
Abstract
In this study, a cocatalyst is supported on the surface of carbon nitride by the following three different methods: thermal phosphorylation, a hydrothermal method, and photochemical reduction. In the first case, the cocatalyst is a cobalt phosphide–cobalt orthophosphate mixture; the use of the hydrothermal and photochemical synthesis methods provides the formation of cobalt orthophosphate. In addition, a sample is synthesized by a hydrothermal treatment of presynthesized carbon nitride and cobalt phosphide. The synthesized samples are studied using the following set of physicochemical methods: X-ray diffraction (XRD) analysis, transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), and diffuse reflectance spectroscopy. All the synthesized photocatalysts are tested in photocatalytic hydrogen evolution from an aqueous alkaline solution of triethanolamine and in a photoelectrochemical cell containing a sodium polysulfide solution. It is shown that, in the case of using a cobalt phosphide–cobalt orthophosphate mixture, the target characteristics are higher than those in the case of cobalt orthophosphate and carbon nitride. The highest catalytic activity is 156 μmol h−1 g−1; it is comparable to the activity of platinized carbon nitride. The highest short-circuit current density obtained in the case of using a photoelectrode containing cobalt phosphide and cobalt phosphate is 2.4 mA/cm2.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
An important task of modern alternative power engineering is the development of materials that provide an effective conversion of the solar radiation energy into other types of energy [1]. This direction is fairly promising, because solar energy is several orders of magnitude higher than the energy demands of mankind. Currently, the following two scientific directions are being extensively developed: (i) photocatalysis [1–5], which provides the production of chemical compounds, such as hydrogen and energy-intensive fuels, and (ii) electric current generation in solar cells and photoelectrochemical cells [1].
The materials that are commonly used to convert solar radiation energy are semiconductors. The first types of catalysts used for photocatalytic hydrogen evolution under the action of visible light were systems based on titania and titanates sensitized by various methods [6] and transition metal chalcogenides [7–9]. In the 2000s, an extensive study of a group of carbon-containing materials for photocatalytic and photoelectrochemical applications began. Particular attention was paid to graphitic carbon nitride. This material is thermally stable and resistant to acids and alkalis. In addition, the position of the conduction band suggests that this sample exhibits strong reducibility [10, 11]. However, the practical application of carbon nitride, in common with that of many other semiconductors, is limited to the high recombination rate of photogenerated electrons and holes. To decrease the recombination rate of charge carriers and increase the catalytic activity of g-C3N4, modifying additives are deposited on the surface of photocatalysts. The most effective of the additives are platinum group metals [12], the use of which is limited to the high cost of the reagents. An equivalent substitute for these metals can be substances with a metal-like structure, among which special attention is paid to transition metal phosphides, for example, cobalt phosphide [13]. It is known that compounds containing phosphorus in higher oxidation states, such as cobalt orthophosphate [14] or cobalt hydroxophosphate [15], also have the ability to increase photocatalytic activity. The following questions naturally arise: which of these cobalt phosphorus salts makes it possible to most efficiently convert light energy into electricity or chemical bond energy? Will the catalytic activity change, if cobalt phosphide and cobalt orthophosphate are deposited on the surface of a photocatalyst simultaneously? Will the laws governing photocatalytic hydrogen evolution be preserved in the case of electric current generation? This study is focused on the exploration of these questions.
EXPERIMENTAL
Photocatalyst Synthesis Procedure
Carbon nitride was synthesized by the thermal polycondensation of melamine at 600°C for 2 h [16]. A cocatalyst based on cobalt phosphide or cobalt phosphate was deposited on the surface of carbon nitride by the following three different methods: thermal phosphorylation [17], a hydrothermal method [18], and photochemical reduction [19]. In the first case, 1.03 wt % of cobalt hydroxide was predeposited on the surface of graphitic carbon nitride by the precipitation method. To this end, 396 mg of carbon nitride was dispersed in 10 mL of distilled water; 444 μL of a 0.1 M cobalt chloride solution was added dropwise; the resulting mixture was stirred for 15 min; after that, 888 μL of a 0.1 M sodium hydroxide solution was added dropwise; the mixture was stirred for 20 min; the precipitate was washed by centrifugation and dried at 50°C. A mixture consisting of 400 mg of 1.03%Co(OH)2/g-C3N4 and 2 g of sodium hypophosphite was heated in a nitrogen atmosphere to 300°С at a rate of 2°С/min, held at this temperature for 1 h, and cooled to room temperature. The resulting mixture was washed several times and dried at a temperature of 50°C. Hereinafter in the text, the sample synthesized by this method is designated as Co(TP)/g-C3N4. For comparison, cobalt phosphide Co(TP) was synthesized from cobalt hydroxide under the same conditions. In accordance with the hydrothermal method, a suspension consisting of 10 mL of water, 400 mg of carbon nitride, and calculated amounts of a 0.1 M cobalt chloride solution and a red phosphorus powder was stirred on a magnetic stirrer for 30 min. The resulting suspension was placed in an autoclave (filling factor of 0.8) and heated at 200°С for 10 h. After that, the suspension was washed several times and dried at a temperature of 50°C. The sample synthesized by this method is designated as Co(HT)/g-C3N4. In the case of the photochemical reduction method, a suspension consisting of 40 mL of an aqueous alcohol solution (10 vol % of ethanol), 400 mg of carbon nitride, and calculated amounts of 0.1 M cobalt chloride and sodium hypophosphite solutions was placed in a reactor, purged with argon until the complete removal of oxygen, and illuminated with a light-emitting diode at a wavelength of 380 nm for 3 h. The resulting suspension was washed several times and dried at a temperature of 50°C. Hereinafter in the text, the sample synthesized by this method is designated as Co(PR)/g-C3N4. For comparison, cobalt phosphide Co(PR) was synthesized under the same conditions from presynthesized cobalt(II) hydroxide and sodium hypophosphite. The Co(TP)/g-C3N4(HT) composite was synthesized by autoclaving a suspension consisting of 396 mg of g‑C3N4 and 4 mg of Co(TP), which was subjected to a 30-min pretreatment in an ultrasonic bath, at 120°С for 2 h. The resulting suspension was washed several times and dried at a temperature of 50°С [17]. For comparison, a 1%Pt/g-C3N4 photocatalyst was synthesized by the chemical reduction method [16].
