
Abstract
Human pluripotent stem cells, which include embryonic stem cells and induced pluripotent cells (iPSCs), are capable of unlimited division and differentiation into all cells of the body. These cells are considered as a potential source of various types of cells for transplantations. The use of autologous iPSCs is not potentially associated with immune rejection and does not require immunosuppression required for allogeneic grafts. However, the high cost of this technology and the duration of obtaining iPSCs and differentiated cells may limit the use of autologous iPSCs in clinical practice. In addition, full equivalence and immunological compatibility of autologous iPSCs and their derivatives have been repeatedly questioned. One approach to solving the problem of the immunological compatibility of allogeneic derivatives of iPSCs can be the establishment of cell lines with reduced immunogenicity. Differentiated derivatives of such iPSCs may be suitable for transplantation to any patient. This review discusses the strategies for evading immune surveillance in normal and tumor processes that can be used to establish stem cell lines with reduced immunogenicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Pluripotent stem cells (PSCs) include embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). PSCs are cell lines that exist only in vitro. The main feature of these cells is their ability to unlimitedly proliferate under conditions that promote their self-renewal and to differentiate into any cells (derivatives of all three germ layers) when these conditions change. Human ESCs are obtained from the inner cell mass of blastocysts that remains unclaimed after in vitro fertilization procedures [1]. Genetic reprogramming of somatic cells (fibroblasts) to a pluripotent state was implemented in 2006 by K. Takahashi and S. Yamanaka [2] with use of the exogenous expression of transcription factors Oct4, Sox2, Klf4, and c-Myc. Since obtaining the first human hESC and iPSC lines, the scientific community has had high hopes for the development of cell replacement therapy. Effective and reproducible protocols for differentiation of various types of PSC, including cardiomyocytes, retinal pigment epithelium (RPE) cells, neurons, and pancreatic β-cells, were developed [3]. Furthermore, iPSCs opened up broad vistas for modeling the so-called “disease in a Petri dish” and drug screening [4–6].
The technology for deriving autologous iPSCs enables personalized cell therapy, which removes the problems associated with immune rejection. However, the high cost, the duration of obtaining clinically certified iPSC lines, and their subsequent differentiation into cells required for transplantation, as well as verification of the therapeutic safety of cell products are the factors that have slowed the introduction of the iPSC technology in clinical practice [3, 7]. The study, the results of which show the existing problems of applying in industry the cell products of autologous iPSCs and their derivatives, is very demonstrative [8]. During obtaining autologous iPSCs of two patients and their differentiation into RPE cells for subsequent transplantation, the authors proposed indicators and quality assessment methods by selecting cell lines at different stages of cultivation and differentiation of iPSC clones by the key characteristics that could potentially affect their safety: morphology, karyotype, growth properties, genetic stability, and the ability for directed differentiation. As many as 12 of 32 iPSC clones derived from the first patient and half of 40 iPSC clones derived from the second patient were culled at the stage of estimation of the morphological characteristics. As a result, two iPSC clones derived from the first patient and only one clone derived from the second patient, which met all the safety-related criteria, were obtained. It is believed that obtaining clinically certified autologous iPSC lines requires expensive and time-consuming analysis of each of the resulting clones, which will make it possible to reduce the risks associated with potential mutagenesis and malignant transformation of cell products during transplantation to a patient. That is why the world scientific community currently holds the opinion that, at least in the short term, the allogeneic transplantation of thoroughly characterized iPSC and ESC derivatives can be considered as a more realistic approach to personalized cell therapy [7, 9].
Histocompatibility is one of the main problems of the therapeutic use of allogeneic cells and tissues, including the cells that are PSC differentiation products. Over 20 000 HLA alleles are known (http://www.ebi.ac.uk/imgt/hla/). This polymorphism creates the problems in selecting donors for transplantation. The incomplete coincidence of the donor and recipient in the major histocompatibility complex (MHC) genes necessitates systemic administration of immunosuppressive drugs to prevent the immune response. Such therapy has serious side effects, increases the risk of infections and tumors, and is often ineffective [10].
Thus, the strong interest in studying the immunogenicity and immunological tolerance mechanisms in health and disease is justified. For example, numerous mechanisms by which the tumor tissue evades immune surveillance are known. It is, therefore, of interest to identify the possible ways and mechanisms involved in tolerance development, which can be used to solve the problem of the histocompatibility of allogeneic differentiated PSC derivatives.
An effective way to solve this problem may be deriving universally histocompatible human iPSC lines suitable for transplanting into any recipient (Fig. 1). The personalized approach to future cell transplantations can be facilitated by using a more universal source of cellular material. It is assumed that the PSC genetic modifications described in our review can be used to develop a safe and effective strategy that will reduce the immune response of a potential recipient to the allogeneic cells differentiated from PSCs without using the standard immunosuppressive drugs.

Advantages and disadvantages of potential use of autologous, hypoimmunogenic, and allogeneic cell products in transplantation.
IMMUNOGENICITY OF PLURIPOTENT STEM CELLS AND THEIR DERIVATIVES
Initially, it was believed that PSCs can evade immune surveillance and will not be rejected after transplantation because of the low expression of HLA class I and II molecules and costimulatory molecules CD80 and CD86, as well as due to the expression of immunomodulatory molecules such as serpin 6 (endogenous granzyme B inhibitor) and TGFβ, which inhibit T-cell proliferation [11]. However, studies in mice with a functional immune system showed that PSCs do not have immunological privileges and cause specific immune response and rejection, as in the case of transplanting mature tissues and organs [12, 13].
Unlike mouse ESCs, on the surface of which MHC I and II molecules are not detected [14], human ESCs express HLA-I [15]. Both the innate and adaptive immunity contribute to the rejection of ESCs. Allogeneic NK cells lyse mouse and human PSCs in vitro [16]. It is believed that PSCs cannot activate allogeneic T cells directly via the interaction of HLA and TCR in vitro or in vivo [17]. However, PSCs express immunogenic antigens, including the Oct 3/4 protein, which can activate T cells via the antigen-presenting cells (APCs) [18].
In one study, human ESCs and their differentiated derivatives were transplanted under the kidney capsule of a humanized mouse [12]. Transplantation of healthy donors skin pieces into these mice led to a generalized transplant rejection, whereas the differentiated ESC derivatives caused only minimal manifestations of inflammation in the form of leukocyte infiltration. However, later these data were not confirmed. For example, the injection of mouse ESCs and their derivatives into the myocardium of allogeneic recipient mice resulted in increased immunogenicity as the degree of differentiation of the transplanted cells was increased [13]. Later, a collection of mouse ESC lines that differed from each other in certain genetic loci with increasing levels of immunological differences was obtained [19]. The degree of the immune response induced by the cells that were differentiated from ESCs was evaluated by implanting these cells under the kidney capsule without the use of immunosuppression. It should be noted that the differentiated iPSC derivatives whose haplotype was almost identical to the haplotype of the recipient mouse line were eliminated much more slowly than those differentiated from the allogeneic ESCs.
In this respect, it is important to clarify the issue of the potential possibility of the rejection of autologous derivatives of pluripotent cells. In 2011, in experiments on autologous transplantation, mouse iPSCs and ESCs were subcutaneously injected into syngeneic recipients [20]. As expected, the syngeneic ESCs formed teratomas; however, the iPSCs with the same genetic background caused the formation of significant T-cell infiltrates, which led to their rejection. Similar results obtained later also showed that the degree of immunogenicity of PSCs decreased in the course of further differentiation [21]. PSCs in an undifferentiated state cannot be regarded as a material for transplantation. Only cells of a certain tissue specificity, directionally differentiated from PSCs in vitro—neurons and their progenitor cells, RPE cells, insulin-producing cells, etc.—can be used for this purpose [3]. The unexpected report of the immunogenicity of the autologous iPSC derivatives in syngeneic recipients caused pessimism about their therapeutic potential. It should be noted that the results of later studies, conversely, indicate the absence of immunogenicity and the safety of the differentiated syngeneic iPSC derivatives [22, 23].
Endothelial cells, hepatocytes, and neuronal cells were derived from ESCs and iPSCs [22]. These cells expressed MHC I and costimulatory molecules; i.e., theoretically, they might activate the T-cell response. However, coculturing with the allogeneic T cells showed no specific T-cell response to the undifferentiated syngeneic iPSCs or their derivatives. The transplantation of three types of cells into syngeneic mice did not lead to the graft’s infiltration by T cells, which is consistent with the data obtained in vitro. The transplantation of the allogeneic cells immediately caused a generalized immune response in vivo and pronounced cytotoxicity in vitro, once again confirming that PSCs and their derivatives are not immunologically inert. These results suggest that the immunogenic proteins are not produced, at least not in the endothelial and neuronal cells and hepatocytes differentiated from iPSCs. Similar conclusions were made by other researchers, who showed that the teratomas formed from syngeneic ESCs and iPSCs do not cause a significant immune response [23].
