N., Wang E.T., Disney M.D. 2019. Precise small-molecule cleavage of an r (CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. U. S. A.116, 7799–7804.
Yuan Y., Compton S.A., Sobczak K., Stenberg M.G., Thornton C.A., Griffith J.D., Swanson M.S. 2007. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res.35, 5474‒5486.
Tawani A., Kumar A. 2015. Structural insights reveal the dynamics of the repeating r (CAG) transcript found in Huntington’s disease (HD) and spinocerebellar ataxias (SCAs). PLoS One.10, e0131788.
Sobczak K., Michlewski G., de Mezer M., Kierzek E., Krol J., Olejniczak M., Kierzek R., Krzyzosiak W.J. 2010. Structural diversity of triplet repeat RNAs. J. Biol. Chem.285, 12755‒12764.
Urbanek M.O., Jazurek M., Switonski P.M., Figura G., Krzyzosiak W.J. 2016. Nuclear speckles are detention centers for transcripts containing expanded CAG repeats. Biochim. Biophys. Acta—Mol.Basis Disease.1862, 1513‒1520.
Mykowska A., Sobczak K., Wojciechowska M., Kozlowski P., Krzyzosiak W.J. 2011. CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res.39, 8938‒8951.
Mizielinska S., Grönke S., Niccoli T., Ridler C.E., Clayton E.L., Devoy A., Moens T., Norona F.E., Woollacott I.O.C., Pietrzyk J., Cleverley K., Nicoll A.J., Pickering-Brown S., Dols J., Cabecinha M., et al. 2014. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science.345, 1192‒1194.
Hodges A., Strand A.D., Aragaki A.K., Kuhn A., Sengstag T., Hughes G., Elliston L.A., Hartog C., Goldstein D.R., Thu D., Hollingsworth Z.R., Collin F., Synek B., Holmans P.A., Young A.B., et al. 2006. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet.15, 965‒977.
Krol J., Fiszer A., Mykowska A., Sobczak K., de Mezer M., Krzyzosiak W.J. 2007. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol. Cell.25, 575‒586.
Bañez-Coronel M., Porta S., Kagerbauer B., Mateu-Huertas E., Pantano L., Ferrer I., Guzmán M., Estivill X., Martí E. 2012. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet.8, e1002481.
Reinhardt A., Feuillette S., Cassar M., Callens C., Thomassin-Bourrel H., Birman S., Lecourtois M., Antoniewski C., Tricoire H. 2012. Lack of miRNA misregulation at early pathological stages in Drosophila neurodegenerative disease models. Front. Genet.3, 226.
Murmann A.E., Gao Q.Q., Putzbach W.E., Patel M., Bartom E.T., Law C.Y., Bridgeman B., Chen S., McMahon K.M., Thaxton C.S., Peter M.E. 2018. Small interfering RNAs based on huntingtin trinucleotide repeats are highly toxic to cancer cells. EMBO Repts.19, e45336.
Sørensen S.A., Fenger K., Olsen J.H. 1999. Significantly lower incidence of cancer among patients with Huntington disease: An apoptotic effect of an expanded polyglutamine tract? Cancer.86, 1342‒1346.
Schilling J., Broemer M., Atanassov I., Duernberger Y., Vorberg I., Dieterich C., Dagane A., Dittmar G., Wanker E., van Roon-Mom W., Winter J., Krauß S. 2019. Deregulated splicing is a major mechanism of RNA-induced toxicity in Huntington’s disease. J. Mol. Biol. 19, 1869‒1877.
Lee J., Hwang Y.J., Boo J.H., Han D., Kwon O.K., Todorova K., Kowall N.W., Kim Y., Ryu H. 2011. Dysregulation of upstream binding factor-1 acetylation at K352 is linked to impaired ribosomal DNA transcription in Huntington’s disease. Cell Death Differ.18, 1726.
Tsoi H., Lau T.C.K., Tsang S.Y., Lau K.F., Chan H.Y.E. 2012. CAG expansion induces nucleolar stress in polyglutamine diseases. Proc. Natl. Acad. Sci. U. S. A.109, 13428‒13433.
Zhang Q., Chen Z.S., An Y., Liu H., Hou Y., Li W., Lau K.F., Koon A.C., Ngo J.C.K., Chan H. Y.E. 2018. A peptidylic inhibitor for neutralizing expanded CAG RNA-induced nucleolar stress in polyglutamine diseases. RNA.24, 486‒498.
Bañez-Coronel M., Ayhan F., Tarabochia A.D., Zu T., Perez B.A., Tusi S.K., Pletnikova O., Borchelt D.R., Ross C.A., Margolis R.L., Yachnis A.T., Troncoso J.C., Ranum L.P. 2015. RAN translation in Huntington disease. Neuron.88, 667‒677.
Volovikov E.A., Davidenko A.V., Lagar’kova M.A. 2019. Molecular mechanisms of type 1 ataxia. Russ. J. Genet.55 (in press).
Krauss S., Griesche N., Jastrzebska E., Chen C., Rutschow D., Achmüller C., Dorn S., Boesch S.M., Lalowski M., Wanker E., Schneider R., Schweiger S. 2013. Translation of HTT mRNA with expanded CAG repeats is regulated by the MID1-PP2A protein complex. Nat. Commun.4, 1511.
Griesche N., Schilling J., Weber S., Rohm M., Pesch V., Matthes F., Auburger G., Krauss S. 2016. Regulation of mRNA translation by MID1: A common mechanism of expanded CAG repeat RNAs. Front. Cell Neurosci.7, 226.
La Spada A.R., Taylor J.P. 2010. Repeat expansion disease: Progress and puzzles in disease pathogenesis. Nat. Rev. Genet.11, 247.