Physicochemical Property Investigation Procedure
The phase composition of the synthesized catalysts was studied on a Bruker D8 X-ray diffractometer (Germany) using CuKα copper radiation at a wavelength of λ = 1.5418 Å. X-ray diffraction patterns were recorded in an angular range of 2θ = 15°–65° by point-by-point scanning in increments of 0.05°; the acquisition time was 10 s per point. The size of the coherent scattering region (CSR) of the samples was determined using the Scherrer equation. The diffuse reflectance spectra of the samples were recorded on a Lambda 35 spectrophotometer (PerkinElmer, United States) equipped with an RSA-PE-20 diffuse reflectance attachment (Labsphere, United States). The reference sample was barium sulfate. The microstructure of the photocatalysts was studied by transmission electron microscopy (TEM) using a ThemisZ microscope (Thermo Fisher Scientific, United States) at an accelerating voltage of 200 kV. The micrographs were recorded using a Ceta 16 CCD sensor (Thermo Fisher Scientific, United States). The chemical composition of the catalysts was studied by X-ray photoelectron spectroscopy (XPS) on a SPECS Surface Nano Analysis GmbH electronic spectrometer (Germany). The spectrometer is equipped with a PHOIBOS-150-MCD-9 hemispherical analyzer and an XR-50 source of characteristic X-ray radiation with an Al/Mg twin anode. The spectra were recorded using nonmonochromatized AlKα radiation (hν = 1486.61 eV). The effect of sample charging was taken into account using the position of the peak corresponding to the carrier, i.e., carbon, in the C3N4 structure (E(C1s) = 288.1 eV). Relative concentrations of elements in the analysis zone were determined from the integral intensities of the XPS peaks taking into account the photoionization cross section of the respective terms [20]. For a detailed analysis, the spectra were decomposed into individual components. After subtracting the background by the Shirley method [21], the experimental curve was decomposed into a series of lines corresponding to the photoemission of electrons from atoms in various chemical environments. The data were processed using the CasaXPS software package [22].
Catalytic Activity Measurement Procedure
The catalytic activity of the synthesized photocatalysts was measured in a static reactor under illumination with a light-emitting diode at a wavelength of 450 nm as described earlier [16]. Ten milliliters of triethanolamine, 90 mL of a 0.11 M sodium hydroxide solution, and 50 mg of a photocatalyst were placed into the reactor. Before reaction, the reactor was purged with argon for about 30 min to remove oxygen; after that, illumination was turned on. The amount of evolved hydrogen was measured on a gas chromatograph (Khromos, Russia).
Photoelectrochemical Property Investigation Procedure
The photoelectrochemical characteristics of the samples were studied in a two-electrode cell [23]. The working electrode was the studied photoelectrode; the counter electrode was brass with a predeposited copper(I) sulfide layer. The working photoelectrodes were prepared by the drop-casting method [20]. Thirty milligrams of a photocatalyst of an appropriate composition was added to 600 μL of ethanol and treated in an ultrasonic bath for 6 min. The resulting suspension was dripped onto conductive FTO glass until the glass surface was completely covered with the solution and then dried at 80°C. The counter electrode was prepared by treating brass with concentrated hydrochloric acid at 70°C for 30 min, rinsing with distilled water, and immersing into a 1 M aqueous solution of sodium sulfide for 15 min [25]. A 1 M aqueous solution of sodium sulfide with dissolved sulfur (1 M) and sodium chloride (0.1 M) was used as the electrolytes. The electrodes were compressed to optimize the contact. Photovoltaic characteristics were determined on a P-45X potentiostat–galvanostat (Russia) equipped with a FRA-24M electrochemical impedance measurement module in a voltage range of −0.8 to +0.8 V; the potential sweep rate was 0.02 V/s. The time dependences of photocurrent density were recorded at a potential of 0 V. Impedance spectroscopy measurements were conducted in a frequency range of 0.8–105 Hz at an amplitude of 10 mV and a constant potential value of 0.2 V. The Mott–Schottky dependences were studied in a voltage range of −0.6 to 0.6 V at an amplitude of 10 mV and a constant frequency of 1000 Hz. The illumination source was a light-emitting diode at a wavelength of 450 nm and a radiation power of 19.2 mW/cm2. The area of the illuminated surface in the cell was 1 cm2.