It was assumed that the immunogenicity of the obtained lines was, most likely, associated with the overexpression of the tumor-associated genes Hormad and Zg16 [24]. However, no differences in the expression of these genes were detected in ESCs and iPSCs [22, 23]. The discrepancy in the results of these studies may be caused by the differences in the technologies used to obtain iPSCs. It is assumed that the immunogenicity of cell lines may be affected by the vector selected for the reprogramming factor delivery [25]. For example, retroviral constructs and plasmids were used for reprogramming [24], iPSCs were obtained using plasmids [22], and ESCs and iPSCs were obtained using both plasmids and retroviruses [23]. Retroviral vectors are integrated primarily into the transcriptionally active sites, causing long-term activation of transgenes or adjacent genes near the integration site, which may lead to the activation of the expression of potentially immunogenic proteins [18]. Therefore, for clinical use, iPSC should be obtained by the methods of reprogramming somatic cells, which do not cause mutagenesis and do not lead to immunogenicity.
The immunogenicity of iPSC derivatives was also studied in primates [26]. It was found that the transplantation of autologous neurons differentiated from iPSCs into the primate brain causes the minimal immune response. Conversely, the allogeneic neurons cause the activation of microglia (IBA-1+/MHC class II+) and infiltration of the graft by T cells. Later, to study the mechanisms that occur during the transplantation of human cells, mice with a reconstituted human immune system were used [24]. After subcutaneous injection of autologous iPSCs, obvious inflammatory and necrotic foci were formed in the teratomas, which indicated immunological rejection of at least some types of the cells differentiated from the iPSCs. In addition, differential immunogenicity of iPSC derivatives of different histotypes was also observed: smooth muscle cells induced a stronger immune response than the differentiated RPEs even when they were transplanted into a region other than the eye. It should be noted that, to date, the only clinical trial of the autologous iPSC derivatives, performed on a patient with age-related macular degeneration, confirmed the absence of immunogenicity in the autologous RPE cells differentiated from iPSCs [8]. In connection with the contradictory data on the immunogenicity of certain types of autologous iPSC derivatives, further development of cell therapy protocols may require an estimation of the immunogenicity of the cells differentiated from iPSCs on animals with a humanized immune system. Since cells of each type contain different sets of proteins, it is likely that, before clinical use, it will be necessary to screen cells of each type for immunogenicity [25].
It is believed that iPSCs, unlike ESCs, largely retain the transitional transcriptional and epigenetic memory of their origin (the so-called somatic memory), especially in the early passages; however, the molecular and functional differences are lost in the course of long-term cultivation [27]. These properties may be specific for each iPSC clone [28]. The epigenetic features can explain the residual expression of immunogenic proteins, which are synthesized during differentiation only in iPSCs but not in hESCs. The contribution of the epigenetic mechanisms to the regulation of expression of immunogenic proteins may be tissue-specific, which was observed in the differentiation of human iPSCs to the RPE and smooth muscle cells [24]. Wang et al. showed how the somatic memory phenomenon may affect the further immunogenicity of the iPSC lines derived [29]. They reprogrammed the mouse testes’ Sertoli cells, which were located in an anatomically immunologically privileged site. The resulting cells were less immunogenic when transplanted into an allogeneic recipient than the iPSCs derived from skin fibroblasts. Similar data were also obtained for the iPSCs reprogrammed from the umbilical mesenchymal cells [30]. The differentiated neuronal progenitors caused a significantly lower proliferation and activation of cytotoxic lymphocytes when cocultured in vitro.
There are also other causes, which are discussed in connection with the potential immunogenicity of iPSCs. Firstly, somatic mutations were identified in many iPSC lines. These mutations can create new immunogenic determinants, similarly to neo-antigens, which occur in tumor cells [31]. Secondly, the genomic translocations detected in iPSCs may lead to the formation of fusion proteins, which may form new immunogenic determinants [31]. Changes in the genome’s functioning may also be caused by long-term cultivation and the reprogramming process itself, which is accompanied by the transgene’s insertion at a random site. Therefore, in addition to the immunogenicity of the cells differentiated from iPSCs, the most serious concern is the possible genomic instability of these cells, which may increase the risk of carcinogenesis. Nevertheless, it should be noted that the data on the genetic instability of iPSCs are ambiguous and contradictory. The results of the comparison of the genetic stability of ESCs and iPSCs probably indicate the absence of differences between ESCs and iPSCs at the complete reprogramming of the latter [32].
CREATING BANKS OF iPSC LINES THAT ARE HOMOZYGOUS FOR HLA GENES
The reprogramming technology makes it possible to obtain autologous patient-specific iPSCs and their derivatives, which largely eliminates the problem of immune rejection. However, this technique has several disadvantages. Firstly, obtaining and characterizing iPSC lines takes a long time calculated in months [33]. Secondly, further production and subsequent clinical use requires the development of specification in accordance with the requirements of the regulations and quality control of the reprogrammed cells, which significantly increases the time of preparation of the cell product and the cost of personalized therapy [34]. Such an approach may be appropriate for persons with chronic diseases, when there is time required to perform all of these processes.
Creating a bank of characterized iPSC lines homozygous for HLA and their derivatives may be an alternative to personalized therapy based on using differentiated iPSC derivatives [35]. It can be assumed that such homozygous lines will be compatible with the heterozygous recipients in which at least one allele coincides with the iPSC line haplotype. Since the incompatibility in the ABO system may be the cause of hyperacute rejection in primarily vascularized tissues and organs, when creating a haplobank, it is necessary to select healthy donors with blood group I (0), which minimizes the risk of rejection [36]. Because of the strong polymorphism of the HLA locus, it will be necessary to create a haplobank including hundreds of donor lines to match the majority of the recipients [35].
Some empirical estimates of the number of the required PSC lines homozygous for HLA are based on the frequency of occurrence and ethnic diversity of the selected population [36]. For example, the PSC bank obtained from 150 homozygotes typed for HLA will coincide with 93% of the population of the United Kingdom, with the necessity of the minimum immunosuppression [35]. For potential matching with 90% of the population of Japan, approximately 50 lines are required [37]; however, to achieve this level of coincidence, over 60 000 volunteers need to be screened [38]. The iPSC bank in Japan was formed in 2012 at the Center for the study and application of iPSCs in Kyoto (http://www.cira.kyoto-u.ac.jp/e/).
For countries with a more ethnically diverse population, including the United States, Brazil, and Russia, the number of such lines will be greater. In 2012, a model was proposed, in which, in order to create a haplobank from 20 donors with the most frequently occurring HLA haplotypes, 26 000 European Americans and 110 000 African Americans must be typed [39]. However, potential clinical use is possible only under the conditions of international cooperation and typing of hundreds of thousands of volunteers. Obviously, the mass-scale creation of specialized cell banks requires huge investments.
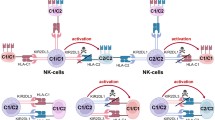
Despite the optimistic forecasts regarding the potential use of haplobanks, it should be borne in mind that the coincidence for the HLA haplotype may be insufficient to prevent allogeneic rejection, and the immunosuppressive therapy may be required for the majority of patients. The minor mH antigens, which will inevitably differ in unrelated donors, and the interaction of inhibitory KIRs (killer cell immunoglobulin-like receptors) on NK cells with different HLA-I alleles may also contribute to alloreactivity. During maturation, NK cells acquire tolerance to their own cells carrying a certain set of HLA-I molecules on the surface. All HLA-C molecules are divided into two groups (C1 and C2) depending on their ability to bind to KIR2DL3 and KIR2DL1 [40]. Therefore, all recipients can be divided into the following groups: C1/C1, C1/C2, and C2/C2, according to the HLA-C genotype. It is assumed that, during transplantation of iPSC derivatives obtained from a homozygous C1/C1 donor to a heterozygous C1/C2 recipient, the licensed NK cells of the recipient will respond to the absence of the KIR ligand (C2) and, eventually, will reject the allograft due to the absence of the KIR2DL1 inhibitory signal in accordance with the conventional mechanism of “friend’s absence” recognition. It was recently found that the NK cells isolated from the HLA-hetero-C1/C2 attack the in vitro T cells and endothelial cells differentiated from HLA-homo-C1/C1 iPSCs [41]. The use of ectopic expression of HLA-C2 on the surface of differentiated cells made it possible to inhibit the NK-cell response. These results confirm that NK cells respond to a mismatched KIR ligand and can kill differentiated cells due to recognition of the absence of the HLA-C expression, which should be taken into account when transplanting iPSC derivatives homozygous for HLA.