Babić Leko M., Župunski V., Kirincich J., Smilović D., Hortobágyi T., Hof P.R., Šimić G. 2019. Molecular mechanisms of neurodegeneration related to C9orf72 hexanucleotide repeat expansion. Behav. Neurol.2019, 2909168
Lee D.Y., McMurray C.T. 2014. Trinucleotide expansion in disease: Why is there a length threshold? Curr. Opin. Genet. Dev.26, 131‒140.
Jain A., Vale R.D. 2017. RNA phase transitions in repeat expansion disorders. Nature. 546, 243.
Fay M.M., Anderson P.J. 2018. The role of RNA in biological phase separations. J. Mol. Biol.430, 4685–4701.
Boeynaems S., Alberti S., Fawzi N.L., Mittag T., Polymenidou M., Rousseau F., Schymkowitz J., Shorter J., Wolozin B., Van Den Bosch L., Tompa P., Fuxreiter M. 2018. Protein phase separation: A new phase in cell biology. Trends Cell Biol.28, 420‒435.
Sabari B.R., Dall’Agnese A., Boija A., Klein I.A., Coffey E.L., Shrinivas K., Abraham B.J., Hannett N.M., Zamudio A.V., Manteiga J.C., Li C.H., Guo Y.E., Day D.S., Schuijers J., Vasile E., et al. 2018. Coactivator condensation at super-enhancers links phase separation and gene control. Science.361, eaar3958.
Larson A.G., Narlikar G.J. 2018. The role of phase separation in heterochromatin formation, function, and regulation. Biochemistry.57, 2540‒2548.
Singatulina A.S., Hamon L., Sukhanova M.V., Desforges B., Joshi V., Bouhss A., Lavrik O.I., Pastré D. 2019. PARP-1 activation directs FUS to DNA damage sites to form PARG-reversible compartments enriched in damaged DNA. Cell Rept.27, 1809‒1821.
Kato M., Yang Y.S., Sutter B.M., Wang Y., McKnight S.L., Tu B.P. 2019. Redox state controls phase separation of the yeast ataxin-2 protein via reversible oxidation of its methionine-rich low-complexity domain. Cell.177, 711‒721.
Harper S.Q., Staber P.D., He X., Eliason S.L., Martins I.H., Mao Q., Yang L, Kotin R.M., Paulson H.L., Davidson B. L. 2005. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. U. S. A.102, 5820‒5825.
http://www.uniqure.com/gene-therapy/huntingtons-disease.php
Bennett C.F., Swayze E.E. 2010. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol.50, 259‒293.
https://clinicaltrials.gov/ct2/show/NCT02519036
Naryshkin N.A., Weetall M., Dakka A., Narasimhan J., Zhao X., Feng Z., Ling K.K., Karp G.M., Qi H., Woll M.G., Chen G., Zhang N., Gabbeta V., Vazirani P., Bhattacharyya A., et al. 2014. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science.345, 688‒693.
Doherty E.M. 2017. Screening approaches to identify small-molecule modulators of huntingtin protein levels. Abstr. CHDI Foundation Annual Therapeutics Conference, Malta, 2017.
Rousseaux M.W., Tschumperlin T., Lu H.C., Lackey E.P., Bondar V.V., Wan Y.W., Tan Q., Adamski C.J., Friedrich J., Twaroski K., Chen W., Tolar J., Henzler C., Sharma A., Bajić A., et al. 2018. ATXN1-CIC complex is the primary driver of cerebellar pathology in spinocerebellar ataxia type 1 through a gain-of-function mechanism. Neuron.97, 1235‒1243.
Takahashi K., Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell.126, 663‒676.
Nekrasov E.D., Vigont V.A., Klyushnikov S.A., Lebedeva O.S., Vassina E.M., Bogomazova A.N., Chestkov I.V., Semashko T.A., Kiseleva E., Suldina L.A., Bobrovsky P.A., Zimina O.A., Ryazantseva M.A., Skopin A.Y., Illarioshkin S.N., et al. 2016. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegeneration.11, 27.
Funding
This work was supported by the Russian Science Foundation (project no. 19-15-00425).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare that they have no conflict of interest. This article does not contain studies performed by the authors involving animals or human subjects.
Additional information
Translated by D. Timchenko
Abbreviations: GABA, gamma-aminobutyric acid; IPSC, induced pluripotent stem cells; ASO, antisense oligonucleotide; DM, myotonic dystrophy; DMPK, myotonic dystrophy protein kinase; DRPA, dentatorubral-pallidoluysian atrophy; FDA, Food and Drug Administration; FISH, fluorescence in situ hybridization; HTT, huntingtin protein; MBNL1, muscleblindlike splicing regulator 1; mCAG-RNA, mRNA with CAG repeat expansion; mCUG-RNA, mRNA with CUG expansion; mHTT, mutant huntingtin; MID1 (Midline 1), E3 ubiquitin-protein ligase Midline-1; OMIM, Online Mendelian Inheritance in Man; PolyQ, polyglutamine; PP2A, protein phosphatase 2A; RAN, repeat associated non-ATG; SBMA, spinal and bulbar muscular atrophy; SCA, spinocerebellar ataxia; sCAG, 21 bp-long RNA duplexes composed of CAG repeats.
Rights and permissions
About this article

Cite this article
Bogomazova, A.N., Eremeev, A.V., Pozmogova, G.E. et al. The Role of Mutant RNA in the Pathogenesis of Huntington’s Disease and Other Polyglutamine Diseases. Mol Biol 53, 838–849 (2019). https://doi.org/10.1134/S0026893319060037
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026893319060037