RESULTS AND DISCUSSION
Physicochemical Properties of the Synthesized Catalysts
The phase composition of all the synthesized catalysts was studied by XRD (Fig. 1). It is evident from Fig. 1a that the X-ray diffraction patterns of all the composite photocatalysts exhibit peaks at ~13° and ~27°, which are characteristic of the (100) and (200) faces of carbon nitride. Since graphitic carbon nitride is a layered material with an ordered two-dimensional (2D) structure, it is characterized by calculating the CSR size in the following two directions: in the plane of 2D La layers (in fact, it shows the distance between tri-s-triazine units [16, 26]) and perpendicular to the 2D layers (shows distance Lc between the material layers). These parameters were calculated using the Scherrer equation; they are listed in Table 1. Note that the deposition of a cocatalyst on the carbon nitride surface leads to a change in the characteristic CSR size in the plane of the La layers, and the higher the synthesis temperature, the more significant the increase in this parameter. For example, in terms of the photochemical reduction method, the reaction occurs at room temperature, and the characteristic size of La remains almost constant. An increase in the synthesis temperature to 200°C (Co(HT)/g-C3N4 sample) leads to an increase in La by 1 nm compared with the value for unmodified carbon nitride. With an increase in the synthesis temperature to 300°C, at which thermal phosphorylation was run (Co(TP)/g-C3N4 sample), the La distance increases to 6.3 nm. At the same time, the temperature regime for the synthesis of cobalt phosphides has hardly any effect on the interlayer spacing in the carbon nitride structure: the Lc parameter is 8–9 nm for the composite photocatalysts on which the cocatalyst was deposited. A different tendency is observed in the case of the Co(TP)/g-C3N4(HT) sample, which was synthesized by subjecting the presynthesized cobalt phosphide and carbon nitride to dispersion and a hydrothermal treatment to provide the formation of an interfacial contact. In this case, the La and Lc distances are almost 2 nm higher than the respective parameters for the original carbon nitride.

X-ray diffraction patterns of photocatalysts: (a) g-C3N4, Co(TP)/g-C3N4, Co(PR)/g-C3N4, Co(HT)/g-C3N4, and Co(TP)/g-C3N4(HT); (b) Co(TP)/g-C3N4 in a range of 2θ = 40°–60°; and (c) Co(TP) and Co(PR).
According to the data shown in Figs. 1a and 1b, the peaks of the phases corresponding to cobalt phosphide were detected only for the sample synthesized by the thermal phosphorylation method. Apparently, this fact can be attributed to the small amount of the deposited component or the high degree of dispersion of the component. To suggest the composition of phosphides that can be formed by the thermal method and photochemical reduction, the synthesis procedure was reproduced in the absence of carbon nitride. X-ray diffraction patterns of the Co(TP) and Co(PR) samples are shown in Fig. 1c. Qualitative analysis showed that, in the case of using the thermal method, it is reasonable to expect the formation of a mixture of phosphides CoP (PDF 01-089-4862) and Co2P (PDF 04-007-15-24) and cobalt oxide (PDF 00-002-0770), which is formed during the calcination of cobalt hydroxide. In the case of photochemical reduction, only peaks of cobalt hydroxide were detected; this fact confirms the necessity for the presence of a photocatalyst in the synthesis of phosphides. Comparison of the estimated results with data obtained by other methods and analysis of the mechanisms of formation of phosphides and reaction byproducts in terms of the different techniques will be described below.
The photocatalysts with deposited components were studied by XPS; the spectra are shown in Figs. 2a–2f. It is evident from Fig. 2a that the spectrum of carbon is adequately described by two peaks with binding energies in the region of 284.6–285.1 and 288.1 eV. The first peak is characteristic of carbon-containing impurities present on the surface of photocatalysts, for example, due to the adsorption of carbon dioxide from the atmosphere. The second peak corresponds to carbon that forms bonds with nitrogen atoms in the structure [27, 28]. The N1s spectrum shown in Fig. 2b exhibits a peak at a binding energy in the region of 398.6 eV, which is attributed to the nitrogen atom that forms the C–N=C bond, a peak at 400.0 eV corresponding to the nitrogen atom that forms a bond with three carbon atoms (N–(C)3), and a peak at 401.0 eV, which confirms the presence of terminal N–H groups [27, 28]. The ratio of carbon-to-nitrogen atom concentrations on the surface of the samples is nearly stoichiometric (Table 1). Thus, the use of the XPS method unambiguously confirms the presence of carbon nitride in the studied photocatalysts.

X-ray photoelectron spectra of photocatalysts: (a) the C1s and (b) N1s lines of g-C3N4, Co(TP)/g-C3N4, Co(PR)/g-C3N4, and Co(HT)/g-C3N4; (c) the Co2p and (d) P2p lines of Co(TP) and Co(PR); (e) the Co2p3/2 lines of Co(TP)/g-C3N4, Co(PR)/g-C3N4, and Co(HT)/g-C3N4; and (f) the P2p lines of Co(PR)/g-C3N4 and Co(HT)/g-C3N4.
In addition, the spectra of cobalt and phosphorus for the Co(TP) and Co(PR) reference samples synthesized by the thermal phosphorylation and photochemical reduction method in the absence of carbon nitride were analyzed. Figure 2c shows that the spectrum of cobalt for the sample synthesized by photochemical reduction can be described by two doublets at a binding energy of the Co2p3/2 peaks in the region of 780.1 and 780.9 eV and the respective “shake-up” satellites in the region of 785.5 and 784.5 eV. The two doublets are attributed to cobalt in the +2 oxidation state. The first doublet corresponds to cobalt contained in cobalt phosphate (6%); the second doublet corresponds to cobalt oxide or hydroxide (94%). The spectrum of phosphorus for the Co(PR) sample exhibits a single peak at a binding energy of 133.5 eV, which is characteristic of the phosphate ion [29–31]. Thus, in the absence of a support, only cobalt phosphate can be synthesized by the photochemical reduction method. A different picture is observed for the Co(TP) sample synthesized by the thermal phosphorylation method. Figure 2c shows that the spectrum of cobalt for this sample is a superposition of two peaks. The first peak has a binding energy of 778.9 eV and corresponds to cobalt in the form of a phosphide [32–35]. The binding energy for the second peak with satellites is 781.8 eV; therefore, it can be attributed to the divalent state of cobalt in the form of a phosphate [36, 37]. The atomic ratio of cobalt in the form of a phosphide and a phosphate is 1 : 4. The presence of a cobalt phosphide–cobalt phosphate mixture in the sample is confirmed by the data shown in Fig. 2d.