In this respect, it is reasonable to consider the possible mechanisms of immunotolerance development, which are implemented by the human immune system not only in health but also in disease, and use these strategies for potentially obtaining immunologically privileged iPSC lines with the minimum risk of malignant transformation.
STRATEGIES USED BY TUMOR CELLS TO EVADE IMMUNE SURVEILLANCE
In an attempt to avoid destruction by the immune cells of the body, tumors can become “invisible” to the immune system. Strategies to evade immune surveillance can be used to derive hypoimmunogenic iPSC lines. Transplanted organs and tissues are rejected by the mechanisms of cellular and humoral responses, which depend primarily on the recognition of foreign HLA antigens by T cells [42]. In order to trick the immune system, tumor cells reduce the expression of the molecules that are required for antigen presentation (HLA), costimulation (CD80 and CD86), and adhesion (CD54), thereby preventing their recognition by immune cells, and increase the expression of the immunosuppressive components such as HLA-G, PD-L1, and CTLA-4 [43]. These mechanisms play a major role in enhancing the activity of regulatory T cells and the subsequent anergy of the cytotoxic T- and NK-cells.
CTLA-4 and PD-L1 are the key immunological checkpoints in maintaining the peripheral tolerance of T cells. Therefore, these molecules can be used to induce immune tolerance to allogeneic grafts [44]. CTLA-4 binds to CD80 and CD86 on the APC surface, thereby blocking the T-cell costimulatory pathways, whereas PD-L1 binds to PD-1, which is expressed on the activated T cells, to induce the inhibitory signaling pathways. Taken together, they control the balance of the coinhibitory and costimulatory signals, which plays an important role in the regulation of the amplitude and duration of the T-cell response. The expression of these molecules is increased in various types of cancer cells [45].
Next, we will consider in more detail the basic strategies used by tumor cells to develop immunotolerance, which can be used to solve the problem of the histocompatibility of allogeneic differentiated derivatives of pluripotent cells.
HLA-I
Evasion of immune surveillance is often associated with the loss of HLA-I molecules on the tumor cell’s surface. HLA-I molecules play the key role in the presentation of peptides (including the tumor-associated antigens) to the cytotoxic T cells. The expression of these antigens and costimulatory receptors, possible signals of the processes occurring in the cell, triggers immune activation and promotes the cytotoxic destruction of the tumor cells [46]. It is assumed that the efficiency of antitumor therapy depends on the expression level of HLA-I molecules on the surface of tumor cells [47]. This primarily depends on the molecular mechanisms underlying the loss of HLA-I expression [48]. In the so-called soft neoplasms, the changes that are responsible for the changes in the expression level of HLA-I disappear after stimulation with cytokines or immunotherapy. In this case, the increasing specific T-cell response leads to a further regression of the neoplasm. In the therapy-resistant, or hard, neoplasms, genomic structural defects are irreversible and cause various mutations. In this case, the mechanisms of evading immune surveillance prevail. According to this idea, the nature of the loss of the HLA-I expression in tumor cells significantly affects the success of anticancer therapy [49]. Therefore, defects in the HLA-I expression by tumor cells often lead to hypoimmunogenicity followed by evasion and the progression of metastasis. The loss of HLA-ABC expression was detected in a number of cancers, including the squamous cell carcinoma of the head and neck (approximately 70% of cases), breast cancer (96%), colon cancer (87%), and melanoma (63%) [50].
HLA-I proteins are heterodimers consisting of the heavy α-chain with a high degree of polymorphism and the conserved β-2-microglobulin light chain (b2m). The latter, a small protein with a molecular weight of approximately 12 kDa without the transmembrane domain, is a member of the immunoglobulin superfamily. The association between b2m and the α3-domain of the heavy α-chain of HLA-I is required to maintain the heterodimer conformation and to form the functional HLA-I complex on the cell surface; it also increases the peptide’s binding affinity [51].
Various mutations in the b2m gene can prevent the synthesis of the b2m protein and, accordingly, the stabilization of the functionally active HLA-I molecule. Such mutations are detected in various cell lines and cancer tissues. They may be represented by insertions and deletions of nucleotides in the motifs with repetitive sequences, as well as by single-letter substitutions in one allele of the b2m gene in combination with the loss of the large 15q21 chromosome segments, including the second allele of the b2m gene [52]. These mutations inhibit the b2m expression, thus preventing transcription, or, more often, lead to a failure of the mRNA translation or to the synthesis of a nonfunctional protein. The occurrence of irreversible functional defects in the b2mf gene facilitates the selection and progression of aggressive tumor-cell clones due to the lack of the HLA-I expression.
Another evasion mechanism (e.g., in the case of hematological tumors) may be changes in the HLA-II expression, including those caused by mutations in the CIITA gene, a HLA-II transactivator. Such mutations were detected in patients with the classical Hodgkin’s lymphoma [53].
HLA-G, an Under-Studied Member of the HLA-I Family
HLA-G, together with HLA-E, HLA-F, and HLA-H, belongs to the non-classical HLA-Ib molecules. HLA-Ib molecules, unlike the classical HLA-Ia molecules with a high level of polymorphism, are fairly conserved and are represented by a small number of alleles. HLA-G not only implements the key function of the HLA-I molecule—presents peptide fragments to specific subpopulations of CD8+ T cells—but also has an immunomodulatory function [54]. Normally, HLA-G is not expressed on the surface of healthy cells and is detected only on the trophoblast cells, thymic epithelial cells, cytokine-activated monocytes, mature myeloid cells, and plasmacytoid dendritic cells (DCs), as well as on inflamed muscle fibers [55]. The main physiological role of this molecule is the formation of immune tolerance in the blood–placenta barrier. Various HLA-G forms are expressed by the trophoblast cells. They can interact with the receptors on the surface of immune cells, thus reducing the maternal immune response to semiallogeneic fetal tissues by reducing the cytotoxicity of the T and NK cells, proliferation of T and B cells, and induction of the apoptosis of the activated CD8+ T cells [56].
HLA-G molecules are present not only on the cell surface but their soluble isoforms also exist. Seven HLA-G isoforms are known, which are formed as a result of the alternative splicing of the same mRNA—the membrane-bound isoforms mHLA-G1, mHLA-G2, mHLA-G3, and mHLA-G4 and the soluble isoforms sHLA-G5, sHL-G6, and sHL-G7 [57]. There is also soluble sHLA-G1, which is identical to sHLA-G5 and is formed as a result of the transmembrane domain excision by metalloproteinases [58]. Only HLA-G1 and HLA-G5 can represent the functional heavy α-chain associated with the b2m molecule that is capable of binding small peptides.
The immunomodulating properties of HLA-G are mediated by the interaction with the immunoglobulin-like transcript 2 (ILT2) on the surface of T and B cells, monocytes/macrophages, DCs, and NK cells, as well as with ILT4, which is expressed only on myeloid cells (DCs), monocytes/macrophages, and neutrophils [54]. In addition, HLA-G interacts with KIR2DL4 on the surface of NK cells [59] and with CD160, which is expressed by T cells, NK cells, and endothelial cells [60]. In quiescent cells, these receptors are expressed at a low level; however, their expression in activated cells in pathological conditions (e.g., in viral infections) increases [61]. By interacting with these receptors, HLA-G affects the function of various cell populations: prevents activation of effector cells and cytokine secretion by B cells, promotes apoptosis, inhibits chemotaxis by reducing the expression of several surface receptors for chemokines [54], and reduces angiogenesis [60].
The increased expression of the membrane-bound and soluble sHLA-G, which is detected in various solid and hematological tumors, is correlated with an increased risk of the progression of tumors and metastasis, as well as a poor prognosis in general [45].
NKG2D Ligands
NKG2D (NK group 2 member D) is the main activatory receptor expressed on the surface of NK cells. It is present on the surface of cytotoxic, CD4+, and γδT cells [62]. Therefore, the NKG2D receptor not only induces the cytotoxicity of effector cells [63] but also promotes cytokine production and affects the differentiation and proliferation of T cells [64]. The ligands of this receptor are MICA and MICB (MHC class I-related chain) as well as the ULBP family (UL-16 binding proteins), which, in contrast to HLA-I, are not associated with b2m. The expression of these so-called stress-induced ligands facilitates the immune surveillance of transformed, infected, or stressed cells. When the expression of the corresponding ligands increases, NKG2D recognizes DNA lesions, high levels of reactive oxygen species, increased proliferation levels, and heat shock. In addition, the ligand–receptor pair is one of the well-known immunological checkpoints. Immune cells expressing NKG2D attack the transformed cells before the beginning of changes in the immunological phenotype of tumor cells, which is a prerequisite for immune evasion. Despite this fact, some data indicate that NKG2D ligands play an ambiguous role in the regulation of tumor development [65].