The Co(TP)/g-C3N4, Co(HT)/g-C3N4, and Co(PR)/g-C3N4 composite catalysts were studied by XPS (Figs. 2e, 2f). For all the studied samples, the Co2p3/2 spectrum is described by a broad peak in the region of 780.8–781.2 eV, which is characteristic of cobalt contained in cobalt oxide and/or cobalt phosphate [32, 33, 35]. For the Co(PR)/g-C3N4 sample, the intensity of the peaks of phosphorus is below the XPS detection limit. In the case of the Co(HT)/g-C3N4 catalyst, phosphorus presents in the form of phosphates. For the Co(TP)/g-C3N4 sample, an extra peak corresponding to cobalt phosphide is identified (Fig. 2f). Thus, in the case of synthesizing photocatalysts by the hydrothermal method and photochemical reduction, cobalt phosphate is formed on the surface of carbon nitride. In the case of thermal phosphorylation, a cobalt phosphate–cobalt phosphide mixture can be formed. It should be noted that, the samples synthesized by the different methods are characterized by different ratios of cobalt-to-carbon atomic surface concentrations (Table 1). Taking into account the fact that the bulk quantity of cocatalysts is identical in all the samples, the above can indicate that the resulting particles have different sizes.
The Co(TP)/g-C3N4 sample was further studied by the TEM method; the images are shown in Fig. 3. It is evident from Fig. 3a that the photocatalyst consists of particles of several types. The first type is carbon nitride support particles, as evidenced by the distribution of carbon and nitrogen atoms over the sample surface (Figs. 3b, 3c). The support particles are stacked in several layers. On the carbon nitride surface, spherical particles with a size of 20–40 nm and cylindrical agglomerates with a size of up to 100–200 nm are detected. To determine the chemical nature of these agglomerates, maps of element distribution over the sample surface were constructed. The cobalt (Fig. 3d) and oxygen atoms (Fig. 3f) are uniformly distributed over the carbon nitride surface. Phosphorus presents in the form of individual clusters of small groups of atoms and agglomerates with a size of 20–150 nm (Fig. 3e). Comparison of the images in Figs. 3d–3f reveals regions where cobalt, phosphorus, and oxygen atoms; cobalt and phosphorus atoms; and phosphorus and oxygen atoms are simultaneously present. This fact confirms the presence of cobalt phosphide and cobalt orthophosphate on the surface. It should be noted that the amount of surface phosphorus is higher than the cobalt concentration. Similar results were obtained by XPS: [P]/[Co] = 47.7. Apparently, a portion of phosphorus remains on the surface after washing the catalyst synthesized in excess sodium hypophosphite.

(a) Transmission electron microscopy image of the Co(TP)/g-C3N4 sample and (b–f) elemental mapping of the TEM image of the Co(TP)/g-C3N4 sample.
The optical properties of the synthesized photocatalysts were studied by diffuse reflectance spectroscopy (Fig. 4). The general shape of the spectra is similar for all the samples, except for Co(TP)/g-C3N4. This fact can be attributed to the presence of cobalt phosphide in the sample. The reflection spectra were used to calculate the Tauc function and determine the absorption edges; the data are summarized in Table 1. The absorption edge of carbon nitride is 444 nm. Upon the deposition of cocatalysts on the sample surface, this parameter is shifted by less than 10 nm. Typically, the shift of the absorption edge of the samples is associated with the transfer of electron density from the carbon nitride to the cobalt phosphide or cobalt phosphate particles. It should be noted that all the synthesized catalysts absorb visible light and are potentially suitable for use in photocatalytic and photovoltaic processes.

Diffuse reflectance spectra of photocatalysts based on carbon nitride.
Cocatalyst Synthesis Mechanism in Terms of the Studied Methods
In this study, the following three different methods for the formation of cobalt salts on the surface of carbon nitride were studied: thermal phosphorylation, photochemical reduction, and the hydrothermal method. According to the published data, thermal phosphorylation is based on the following two sequential processes: the decomposition of sodium hypophosphite to form phosphine (in this case, the solid-phase decomposition product is various salts of oxygen-containing phosphoric acids [17, 38], Eqs. (I)–(III)) and the reaction of phosphine with cobalt hydroxide (Eq. (IV)). The process occurs at 300°C; this factor eliminates the occurrence of solid-phase transformations:
According to XRD, in the case of the Co(TP) sample synthesized by the above method, a mixture of cobalt phosphides is actually formed. However, the results of the XPS studies of Co(TP) show the presence of a cobalt phosphide–cobalt phosphate mixture on the surface. Apparently, during the storage of the photocatalysts, fine cobalt phosphide particles are oxidized by atmospheric oxygen.
In terms of hydrothermal synthesis, a reaction of phosphorus, water, and cobalt hydroxide takes place. Under autoclaving conditions, phosphorus disproportionation (Eq. (V)) and the subsequent reaction of the disproportionation products with cobalt hydroxide can occur (Eqs. (IV), (VI)). It should be noted that a byproduct of the disproportionation is phosphoric acid; as a consequence, cobalt phosphate is formed in the system. X-ray photoelectron spectroscopy analysis of the Co(HT)/g-C3N4 sample showed that only cobalt orthophosphate is presented on the surface. Apparently, the reaction of phosphine with cobalt hydroxide occurs more slowly than the formation of phosphate does or the resulting cobalt phosphide particles are oxidized by atmospheric oxygen during storage. To increase the phosphide yield in terms of this method, phosphorus precursors of a more complex composition should be apparently used:
The cocatalyst synthesis method based on the photochemical reduction of precursors with electrons formed upon the illumination of a semiconductor has been effectively used earlier to synthesize particles of noble metals [39]. The case of phosphides is more complicated: both cobalt and phosphorus undergo reduction (Eqs. (VII), (VIII)). According to the published data, the main product of the above reaction is a CoP–Co2P mixture [19]. A major problem that arises in using the photochemical reduction method is the low yield of reaction products due to the recombination of electron–hole pairs. Since a side process involving holes is the formation of phosphates (Eq. (IX)), it was decided to implement the process in an aqueous alcohol solution, rather than in water. In this case, the photogenerated holes will partly oxidize ethanol. Analysis of the Co(PR) sample, which was synthesized by illuminating a mixture of cobalt hydroxide and an aqueous alcoholic solution of sodium hypophosphite with a light-emitting diode, showed the formation of cobalt orthophosphate under these conditions. Further, the procedure was supplemented with the introduction of a fivefold excess of barium ions into the reaction mixture; barium ions are capable of precipitating the orthophosphate ion without forming insoluble hypophosphites. However, even in this case, cobalt phosphide was not identified. Thus, the use of the photochemical reduction method provides the formation exclusively of cobalt orthophosphate:
Photocatalytic Activity and Photoelectrochemical Properties of the Synthesized Photocatalysts
The photocatalytic activity of catalysts based on graphitic carbon nitride is most commonly studied in aqueous alkaline solutions of triethanolamine. This reagent, being adsorbed on the surface, protects the material from photocorrosion processes and thereby extends the preservation of the catalytic properties of carbon nitride [16]. However, unmodified carbon nitride does not exhibit catalytic properties. Cocatalysts—cobalt phosphide and cobalt orthophosphate—were deposited on the surface of unmodified C3N4. The kinetic curves recorded for the studied samples are shown in Fig. 5; for comparison, the kinetic curve of hydrogen evolution recorded in the presence of the 1%Pt/g-C3N4 photocatalyst is also shown there. It is evident from Fig. 5 that photocatalytic hydrogen evolution in the presence of the studied catalysts is characterized by an induction period of 45–60 min, after which the amount of hydrogen depends linearly on time. The linear portion of the recorded curves was subsequently used to calculate the reaction rate. It should be noted that the induction period in the presence of photocatalysts with deposited metal salts was described earlier [40, 41]; it is associated with the partial reduction of the cocatalyst to the metallic state [41]. A similar effect can occur for platinum, because deposition by reduction with sodium borohydride leads to the formation of platinum particles in the 0 and +2 oxidation states on the surface; the metallic state is more preferable for hydrogen evolution [39].

Time dependences of the amount of evolved hydrogen recorded in the presence of the Co(TP)/g-C3N4, Co(PR)/g-C3N4, Co(HT)/g-C3N4, Co(TP)/g-C3N4(HT), and 1%Pt/g-C3N4 photocatalysts.
Analysis of the data in Fig. 5 shows that nonzero reaction rates were obtained for Co(TP)/g-C3N4 synthesized by thermal phosphorylation and the reference sample containing platinum. Apparently, the presence of a small amount of cobalt phosphide in the cocatalyst favorably affects the reaction rate, because CoP, in common with noble metals, is capable of attracting electron density to itself and thereby improving the degree of spatial charge separation of the semiconductor. It should be noted that the activity of the Co(TP)/g-C3N4 catalyst (156 μmol h−1 g−1) is comparable to the activity of platinized carbon nitride 1%Pt/g-C3N4 (216 μmol h−1 g−1).
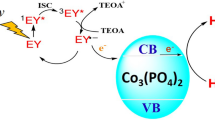
The samples synthesized by the hydrothermal method and photochemical reduction, in which cobalt is presented only in the form of orthophosphate, do not exhibit activity in the target reaction. It is known that cobalt orthophosphate is a semiconductor; the conduction and valence band edge potential is –0.73 and 1.62 V, respectively [42]. Since the conduction band potential of cobalt orthophosphate is less than 0 V, this semiconductor can theoretically be used as a photocatalyst for hydrogen evolution [43]. Apparently, heterojunctions can be formed in a composite photocatalyst consisting of cobalt orthophosphate and graphitic carbon nitride. Upon the illumination of this sample, electron–hole pairs are formed inside the particles of the two semiconductors. Since the potentials of the conduction (–1.3 V) and valence band (1.46 V) of carbon nitride [44] are higher than those of cobalt phosphate, the presence of electrons in the conduction band of cobalt orthophosphate and the presence of holes in the valence band of carbon nitride are thermodynamically most favorable. The transfer of photogenerated charges to the particles of the semiconductors in accordance with the scheme shown in Fig. 6 provides an increase in the lifetime of electrons and holes and the degree of spatial separation of charge carriers and an improvement in the catalytic activity of carbon nitride. However, the amount of deposited cobalt orthophosphate is too small for the full manifestation of this effect; therefore, the catalytic activity of these samples does not exceed 1 μmol h−1 g−1. An example to confirm the validity of this statement is provided by a system of semiconductors consisting of a solid solution of cadmium and zinc sulfides—Cd0.3Zn0.7S and Zn(OH)2—inside which heterojunctions are formed: the deposition of 1 wt % of zinc hydroxide leads to a change in catalytic activity by 10%; a significant effect is observed in the case of depositing 10–30 wt % of the cocatalyst [45]. Thus, to provide the formation of active photocatalysts, it is necessary to increase either the cobalt phosphide content in the cocatalyst or the amount of deposited cobalt orthophosphate.