Cancer cells are capable of governing the expression of NKG2D ligands at the posttranscriptional and posttranslational levels. The regulation of the expression of NKG2D ligands has been insufficiently studied. Recently, however, multiple mechanisms were revealed that are used by cancer cells to reduce the expression of stress-induced ligands, thus evading immune recognition [66]. The most frequently used mechanism is to shed molecules from the cell-membrane surface. Metalloproteases (particularly, ADAM10, ADAM17, and MMP14), which are often detected in tumor microenvironment, cut the transmembrane domain, thus removing MICA, MICB, and ULBP1-6 proteins from the surface of malignant cells [67–69]. In addition, the glycosylphosphatidylinositol (GPI)-anchored ligands, such as ULBP1, ULBP3, and MICA*008, are often released in exosomal vesicles [70, 71], similarly to the tumor-associated antigens, HLA-I/ HLA-II molecules, “death receptor” ligands, and adhesion molecules [72]. Soluble and exosomal receptors bind to the corresponding sites on NK and T cells with subsequent internalization and degradation [73]. While various cellular stress signals can be recognized by the functional receptor, shedding ligands of one type is sufficient to make immune cells “blind” with respect to the entire family of ligands [66].
The level of stress-induced soluble ligands in the serum of cancer patients is regarded as a significant prognostic factor. An inverse correlation between the level of these ligands and the activity of NK and T cells, correlation with the cancer grade, and a negative effect on the patient survival were found [66].
CD47
CD47, or integrin-associated protein (IAP), is a transmembrane protein of the immunoglobulin superfamily. CD47, which is widely represented in different tissues of the adult organism, performs many functions: it regulates various processes, including apoptosis, proliferation, adhesion, and migration, as well as immune and angiogenic responses. CD47’s expression in tumor cells is significantly increased [74, 75].
Experiments with blocking antibodies showed that CD47 knockout inhibits migration and metastasizing of melanoma cells, as well as prostate and ovarian cancers [76]. In CD47-deficient mice, which are used as multiple myeloma models, the number of metastases into bones was less than in the control [77]. Similar results were obtained in mice with non-Hodgkin’s lymphoma xenografts: the blockade of the CD47 function with antibodies drastically reduced the number of metastases [78].
According to the results published in 2012, CD47 prevents the phagocytosis of cancer cells. The blockade of the interaction of CD47 with its receptor SIRPα (signal regulatory proteins) by anti-CD47 antibodies made it possible to slow down the progression of leukosarcoma in mice [79, 80]. Moreover, it was found that phagocytosis of cancer cells by macrophages, mediated by blocking anti-CD47 antibodies, may initiate the antitumor immune response of T cells (in particular, in tumors whose immunotherapy was previously ineffective) [81–83]. In addition, the binding of CD47 to SIRPα blocks maturation of the immature DCs and inhibits the synthesis of cytokines by the mature DCs, thus suppressing their antigen-presenting functions. The interaction between CD47 on endothelial cells and SIRP on leukocytes regulates the transendothelial migration of T cells. In mice with the CD47 knockout, the proportion of T cells, neutrophils, and monocytes in the inflammation sites is reduced [84]. CD47 may also decrease the cytotoxicity of NK cells in tumor cell cultures in vitro [85].
The important role of CD47 in the regulation of phagocytosis was shown not only in neoplastic diseases but also in the normal state. In particular, the interaction of CD47 with the soluble thrombospondin 1 (THBS1) and SIRPα on the surface of macrophages downregulates the phagocytosis of normal erythrocytes [86].
Thus, a high level of CD47 allows cancer cells to evade phagocytosis by macrophages during the interaction with SIRPα and suppresses the recognition by cytotoxic T cells and, possibly, NK cells. Currently, CD47 is considered as another target for obtaining hypoimmunogenic iPSC lines [87].
The main molecules that determine the development of immunological tolerance in health and disease are shown in Fig. 2.

Generalized immunological tolerance induction strategies used by tumor or normal cells. Immunological checkpoints CTLA-4 and PD-1L prevent T-cell activation. Suppression of HLA-I expression (e.g., due to mutations in b2m gene) hampers presentation of antigens (including tumor-associated antigens) to cytotoxic T cells. Soluble ligands MICA, MICB, and ULBP1-6 inhibit NK cell-mediated immune response. Due to interaction with ILT2 and KIR2DL4 receptors, HLA-G prevents activation of various effector cells, including T, B, and NK cells. Interaction between CD47 and SIRPα suppresses phagocytosis by macrophages and reduces cytotoxicity of NK cells. However, mechanism of interaction of CD47 with receptors on NK cells remains obscure.
PROSPECTS OF OBTAINING PSC LINES WITH REDUCED IMMUNOGENICITY
Genomic editing in PSCs is regarded as a strategy to minimize the immunogenicity of stem cells, which may contribute to the use of cell technologies in regenerative medicine, because the tissues differentiated from the stem cells that contain altered genes will also contain this modification. Probably, genome editing technology can be used to create a universal stem cell line, suitable for transplantation into any patient. For this purpose, the HLA complex genes or the genes required for their expression are suppressed or eliminated and the immunosuppressive molecules are induced. Possible strategies for preventing immune rejection are listed in detail in a recent review [88].
HLA-I expression on the surface of PSCs can be suppressed by inhibiting transcription or by deleting the genes encoding the heavy α-chain or the light chain (b2m). The inactivation of b2m, which is required to maintain the heterodimer conformation, disturbs the formation of the functional HLA-I complex. The cells that do not express HLA-I on their surface become invisible to the DCs and T cells of the recipient. Therefore, they should exhibit a reduced immunogenicity with respect to the allogeneic CD8+ T cells of the recipient. It should be noted that the cells that do not contain HLA-I molecules on their surface may become targets for NK cells. Although CD8+ T cells make a more significant contribution to a graft’s rejection than NK cells, the NK-mediated cell death may become a problem for the potential therapeutic use of iPSC lines and their differentiated derivatives with the b2m deletion.
The first attempt to derive pluripotent cell lines (hESCs) with a reduced immunogenicity was implemented in 2011. This attempt was based on the HLA-I knockdown with the use of small interfering RNAs (siRNA) and intracellular antibodies to inhibit the gene expression at the posttranscriptional and posttranslational levels [89]. HLA-I-knockdown human ESCs (ESCKD) were characterized by a reduced sensitivity to the cytotoxicity mediated by the CD3+ fraction of peripheral blood mononuclear cells in vitro. After transplantation into immunocompetent BALB/c mice, four of the ten teratomas formed by ESCKD survived for 42 days, whereas the control ESCs (six mice) were eliminated within 10 days. Despite the HLA-I knockdown, ESCKD did not initiate significant NK-cell activity, which may be due to a low expression of the stimulatory ligands of these cells. In 2013, to disturb HLA-A expression in the WIBR3 ESC line, nucleases with the zinc-finger motif (ZFN) were used [90]. However, because of the HLA-I heavy chain polymorphism, it is technically easier to edit the genes that are required for the expression of the HLA-I complex (e.g., the conserved b2m gene).
To inactivate both copies of the b2m gene in ESCs, adeno-associated viral vectors (AAVs) were also used [91]. The b2m–/– ESCs were derived using TALEN nucleases [92] and a homologous recombination [93]. The cell lines derived in these experiments did not express b2m and HLA-I on their surface, had a normal karyotype, retained the characteristic expression pattern of pluripotency markers, and were able to differentiate into the derivatives of the three germ layers in vitro and form teratomas in vivo. Transcriptome analysis showed that the gene expression pattern in the b2m–/– cell lines did not differ from the expression pattern in the original parental ESC line [91]. The peripheral blood mononuclear cells showed reduced reactivity against the embryoid bodies (EBs) derived from the b2m–/– ESCs in the MLR (mixed lymphocyte reaction) assay. In addition, the b2m–/– EBs that were cultured with the primed HLA-A*0201-CD8+ T cells expressed IFN-γ at lower levels than the control cell line. However, the incubation of b2m+/+ or b2m–/– EBs with NK cells did not lead to an enhanced expression of CD107a, an indicator of degranulation and cytotoxicity of NK cells. It is assumed that, in order to demonstrate the phenomenon of a “friend’s absence” recognition in the cells differentiated from ESCs, further differentiation in the hematopoietic direction is required.
Chinese researchers tested the immunogenicity of b2m–/– ESCs by coculturing them in vitro with the peripheral blood mononuclear cells [92]. Using the ELISPOT technique, they showed an increased IFN-γ secretion by the mononuclear cells in response to the wild-type ESCs compared to the b2m–/– ESCs. Hypoimmunogenicity in vivo was also shown: during the injection of b2m–/– ESCs into BALB/c mice, the infiltration of the implantation area with lymphocytes was significantly less pronounced than in the mice that were inoculated with the wild-type ESCs.