Schematic charge transfer that occurs under illumination of a composite photocatalyst consisting of carbon nitride and cobalt orthophosphate.
In addition, in this study, a Co(TP)/g-C3N4(HT) photocatalyst was synthesized by the hydrothermal treatment of a preprepared cobalt orthophosphate–cobalt phosphide mixture (Co(TP) sample) and graphitic carbon nitride, which did not exhibit catalytic activity in the hydrogen evolution reaction. Apparently, the formation of a composite sample from individual phases of carbon nitride and a cobalt phosphate–cobalt phosphide mixture does not provide the formation of sufficiently tight contacts between the phases, which are provided by the phosphorylation of samples containing cobalt hydroxide preadsorbed on the surface.
All the photocatalysts containing cobalt phosphide and cobalt phosphate were further studied by electrochemical methods. The main photovoltaic characteristics were calculated from voltammetry data and summarized in Table 2. It is evident from Table 2 that the short-circuit current density for the sample synthesized by thermal phosphorylation is higher than respective parameter of the other catalysts. The photocurrent value decreases in the case of using the hydrothermal method. The use of the photodeposition technique and the assembly of a composite from presynthesized photocatalysts give lower results: the short-circuit current density is three times lower than that for the samples synthesized by the hydrothermal method. Calculations of the efficiency confirm that thermal phosphorylation is the most effective of all the described techniques. Analysis of the voltammetry data shows that, for the Co(TP) sample synthesized by thermal phosphorylation, the short-circuit current density and efficiency values are higher than those obtained for the sample synthesized by the photochemical reduction method. Apparently, the presence of a small amount of cobalt phosphide in the sample makes it possible to significantly improve the photovoltaic characteristics.
For a more fundamental understanding of the causes of changes in the key characteristics of photoelectrodes, the samples were studied by impedance spectroscopy. Figure 7 shows plots in Nyquist coordinates. It is evident that the type of cocatalyst synthesis method does not affect the general form of the dependence of the imaginary part of the impedance on the real part. Moreover, the resistance in the system decreases in the same order in which the short-circuit current density increases. Thus, the results obtained by impedance spectroscopy and voltammetry are in agreement with each other. In addition, the impedance data were used to calculate the electron lifetimes (Table 2). It should be noted that the values of this quantity decrease in the same order in which the short-circuit current density decreases. For the Co(TP) and Co(PR) samples synthesized by the thermal phosphorylation and photodeposition methods, the electron lifetime is longer than that of composite photocatalysts deposited on conductive glass. For the same samples, higher short-circuit current densities were obtained.

Nyquist hodographs plotted for the studied photoelectrodes.
To determine the type of conductivity of the synthesized samples, the Mott–Schottky plots were constructed; they are shown in Fig. 8a. The positive slope of the straight line suggests that the synthesized composite photocatalysts, in common with graphitic carbon nitride, are n-type semiconductors [46]. Thus, the deposition of 1 wt % of the cocatalyst does not change the conductivity type of the material. Mott–Schottky plots can be used to calculate the flat band potential of the studied samples. To this end, the linear portion of the recorded dependences was approximated by a straight line and the flat band potential value was calculated using the following equation [47]:
where ε is the dielectric permeability of the medium, ε0 is the dielectric constant (F/m2), C is the capacitance (F), N is the electron concentration (m−3), S is the area of the illuminated surface (m2), V is the potential applied to the cell (V), e is the electron charge (C), k is the Boltzmann constant (J/K), T is the absolute temperature (K), and VFB is the flat band potential (V).

Mott–Schottky plots constructed for (a) the composite photocatalysts and (b) the Co(TP) and Co(PR) reference samples.
According to Table 2, the flat band potential of the composite photocatalysts lies in a range of –258 to ‒325 mV; for all the samples, except for the catalyst synthesized by the photodeposition method, this parameter is about –320 mV. Since the flat band potential value hardly changes with a change in the cocatalyst synthesis method, it can be concluded that, in the case under consideration, VFB does not significantly affect the catalytic and electrochemical activity of the synthesized samples.
Similar dependences were plotted for the samples synthesized by the thermal phosphorylation and photodeposition methods (Fig. 8b). Unlike carbon nitride and the composite photocatalysts based on it, cobalt phosphide and cobalt phosphate are p-type semiconductors; this finding is consistent with the published data [48]. The flat band potential of the samples synthesized by the thermal phosphorylation and photodeposition methods is –362 and –293 mV, respectively. The differences between these values are associated with the different phase compositions of the synthesized samples. It should be noted that the formation of composite photocatalysts leads to a shift of the flat band potential of the cocatalysts by almost 40 mV, regardless of the synthesis method.
Another parameter that can be calculated from the Mott–Schottky plot is charge carrier concentration. Since the dielectric permeability of the composite photocatalysts is unknown, the product of the electron concentration by the dielectric permeability of the medium was found for the estimates (Table 2). Comparison of these quantities is assumed to be reasonable, because it is scarcely probable that the difference in the phase composition of the component deposited in an amount of 1 wt % can significantly change the dielectric permeability of the material. It is evident from Table 2 that the products of the charge carrier concentration by the dielectric permeability of photocatalysts—N × ε—for the synthesized samples are of the same order of magnitude and decrease in the same order in which the electron lifetime and the short-circuit current density decrease.