The cytotoxic activity of allogeneic CD8+ T cells against ESCs, B2M-null ESCs, and their derivatives at different ratios of the effector and target cells (E/T-effector/target) was analyzed in [93]. As expected, the B2M-null ESCs and their derivatives were tolerant to the cytotoxicity mediated by the CD8+ T-lymphocytes in vitro. It is known that the IFN-γ produced during the immune response can regulate the b2m expression, thereby facilitating rejection [15]. The survival assessment after the treatment with the proinflammatory cytokine showed that the control ESC line and its derivatives became more sensitive to the allogeneic CD8+ cells. However, IFN-γ had no effect on the tolerance of the B2M-null ESCs to the immune response. The sensitivity of the B2M-null ESCs to the response to NK cells was also tested. It was confirmed that the knockout ESC line and its derivatives, which do not carry HLA-I on their surface, are recognized and eliminated by the NK cells. To test the immunogenicity of B2M-null ESCs in vivo, cells were intramuscularly injected into the hind limbs of SCID mice. Teratomas were allowed to form for 10 weeks, after which they were extracted and analyzed. The size of the tumors derived from the B2M-null ESCs was significantly smaller than that of the tumors derived from the control ESCs. It was assumed that this might be due to the fact that SCID mice have functional NK cells, which could lyse the teratomas that did not express HLA-I. Next, the ability of B2M-null ESCs to form teratomas in SCID mice with a reduced component of the NK cell-mediated immunity was studied. As expected, the size of these tumors was comparable to that of the control teratomas formed from the ESCs in the SCID mice that had functional NK cells.
iPSC lines with biallelic knockout of the b2m gene were obtained in our laboratory. The fibroblast-like CD105+ derivatives exhibited an increased resistance to the allogeneic CD8+ T cells in vitro [94].
HLA-I-negative cells become sensitive to NK cell-mediated cytotoxicity, which is an important limitation of simply editing the b2m gene [93]. It is assumed that the NK-cell response can be reduced by inhibiting the activatory signal or expression of the soluble MHC homologue (MICA) [90, 93].
In addition, a potential defense against the NK-cell response may be the expression of the nonclassical HLA-I molecules—HLA-E and HLA-G. A decrease in the NK cell-mediated lysis of the transgenic ESC line ectopically expressing mutant mHLA-G and of the differentiated epidermal progenitors, unlike the control cell lines, was observed [95]. The recently performed HLA-E knock-in into the b2m locus in an ESC line allowed stable expression of a single-chain HLA-E without the expression of HLA-A, HLA-B, or HLA-C [96]. HLA-E is a ligand of the CD94/NKG2A inhibitory receptor. The resulting ESC line and its differentiated derivatives were not recognized by the allogeneic CD8+ T cells and were resistant to the NK cell-mediated lysis. HLA-E also increases the survival rate of in vivo differentiated CD45+ cells and teratomas by suppressing the cytotoxicity implemented by the NK-92 line expressing CD94/NKG2A.
In 2016, using the TALEN genome editing technology, TAP1 (transporter associated with antigen presentation 1) and TAPBP (TAP-associated glycoprotein) genes, which are involved in regulating the HLA-I expression, were deleted [97]. The HLA-I expression on the cell surface and the immunogenicity of the lines deficient in TAP1 and TAPBP was lower than in the control cell lines, whereas the normal karyotype and pluripotency were retained.
To ensure adequate protection against viruses, viral peptides should be presented in the HLA-I molecule complex. In view of this, the absence of HLA-I substantially increases the risk of infection of the allogeneic grafts derived from PSCs. This potential risk should be taken into account in the future, especially in the case of diseases associated with viral infection. Dimeric HLA-E molecules can provide some protection under these conditions, because they are able to present pathogen-specific peptides and, possibly, tumor peptides for subsequent recognition by the cytotoxic T cells [96]. It should be noted that NK cells are an important component of the antiviral and antitumor immune defense. Therefore, long-term systemic suppression of NK cells may significantly increase the risk of opportunistic infections and tumorigenesis.
Repression or deletion of HLA-II molecules to prevent the immune rejection reactions has attracted much less attention, because the HLA-II expression is limited to the professional APCs. However, allogeneic HLA-II molecules can induce the response of CD4+ T helpers and activate primarily B cells and macrophages. There are several known transcription factors that regulate the HLA-II expression, including RFX, X2BP, NFY, and CIITA. Using the TALEN genome editing technology, CIITA in ESCs was deleted [98]. The CIITA–/– ESC line retained the characteristics of a pluripotent cell line. Differentiated fibroblasts and DCs did not express HLA-II. The CIITA–/– T cells and DCs can be used in cell therapy because they are able to evade the CD4+ T-cell response of the recipient. However, the CIITA knockout alone is insufficient to prevent immune rejection. To ensure the maximum protection against the immune response, it is better to use the double knockout—CIITA and b2m. The first iPSC lines with the biallelic b2m and CIITA knockout were derived in December 2018 [99]. The b2m/CIITA–/– cardiomyocytes differentiated from iPSCs did not cause activation of T cells in vitro and, in contrast to the wild-type isogenic control, exhibited an increased resistance to the activity of peripheral blood mononuclear cells, including NK cells.
The possibility of using CD47 as an immunosuppressive agent was tested in [87]. For this purpose, the b2m–/– CIITA–/– iPSC lines expressing CD47 were derived. The formation of teratomas in the case of allogeneic transplantation of mouse b2m–/– CIITA–/–Cd47tg+ iPSCs was shown. It should be noted that the survival rate of the b2m–/– CIITA–/– iPSCs was higher than the corresponding control. Then, the immunogenicity of the differentiated derivatives of the modified line was analyzed. In the case of the allogeneic transplantation of b2m–/– CIITA–/– Cd47tg+ iPSCs into the mice with a functional immune system, the endothelial cells, smooth muscle cells, and cardiomyocytes did not cause any immune response and did not produce antibodies, whereas the wild-type differentiated derivatives induced an acute rejection of the graft. A similar approach was used to derive human b2m–/– CIITA–/– Cd47tg+ iPSCs with subsequent testing of the immune response on the humanized mice. The absence of cellular immune responses and production of antibodies was observed. It was concluded that CD47 alone is sufficient to “blind” the recipient’s immune system. It should be noted that the authors of the cited paper do not discuss how CD47 interacts with NK cells, because CD47 receptors (SIRPA (SIRPα, CD172a) and CD61) are not expressed on the NK cells [85, 100]. The duration of this immunotolerance has not yet been established. It remains unclear whether the immune response to the allogeneic b2m–/– CIITA–/– Cd47tg+ iPSCs will develop in a remote period (up to several months) or the proposed technique is sufficient and CD47 switches off the NK cells by another mechanism. To answer this question, further studies are required.
Other strategies to ensure immune tolerance to allogeneic grafts were also developed. The modified CP ESC line was obtained by increasing the CTLA4-Ig and PD-L1 expression [44]. Teratomas derived from the CP ESCs, as well as fibroblasts and cardiomyocytes were not rejected after being transplanted into humanized mice, in contrast to the control cell lines. Significantly less pronounced T-cell infiltration was detected in the grafts, which suggests the ability of the edited cell lines to create a local immunosuppressive niche. However, the immunogenicity of all the lines mentioned above should be tested in vivo.
A decrease in the immunogenicity of PSCs is a “sword of Damocles,” because hypoimmunogenicity can not only reduce the rejection of the cells derived from PSCs but also increase the risk of tumorigenesis. For example, the pluripotent stem cells that can remain in the graft among the differentiated cells, especially in the recipients receiving immunosuppressive drugs, may form teratomas. The residual pluripotent cells can be eliminated from the differentiated cell culture prior to transplantation by using cell sorting, density gradient centrifugation, or selection of antibiotics if the iPSCs were generated using the respective vectors. It should also be noted that PSCs and their derivatives with genetic aberrations may also form malignant tumors in model animals; however, such facts are reported very rarely [88].
The insertion of “suicide” cassettes into the PSC genome is a possible method for eliminating malignant neoplasms and teratomas. A group of Japanese researchers obtained an ESC line carrying the HSV-TK (herpes simplex virus–thymidine kinase) transgene under the promoter of the Oct4 gene, whose expression is characteristic only of pluripotent cells [101]. Selective elimination of undifferentiated ESCs expressing HSV-TK was achieved by the treatment of cells with ganciclovir. Another strategy to overcome the problem of tumorigenicity is the use of the suicide gene of inducible caspase 9 (iC9) [102]. iC9 is activated upon the interaction with the chemical activator of dimerization, which triggers the caspase cascade destroying iC9 iPSCs, their derivatives, and iC9 teratomas in vivo. It should be noted that 94–99% of the target cells were eliminated within 1 day, which makes iC9 a promising candidate for a suicide gene for a possible increase in the safety of PSC therapy.