Stability of the Synthesized Photocatalysts and Photoelectrodes
It is known that one of the requirements for photocatalysts and photoelectrodes is the preservation of their activity in several illumination runs. Figure 9a shows the kinetic curves of the Co(TP)/g-C3N4 sample recorded during four 1.5-h illumination runs. Note that, during the 6-h illumination of the photocatalyst, the initial catalytic activity of the sample decreases by 38%. The time variation of the maximum amount of generated photocurrent is shown in Fig. 9b. During the first three illumination on/off runs, the short-circuit current density decreases and achieves a constant value, which does not change during the next six runs. This tendency is characteristic of all the studied photoelectrodes in the system. In general, after eight illumination on/off runs, the short-circuit current density decreases by 20–25%.

Study of the photostability of (a) the Co(TP)/g-C3N4 photocatalyst and (b) photoelectrodes synthesized by depositing composite photocatalysts on conductive glass.
CONCLUSIONS
In this study, a cocatalyst was deposited on the surface of carbon nitride by the following three different methods: thermal phosphorylation, the hydrothermal method, and photochemical reduction. In the first case, the cocatalyst was a cobalt phosphide–cobalt orthophosphate mixture; the use of the hydrothermal and photochemical synthesis methods provided the formation of cobalt orthophosphate. In addition, a sample was synthesized by a hydrothermal treatment of presynthesized carbon nitride and cobalt phosphide. The only sample that exhibited catalytic activity in photocatalytic hydrogen evolution from an aqueous alkaline solution of triethanolamine was the sample synthesized by thermal phosphorylation. The catalytic activity of this sample was 156 μmol h−1 g−1; this value is comparable to that of platinized carbon nitride 1%Pt/g-C3N4 (216 μmol h−1 g−1). The photoelectrochemical tests showed that all the synthesized photoelectrodes are capable of generating photocurrent. The highest photocurrent was recorded for the cobalt phosphide-containing sample synthesized by thermal phosphorylation. Further tests showed that the current density of the photoelectrode containing 20 mol % of cobalt (of total cobalt) in the form of CoP is 1.8 times higher than the value for the sample containing exclusively cobalt orthophosphate. Thus, an efficient system for photocatalytic and photovoltaic applications should contain a cocatalyst with a maximum content of cobalt phosphide.
REFERENCES
Kozlova, E.A. and Parmon, V.N., Russ. Chem. Rev., 2017, vol. 86, no. 9, p. 870. )https://doi.org/10.1070/RCR4739
Markovskaya, D.V., Lyulyukin, M.N., Zhurenok, A.V., and Kozlova, E.A., Kinet. Catal., 2021, vol. 62, no. 4, p. 488. https://doi.org/10.1134/s002315842104008x
An, C.W., Liu, T., Zhang, D.F., and Yan, J.S., Kinet. Catal., 2020, vol. 61, no. 6, p. 854.
Jain, A. and Ameta, C., Kinet. Catal., 2020, vol. 61, no. 2, p. 242.
Krasnyakova, T.V., Yurchilo, S.A., Morenko, V.V., Nosolev, I.K., Glazunova, E.V., Khasbulatov, S.V., Verbenko, I.A., and Mitchenko, S.A., Kinet. Catal., 2020, vol. 61, no. 3, p. 384.
Carp, O., Huisman, C.L., and Reller, A. Prog. Solid State Chem., 2004, vol. 32, p. 33.
Ran, J., Zhang, J., Yu, J., and Qiao, S.Z., ChemSusChem., 2014, vol. 7, p. 3426.
Huan, Q.-Z., Tao, Z.-J., Ye, L.Q., Yao, H.-C., and Li, Z-J., Appl. Catal., B, 2018, vol. 237, p. 689.
Mansour, A.S., Gaddalah, A.-S., Al-Sherbini, A.-S., Youssef, T., and Mohamed, M.B., J. Mol. Struct., 2017, vol. 1149, p. 626.
Cao, S. and Yu, J., J. Photochem. Photobiol. C, 2016, vol. 27, p. 72.
Kahng, S., Yoo, H., and Kim, H., Adv. Powder. Technol., 2020, vol. 31, p. 11.
Wen, J., Xie, J., Chen, X., and Li, X., Appl. Surf. Sci., 2017, vol. 391, p. 72.
Xu, J., Qi, Y., Wang, W., and Wang, L., Int. J. Hydrogen Energy, 2019, vol. 44, p. 4114.
Ge, L., Han, C., Xiao, X., and Guo, L., Appl. Catal., B, 2013, vols. 142–143, p. 414.
Lakhera, S.K., Vijayarajan, V.S., Krishna, B.S.R., Veluswamy, P., and Neppolian, B., Int. J. Hydrogen Energy, 2020, vol. 45, p. 7562.
Zhurenok, A.V., Larina, T.V., Markovskaya, D.V., Cherepanova, S.V., Mel’gunova, E.A., and Kozlova, E.A., Mendeleev Commun., 2021, vol. 31, p. 157.
Li, H., Zhao, J., Geng, Y., Li, Z., Li, Y., and Wang, J., Appl. Surf. Sci., 2019, vol. 496, p. 143738.
Wang, B., Huang, X., Zhu, Z., Huang, H., and Dai, J., Appl. Nanosci., 2012, vol. 2, p. 481.
Dong, Y., Kong, L., Wang, G., Jiang, P., Zhao, N., and Zhang, H., Appl. Catal., B, 2017, vol. 211, p. 245.
Scofield, J.H., J. Electron Spectrosc. Relat. Phenom., 1976, vol. 8, p. 129.
Shirley, D.A., Phys. Rev. B, 1972, vol. 5, p. 4709.
N. Fairley, www.casaxps.com
Markovskaya, D.V., Zhurenok, A.V., Cherepanova, S.V., and Kozlova, E.A., Appl. Surf. Sci. Adv., 2021, vol. 4, p. 100076:1.