CONCLUSIONS
The pathways and mechanisms of immunotolerance development considered in this review are quite diverse. Which of them is realized often depends on the tissue into which transplantation is performed or in which a tumor develops, and is determined by the local homeostasis of the signaling molecules and immune system cells. Suppression or inactivation of even one pathway responsible for immune surveillance may result in a significant reduction in the recognition by the T or NK cells or in a decrease in the production of cytotoxic antibodies by the B-cell system, which is initiated by the APCs. In deriving a universal immunotolerant line or lines of iPSCs, the most reasonable approach is to switch off all the main components that control tolerance. However, the high risk of tumorigenesis or increased proliferation of hypoimmunogenic iPSCs and their derivatives should not be disregarded. Approaches to reducing these risks only recently started being developed, and before using these “universal” lines in clinical practice, numerous studies of their biosafety and efficacy in animals, including those with a humanized immune system, should be performed.
REFERENCES
Thomson J.A., Itskovitz-Eldor J., Shapiro S.S., Waknitz M.A., Swiergiel J.J., Marshall V.S., Jones J.M. 1998. Embryonic stem cell lines derived from human blastocysts. Science. 282, 1145–1147.
Takahashi K., Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 126, 663–676.
Sayed N., Liu C., Wu J.C. 2016. Translation of human iPSCs: from clinical trial in a dish to precision medicine. J. Am. Coll. Cardiol. 67, 2161–2176.
Bogomazova A.N., Vasina E.M., Kiselev S.L., Lagarkova M.A., Lebedeva O.S., Nekrasov E.D., Panova A.V., Filonenko E.S., Khomyakova E.A., Tskhovrebova L.V., Chestkov I.V., Shutova M.V. 2015. Genetic cell reprogramming: A new technology for basic research and applied usage. Russ. J. Genet. 51 (4), 386–396.
Lebedeva O.S., Lagarkova M.A. 2018. Pluripotent stem cells for modelling and cell therapy of Parkinson’s disease. Biochemistry (Moscow). 83 (9), 1046–1056.
Kharitonov A.E., Surdina A.V., Lebedeva O.S., Bogomazova A.N., Lagarkova M.A. 2018. Possibilities of using pluripotent stem cells for restoring damaged retinal pigment epithelium. Acta Naturae. 10, 30–39.
Neofytou E., O’Brien C.G., Couture L.A, Wu J.C. 2015. Hurdles to clinical translation of human induced pluripotent stem cells. J. Clin. Invest. 125, 2551–2557.
Mandai M., Watanabe A., Kurimoto Y., Hirami Y., Morinaga C., Daimon T., Fujihara M., Akimaru H., Sakai N., Shibata Y., Terada M., Nomiya Y., Tanishima S., Nakamura M., Kamao H., et al. 2017. Autologous induced stem-cell–derived retinal cells for macular degeneration. N. Engl. J. Med. 376, 1038–1046.
Martin U. 2017. Therapeutic application of pluripotent stem cells: Challenges and risks. Front. Med. 4, 229.
Bolton E.M., Bradley J.A. 2015. Avoiding immunological rejection in regenerative medicine. Regen. Med. 10, 287–304.
de Almeida P.E., Ransohoff J.D., Nahid M.A., Wu J.C. 2013. Immunogenicity of pluripotent stem cells and their derivatives. Circulation Res. 112, 549–561.
Drukker M., Katchman H., Katz G., Even-Tov Friedman S., Shezen E., Hornstein E., Mandelboim O., Reisner Y., Benvenisty N. 2006. Human embryonic stem cells and their differentiated derivatives are less susceptible to immune rejection than adult cells. Stem Cells. 24, 221–229.
Swijnenburg R.J., Schrepfer S., Cao F., Pearl J.I., Xie X., Connolly A.J., Robbins R.C., Wu J.C. 2008. In vivo imaging of embryonic stem cells reveals patterns of survival and immune rejection following transplantation. Stem Cells Dev. 17, 1023–1029.
Jaffe L., Robertson E.J., Bikoff E.K. 1991. Distinct patterns of expression of MHC class I and beta 2-microglobulin transcripts at early stages of mouse development. J. Immunol. 147, 2740–2749.
Drukker M., Katz G., Urbach A., Schuldiner M., Markel G., Itskovitz-Eldor J., Reubinoff B., Mandelboim O., Benvenisty N. 2002. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc. Natl. Acad. Sci. U. S. A. 99, 9864–9869.
Perez-Cunningham J., Ames E., Smith R.C., Peter A.K., Naidu R., Nolta J.A., Murphy W.J. 2014. Natural killer cell subsets differentially reject embryonic stem cells based on licensing. Transplantation. 97, 992–998.
Li L., Baroja M.L., Majumdar A., Chadwick K., Rouleau A., Gallacher L., Ferber I., Lebkowski J., Martin T., Madrenas J., Bhatia M. 2004. Human embryonic stem cells possess immune-privileged properties. Stem Cells. 22, 448–456.
Dhodapkar K.M., Feldman D., Matthews P., Radfar S., Pickering R., Turkula S., Zebroski H., Dhodapkar M.V. 2010. Natural immunity to pluripotency antigen OCT4 in humans. Proc. Natl. Acad. Sci. U. S. A. 107, 8718–8723.
Robertson N.J., Brook F.A., Gardner R.L., Cobbold S.P., Waldmann H., Fairchild P.J. 2007. Embryonic stem cell-derived tissues are immunogenic but their inherent immune privilege promotes the induction of tolerance. Proc. Natl. Acad. Sci. U. S. A. 104, 20920–20925.
Zhao T., Zhang Z.N., Rong Z., Todorova D., Hu Z., Lin T., Rong Z., Kim J., He J., Wang M., Clegg D.O., Yang Y.G9. Zhang K., Friedlander M., Xu Y. 2011. Immunogenicity of induced pluripotent stem cells. Nature. 474, 212–215.
de Almeida P.E., Meyer E.H., Kooreman N.G., Diecke S., Dey D., Sanchez-Freire V., Hu S., Ebert A., Odegaard J., Mordwinkin N.M., Brouwer T.P., Lo D., Montoro D.T., Longaker M.T., Negrin R.S., Wu J.C. 2014. Transplanted terminally differentiated induced pluripotent stem cells are accepted by immune mechanisms similar to self-tolerance. Nat. Commun. 5, 3903.
Guha P., Morgan J.W., Mostoslavsky G., Rodrigues N.P., Boyd A.S. 2013. Lack of immune response to differentiated cells derived from syngeneic induced pluripotent stem cells. Cell Stem Cell. 12, 407–412.
Araki R., Uda M., Hoki Y., Sunayama M., Nakamura M., Ando S., Sugiura M., Ideno H., Shimada A., Nifuji A., Abe M. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature. 494, 100–104.
Zhao T., Zhang Z.N., Westenskow P.D., Todorova D., Hu Z., Lin T., Rong Z., Kim J., He J., Wang M., Clegg D.O., Yang Y.G., Zhang K., Friedlander M., Xu Y. 2015. Humanized mice reveal differential immunogenicity of cells derived from autologous induced pluripotent stem cells. Cell Stem Cell. 17, 353–359.
Kaneko S., Yamanaka S. 2013. To be immunogenic, or not to be: That’s the iPSC question. Cell Stem Cell. 12, 385–386.
Morizane A., Doi D., Kikuchi T., Okita K., Hotta A., Kawasaki T., Hayashi T., Onoe H., Shiina T., Yamanaka S., Takahashi J. 2013. Direct comparison of autologous and allogeneic transplantation of iPSC-derived neural cells in the brain of a nonhuman primate. Stem Cell Rep. 1, 283–292.
Polo J.M., Liu S., Figueroa M.E., Kulalert W., Eminli S., Tan K.Y., Apostolou E., Stadtfeld M., Li Y., Shioda T., Natesan S., Wagers A.J., Melnick A., Evans T., Hochedlinger K. 2010. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 28, 848–855.
Shutova M.V., Surdina A.V., Ischenko D.S., Naumov V.A., Bogomazova A.N., Vassina E.M., Alekseev D.G., Lagarkova M.A., Kiselev S.L. 2016. An integrative analysis of reprogramming in human isogenic system identified a clone selection criterion. Cell Cycle. 15, 986–997.
Wang X., Qin J., Zhao R.C., Zenke M. 2014. Reduced immunogenicity of induced pluripotent stem cells derived from Sertoli cells. PLoS One. 9, e106110.
Liu P., Chen S., Li X., Zenke M. 2013. Low immunogenicity of neural progenitor cells differentiated from induced pluripotent stem cells derived from less immunogenic somatic cells. PLoS One. 8, e69617.
Liu X., Li W., Fu X., Xu Y. 2017. The immunogenicity and immune tolerance of pluripotent stem cell derivatives. Front. Immunol. 8, 645.