Markovskaya, D.V., Gribov, E.N., Kozlova, E.A., Kozlov, D.V., and Parmon, V.N., Renew. Energy, 2020, vol. 151, p. 286.
Kamaja, C.K., Devarapalli, R.R., Dave, Y., Debgupta, J., and Shelke, M.V., J. Power Sources, 2016, vol. 315, p. 277.
Wang, X., Maeda, K., Thomas, A., Takanabe, K., Xin, G., Carlsson, J.M., Domen, K., and Antonietti, M., Nat. Mater., 2009, vol. 8, p. 76.
Dong, F., Zhao, Z., Xiong, T., Ni, Z., Zhang, W., Sun, Y., and Ho, W.K., ACS Appl. Mater. Int., 2013, vol. 5, p. 11392.
Liu, H., Chen, D., Wang, Z., Jing, H., and Zhang, R., Appl. Catal., B, 2017, vol. 203, p. 300.
Guo, L., Zhao, Y., and Yao, Z., Dalton Trans., 2016, vol. 45, p. 1225.
Yao, Z., Wang, G., Shi, Y., Zhao, Y., Jiang, J., Zhang, Y., and Wang, H., Dalton Trans., 2015, vol. 44, p. 14122.
Burns, A.W., Gaudette, A.F., and Bussell, M.E., J. Catal., 2008, vol. 260, p. 262.
Khassin, A.A., Yurieva, T.M., Kaichev, V.V., Bukhtiyarov, V.I., Budneva, A.A., Paukshtis, E.A., and Parmon, V.N., J. Mol. Catal. A: Chem., 2001, vol. 175, p. 189.
Biesinger, M.C., Payne, B.P., Grosvenor, A.P., Lau, L.W.M., Gerson, A.R., and Smart, R.S.C., Appl. Surf. Sci., 2011, vol. 257, p. 2717.
Venezia, A.M., Murania, R., Pantaleo, G., and Deganello, G., J. Mol. Catal. A: Chem., 2007, vol. 271, p. 238.
Kosova, N.V., Devyatkina, E.T., and Kaichev, V.V., J. Power Sources, 2007, vol. 174, p. 965.
Liu, B., Chai, Y., Li, Y., Wang, A., Liu, Y., and Liu, C., Appl. Catal., A, 2014, vol. 471, p. 70.
Guan, Q., Cheng, J., Li, X., Wang, B., Huang, L., Nie, F., and Ni, W., Sci. Rep., 2015, vol. 5, p. 10017.
Liu, D., Li, X., Wei, L., Zhang, T., Wang, A., Liu, C., and Prins, R., Dalton Trans., 2017, vol. 46, p. 6366.
Kozlova, E.A., Markovskaya, D.V., Cherepanova, S.V., Saraev, A.A., Gerasimov, E.Y., Perevalov, T.V., Kaichev, V.V., and Parmon, V.N., Int. J. Hydrogen Energy, 2014, vol. 39, p. 18758.
Alam, K.M., Kumar, P., Kar, P., Thakur, U.K., Zeng, S., Cui, K., and Shankar, K., Nanoscale Adv., 2019, vol. 1, p. 1460.
Jones, W., Martin, D.J., Caravaca, A., Beale, A.M., Bowker, M., Maschmeyer, T., Hartley, G., and Masters, A., Appl. Catal., B, 2019, vol. 240, p. 373.
Kalousek, V., Kikuta, K., Vagvala, T.C., and Ikeue, K., Mater. Lett., 2017, vol. 199, p. 65.
Samal, A., Swain, S., Satpati, B., Das, D.P., and Mishra, B.K., ChemSusChem, 2016, vol. 9, p. 3150.
Liu, X., Lai, H., Li, J., Peng, G., and Zeng, R., Photochem. Photobiol. Sci., 2021 (in press). https://doi.org/10.1007/s43630-021-00139-2
Guo, F., Shi, W., Wang, H., Han, M., Li, H., Huang, H., Liu, Y., and Kang, Z., Catal. Sci. Technol., 2017, vol. 7, p. 3325.
Markovskaya, D.V., Kozlova, E.A., Cherepanova, S.V., Saraev, A.A., Gerasimov, E.Yu., and Parmon, V.N., Top. Catal., 2016, vol. 59, p. 1297.
Liu, Y., Yu, Y.-X., and Zhang, W.-D., J. Phys. Chem. C, 2013, vol. 117, p. 29549.
Liang, R., Wang, Y., Qin, C., Chen, X., and Chen, X., Langmuir, 2021, vol. 37, p. 3321.
ACKNOWLEDGMENTS
The authors thank Dr. I.V. Shamanaev for his assistance in synthesizing the samples by the thermal phosphorylation method and Dr. T.V. Larina for analysis of the samples by diffuse reflectance spectroscopy.
Funding
This work was supported by the Russian Foundation for Basic Research and the Novosibirsk oblast (research project no. 20-43-543012).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by M. Timoshinina
Abbreviations and notation: XRD, X-ray diffraction analysis; TEM, transmission electron microscopy; XPS, X-ray photoelectron spectroscopy; CSR, coherent scattering region; NHE, normal hydrogen electrode.
Rights and permissions
About this article

Cite this article
Zhurenok, A.V., Markovskaya, D.V., Potapenko, K.O. et al. A Study of the Photocatalytic and Photovoltaic Properties of Photocatalysts Based on Carbon Nitride, Cobalt Phosphide, and Cobalt Phosphate. Kinet Catal 63, 248–260 (2022). https://doi.org/10.1134/S0023158422030107
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0023158422030107