Bhutani K., Nazor K.L., Williams R., Tran H., Dai H., Džakula Ž., Cho E.H., Pang A.W., Rao M., Cao H., Schork N.J., Loring J.F. 2016. Whole-genome mutational burden analysis of three pluripotency induction methods. Nat. Commun. 7, 10536.
Hanna J., Saha K., Jaenisch R. 2010. Pluripotency and cellular reprogramming: Facts, hypotheses, unresolved issues. Cell. 143, 508–525.
Robinson D.A., Daley G.Q. 2012. The promise of induced pluripotent stem cells in research and therapy. Nature. 481, 295–305.
Taylor C.J., Peacock S., Chaudhry A.N., Bradley J.A., Bolton E.M. 2012. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell. 11, 147–152.
Solomon S., Pitossi F., Rao M.S. 2015. Banking on iPSC—is it doable and is it worthwhile. Stem Cell Rev. 11, 1–10.
Nakatsuji N., Nakajima F., Tokunaga K. 2008. HLA-haplotype banking and iPS cells. Nat. Biotechnol. 26, 739–740.
Okita K., Matsumura Y., Sato Y., Okada A., Morizane A., Okamoto S., Hong H., Nakagawa M., Tanabe K., Tezuka K., Shibata T., Kunisada T., Takahashi M., Takahashi J., Saji H., Yamanaka S. 2011. A more efficient method to generate integration-free human iPS cells. Nat. Methods. 8, 409–412.
Gourraud P.-A., Gilson L., Girard M., Peschanski M. 2012. The role of human leukocyte antigen matching in the development of multiethnic “haplobank” of induced pluripotent stem cell lines. Stem Cells. 30, 180–186.
Moffett A., Colucci F. 2015. Co-evolution of NK receptors and HLA ligands in humans is driven by reproduction. Immunol. Rev. 267, 283–297.
Ichise H., Nagano S., Maeda T., Miyazaki M., Miyazaki Y., Kojima H., Yawata N., Yawata M., Tanaka H., Saji H., Masuda K., Kawamoto H. 2017. NK cell alloreactivity against KIR-ligand-mismatched HLA-haploidentical tissue derived from HLA haplotype-homozygous iPSCs. Stem Cell Rep. 9, 853–867.
Wood K.J., Goto R. 2012. Mechanisms of rejection: Current perspectives. Transplantation. 93, 1–10.
de Charette M., Houot R. 2018. Hide or defend, the two strategies of lymphoma immune evasion: Potential implications for immunotherapy. Haematologica. 103, 1256–1268.
Rong Z., Wang M., Hu Z., Stradner M., Zhu S., Kong H., Yi H., Goldrath A., Yang Y.G., Xu Y., Fu X. 2014. An effective approach to prevent immune rejection of human ESC-derived allografts. Cell Stem Cell. 14, 121–130.
Menter T., Tzankov A. 2018. Mechanisms of immune evasion and immune modulation by lymphoma cells. Front. Oncol. 8, 54.
Stewart T.J., Abrams S.I. 2008. How tumours escape mass destruction. Oncogene. 27, 5894–5903.
Garrido F., Aptsiauri N., Doorduijn E.M, Garcia Lora A.M., van Hall T. 2016. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 39, 44–51.
Garrido F., Cabrera T., Aptsiauri N. 2010. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int. J. Cancer. 127, 249–256.
Garrido C., Romero I., Berruguilla E., Cancela B., Algarra I., Collado A., García-Lora A., Garrido F. 2011. Immunotherapy eradicates metastases with reversible defects in MHC class I expression. Cancer Immunol. Immunother. 60, 1257–1268.
Angell T.E., Lechner M.G., Jang J.K., LoPresti J.S., Epstein A.L. 2014. MHC class I loss is a frequent mechanism of immune escape in papillary thyroid cancer that is reversed by interferon and selumetinib treatment in vitro. Clin. Cancer Res. 20, 6034–6044.
Li L., Dong M., Wang X.G. 2016. The implication and significance of beta 2 microglobulin: A conservative multifunctional regulator. Chin. Med. J. 129, 448–455.
Bernal M., Ruiz-Cabello F., Concha A., Paschen A., Garrido F. 2012. Implication of the β2-microglobulin gene in the generation of tumor escape phenotypes. Cancer Immunol. Immunother. 61, 1359–1371
Steidl C., Shah S.P., Woolcock B.W., Rui L., Kawahara M., Farinha P., Johnson N.A., Zhao Y., Telenius A., Neriah S.B., McPherson A., Meissner B., Okoye U.C., Diepstra A., van den Berg A., et al. 2011. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 471, 377–381.
Morandi F., Rizzo R., Fainardi E., Rouas-Freiss N., Pistoia V. 2016. Recent advances in our understanding of HLA-G biology: lessons from a wide spectrum of human diseases. J. Immunol. Res. 2016, 4326495.
Rouas-Freiss N., Moreau P., LeMaoult J., Carosella E.D. 2014. The dual role of HLA-G in cancer. J. Immunol. Res. 2014, 359748.
Le Bouteiller P. 2015. HLA-G in human early pregnancy: Control of uterine immune cell activation and likely vascular remodeling. Biomed. J. 38, 32–38.
González A., Rebmann V., LeMaoult J, Horn P.A., Carosella E.D., Alegre E. 2012. The immunosuppressive molecule HLA-G and its clinical implications. Crit. Rev. Clin. Lab. Sci. 49, 63–84.
Pistoia V., Morandi F., Wang X., Ferrone S. 2007. Soluble HLA-G: Are they clinically relevant? Semin. Cancer Biol. 17, 469–479.
Le Page M.E., Goodridge J.P., John E., Christiansen F.T., Witt C.S. 2014. Killer Ig-like receptor 2DL4 does not mediate NK cell IFN-γ responses to soluble HLA-G preparations. J. Immunol. 192, 732–740.
Fons P., Chabot S., Cartwright J.E., Lenfant F., L’Faqihi F., Giustiniani J., Herault J.P., Gueguen G., Bono F., Savi P., Aguerre-Girr M., Fournel S., Malecaze F., Bensussan A., Plouët J., Le Bouteiller P. 2006. Soluble HLA-G1 inhibits angiogenesis through an apoptotic pathway and by direct binding to CD160 receptor expressed by endothelial cells. Blood. 108, 2608–2615.
Nakajima H., Asai A., Okada A., Ping L., Hamajima F., Sata T., Isobe K. 2003. Transcriptional regulation of ILT family receptors. J. Immunol. 171, 6611–6620.
Ogasawara K., Lanier L.L. 2005. NKG2D in NK and T cell-mediated immunity. J. Clin Immunol. 25, 534–540.
Wu J., Song Y., Bakker A.B., Bauer S., Spies T., Lanier L.L., Phillips J.H. 1999. An activating immunoreceptor complex formed by NKG2D and DAP10. Science. 285, 730–732.
Verneris M.R., Karami M., Baker J., Jayaswal A., Negrin R.S. 2004. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood. 103, 3065–3072.
Zhang J., Basher F., Wu J.D. 2015. NKG2D ligands in tumor immunity: Two sides of a coin. Front Immunol. 6, 97.
Schmiedel D., Mandelboim O. 2018. NKG2D ligands: Critical targets for cancer immune escape and therapy. Front. Immunol. 9, 2040.
Salih H.R., Rammensee H.G., Steinle A. 2002. Cutting edge: Down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol. 169, 4098–4102.
Waldhauer I., Steinle A. 2006. Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res. 66, 2520–2526.
Salih H.R., Goehlsdorf D., Steinle A. 2006. Release of MICB molecules by tumor cells: Mechanism and soluble MICB in sera of cancer patients. Hum Immunol. 67, 188–195.
Fernandez-Messina L., Ashiru O., Boutet P., Agüera-González S., Skepper J.N., Reyburn H.T., Valés-Gómez M. 2010. Differential mechanisms of shedding of the glycosylphosphatidylinositol (GPI)-anchored NKG2D ligands. J. Biol. Chem. 285, 8543–8551.
Ashiru O., Boutet P., Fernandez-Messina L., Agüera-González S., Skepper J.N., Valés-Gómez M., Reyburn H.T. 2010. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res. 70, 481–489.
Whiteside T.L. 2013. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 41, 245–251.
Groh V., Wu J, Yee C, Spies T. 2002. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 419, 734–738.
Sick E., Jeanne A., Schneider C., Dedieu S., Takeda K., Martiny L. 2012. CD47 update: A multifaceted actor in the tumour microenvironment of potential therapeutic interest. Br. J. Pharmacol. 167, 1415–1430.
Chao M.P, Weissman I.L, Majeti R. 2012). The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 24, 225–232.
Shahan T.A, Fawzi A., Bellon G., Monboisse J.C, Kefalides N.A. 2000. Regulation of tumor cell chemotaxis by type IV collagen is mediated by a Ca(2+)-dependent mechanism requiring CD47 and the integrin alpha(V)beta(3)". J. Biol. Chem. 2757, 4796–4802.
Uluçkan O., Becker S.N., Deng H., Zou W., Prior J.L., Piwnica-Worms D., Frazier W.A., Weilbaecher K.N. 2009. CD47 regulates bone mass and tumor metastasis to bone. Cancer Res. 69, 3196–3204.
Chao M.P., Tang C., Pachynski R.K., Chin R., Majeti R., Weissman I.L. 2011. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood. 118, 4890–4901.
Edris B., Weiskopf K., Volkmer A.K, Volkmer J.P., Willingham S.B., Contreras-Trujillo H., Liu J., Majeti R., West R.B., Fletcher J.A., Beck A.H., Weissman I.L., van de Rijn M. 2012. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. U. S. A. 109, 6656–6661.
Willingham S.B., Volkmer J.P., Gentles A.J., Sahoo D., Dalerba P., Mitra S.S., Wang J., Contreras-Trujillo H., Martin R., Cohen J.D., Lovelace P., Scheeren F.A., Chao M.P., Weiskopf K., Tang C., et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. U. S. A. 109, 6662–6667.
Tseng D., Volkmer J.P., Willingham S.B., Contreras-Trujillo H., Fathman J.W., Fernhoff N.B., Seita J., Inlay M.A., Weiskopf K., Miyanishi M., Weissman I.L. 2013. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. U. S. A. 110, 11103–11108.
Unanue E. R. 2013. Perspectives on anti-CD47 antibody treatment for experimental cancer. Proc. Nat. Acad. Sci. U. S. A. 110, 10886–10887.
Matlung H.L, Szilagyi K., Barclay N.A, van den Berg T.K. 2017. The CD47-SIRP alpha signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 276, 145–164.
Azcutia V., Stefanidakis M., Tsuboi N., Mayadas T., Croce K.J., Fukuda D., Aikawa M., Newton G., Luscinskas F.W. 2012. Endothelial CD47 promotes vascular endothelial-cadherin tyrosine phosphorylation and participates in T cell recruitment at sites of inflammation in vivo. J. Immunol. 189, 2553–2562.
Kim M.J., Lee J.C, Lee J.J, Kim S., Lee S.G., Park S.W., Sung M.W., Heo D.S. 2008. Association of CD47 with natural killer cell-mediated cytotoxicity of head-and-neck squamous cell carcinoma lines. Tumor Biol. 29, 28–34.
Soto-Pantoja D.R, Terabe M., Ghosh A., Ridnour L.A., DeGraff W.G., Wink D.A., Berzofsky J.A., Roberts D.D. 2014. CD47 in the tumor microenvironment limits cooperation between antitumor T-cell immunity and radiotherapy. Cancer Res. 74, 6771–6783.
Deuse T., Hu X., Gravina A., Wang D., Tediashvili G., De C., Thayer W.O., Wahl A., Garcia J.V., Reichenspurner H., Davis M.M., Lanier L.L., Schrepfer S. 2019. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 37 (3), 252‒258.
Zheng D., Wang X., Xu R.-H. 2016. Concise review: One stone for multiple birds: generating universally compatible human embryonic stem cells. Stem Cells. 34, 2269–2275.
Deuse T., Seifert M., Phillips N., Tsao P.S., Hua X., Velden J., Eiermann T., Volk H.D., Reichenspurner H., Robbins R.C., Schrepfer S. 2011. Immunobiology of naïve and genetically modified HLA-class-I-knockdown human embryonic stem cells. J. Cell Sci. 124, 3029–3037.
Torikai H., Reik A., Soldner F., Warren E.H., Yuen C., Zhou Y., Crossland D.L., Huls H., Littman N., Zhang Z., Tykodi S.S., Kebriaei P., Lee D.A., Miller J.C., Rebar E.J., et al. 2013. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. 122, 1341–1349.
Riolobos L., Hirata R.K., Turtle C.J., Wang P.R., Gornalusse G.G., Zavajlevski M., Riddell S.R., Russell D.W. 2013. HLA engineering of human pluripotent stem cells. Mol. Ther. 21, 1232–1241.
Lu P., Chen J., He L., Ren J., Chen H., Rao L., Zhuang Q., Li H., Li L., Bao L., He J., Zhang W., Zhu F., Cui C., Xiao L. 2013. Generating hypoimmunogenic human embryonic stem cells by the disruption of beta 2-microglobulin. Stem Cell Rev. 9, 806–813.
Wang D., Quan Y., Yan Q., Morales J.E., Wetsel R.A. 2015. Targeted disruption of the β2-microglobulin gene minimizes the immunogenicity of human embryonic stem cells. Stem Cells Transl. Med. 4, 1234–1245.
Bogomiakova M.E., Bobrovsky P.A., Zhukova Y.N., Lazarev V.N., Lagarkova M.A. 2018. Derivation and characterization of induced pluripotent stem cells lines with inactivation of the beta-2-microglobulin gene by CRISPR/Cas9 genome editing. FEBS Open Biol. 8, 152–153.
Zhao L., Teklemariam T., Hantash B.M. 2014. Heterologous expression of mutated HLA-G decreases immunogenicity of human embryonic stem cells and their epidermal derivatives. Stem Cell Res. 13, 342–354.
Gornalusse G.G., Hirata R.K., Funk S., Riolobos L., Lopes V.S., Manske G., Prunkard D., Colunga A.G., Hanafi L.A., Clegg D.O., Turtle C., Russell D.W. 2017. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 35, 765–772.
Cui D., Wang J., Zeng Y., Rao L., Chen H., Li W., Li Y., Li H., Cui C., Xiao L. 2016. Generating hESCs with reduced immunogenicity by disrupting TAP1 or TAPBP. Biosci. Biotechnol. Biochem. 80, 1484–1491.
Chen H., Li Y., Lin X., Cui D., Cui C., Li H., Xiao L. 2015. Functional disruption of human leukocyte antigen II in human embryonic stem cell. Biol. Res. 48, 59.
Matapally S., Pawlik K.M, Fast V.G, Zumaquero E., Lund F.E., Randall T.D., Townes T.M., Zhang J. 2018. Human leukocyte antigen class I and II knockout human induced pluripotent stem cell-derived cells: Universal donor for cell therapy. J. Am. Heart Assoc. 7, e010239.
Nath P.R., Gangaplara A., Pal-Nath D., Mandal A., Maric D., Sipes J.M., Cam M., Shevach E.M., Roberts D.D. 2018. CD47 expression in natural killer cells regulates homeostasis and modulates immune response to lymphocytic choriomeningitis virus. Front Immunol. 9, 2985.
Hara A., Aoki H., Taguchi A., Niwa M., Yamada Y., Kunisada T., Mori H. 2008. Neuron-like differentiation and selective ablation of undifferentiated embryonic stem cells containing suicide gene with Oct-4 promoter. Stem Cells Dev. 17, 619–627.
Yagyu S., Hoyos V., Del Bufalo F., Brenner M.K. 2015. An inducible caspase-9 suicide gene to improve the safety of therapy using human induced pluripotent stem cells. Mol. Ther. 23, 1475–1485.
Funding
This work was supported by the Russian Science Foundation (project no. 17-75-10206).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Translated by M. Batrukova
Abbreviations: APCs, antigen-presenting cells; DCs, dendritic cells; IPSCs, induced pluripotent stem cells; NHL, non-Hodgkin’s lymphoma; PSCs, pluripotent stem cells; RPE, retinal pigment epithelium; ESCs, embryonic stem cells; b2m, beta-2 microglobulin; EBs, embryoid bodies; CD, cluster of differentiation; CIITA, class II major histocompatibility complex transactivator; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; HLA, human leukocyte antigen; IAP, integrin associated protein; IFN-γ, interferon γ; ILT, immunoglobulin-like transcript; KIR, killer cell immunoglobulin-like receptor; MHC, major histocompatibility complex; NK cells, natural killer cells; NKG2D, NK group 2 member D; PD-1, programmed cell death 1; PD-L1, programmed cell death-ligand 1; SCID, severe combined immunodeficiency; TALEN, transcription activator-like effector nucleases; TAP1, transporter associated with antigen presentation 1; TAPBP, TAP-associated glycoprotein); TCR, T-cell receptor; TGFβ, transforming growth factor β; ULBP, UL-16 binding protein.
Rights and permissions
About this article

Cite this article
Bogomiakova, M.E., Eremeev, A.V. & Lagarkova, M.A. At Home among Strangers: Is It Possible to Create Hypoimmunogenic Pluripotent Stem Cell Lines?. Mol Biol 53, 638–652 (2019). https://doi.org/10.1134/S0026893319050042
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026893319050042