
Abstract
The kinetics of oxidation of diethyl sulfide (Et2S) with hydrogen peroxide (H2O2) in aqueous solutions of acetonitrile (MeCN) was studied using a kinetic distribution method. It was found that the order of the reaction with respect to the substrate depends on pH and changes from the first at pH 8.06 to nearly zero at pH 11.02. The initial rates of Et2S consumption increase with pH and linearly depend on the concentrations of the hydroperoxide anion (HOO–) and MeCN. It was assumed that the reaction involves the nonequilibrium formation of peroxyimidate in the slow stage of the interaction of HOO– with MeCN followed by the rapid oxidation of Et2S.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The oxidation of organic sulfides (RSR′) is widely used for the decomposition of active components of pesticides and toxic substances [1], the desulfurization of hydrocarbons and fuels [2], and the synthesis of sulfones and sulfoxides, which are important intermediates in the synthesis of biologically active compounds and drugs [3]. The development of systems for the rapid selective oxidation of RSR' includes the solution of two main problems: (1) the search for new effective oxidants that meet the requirements of green chemistry and (2) the choice of solvents that significantly increase the solubility of sulfides, which are often almost insoluble in water, on the one hand and retain a high rate of the oxidation on the other hand.
Among the numerous oxidants [4] used for the oxidation of RSR', hydrogen peroxide (Н2О2) is the most environmentally friendly and cheap one. However, hydrogen peroxide itself has a low activity in the oxidation of sulfides and often requires specific activation. One of the ways to activate Н2О2 is its conversion into peroxo acids using activators such as bicarbonates [5, 6], borates [7–10], molybdates [11], silicates [12], nitrites [13, 14], and other compounds [4].
Water–alcohol mixtures are used to increase the solubility of sulfides and, consequently, the rate of their oxidation [5, 6, 10]. An alternative method for increasing the rates of reactions of hydrophobic substrates in water consists in their solubilization with surfactants.
In this regard, water/acetonitrile (MeCN) solutions are of particular interest. The use of a Н2О/МеСN mixture should lead to an increase in the solubility of RSR'. In addition, it is well known that nitriles (RCN) interact with Н2О2 in alkaline media to form short-lived peroxyimidic acids RC(O2H)=NH2 or peroxyimidates RC(O2H)=NH– [15–17], which efficiently oxidize alkenes to epoxides [18], amines to N-oxides [15], and sulfides to sulfoxides and sulfones [16, 17].
The oxidation of aryl methyl sulfides (ArSMe) by hydrogen peroxide in the presence of МеСN was studied in methanol–K2CO3 solutions [16] and in aqueous NaOH solutions (0.001–0.002 M) at [H2O2] \( \gg \) [NaOH] [17]. In the latter system, the rate of thioanisole oxidation does not depend on the substrate concentration (zero order with respect to ArSMe), but it linearly depends on [HOO–] and [МеCN]. The kinetic data were interpreted within the framework of a mechanism involving the formation of peroxyimidate (or peroxyimidic acid) in a slow step and in their rapid competitive reactions with ArSMe and Н2О2 with the formation of sulfoxide and amide, respectively [17].
The aim of this work was to establish the reaction mechanism and the nature of the active species in the oxidation of diethyl sulfide by the Н2О2/МеСN system. For this purpose, we studied the dependence of the initial rates of Et2S reactions with hydrogen peroxide on the acidity of solution over a wide range of pH and on the concentrations of Н2О2 and МеСN.
EXPERIMENTAL
Starting Materials and Reagents
Diethyl sulfide (Et2S) was synthesized in accordance with a published procedure [19]. Twice-distilled water, a 30% solution of Н2О2, H3PO4 (chemically pure grade), and NaOH (chemically pure grade) were used to prepare working solutions. Acetonitrile was purified as described elsewhere [20].
Kinetic Measurements
The kinetics of Et2S oxidation in the Н2О/MeCN system was studied by the kinetic distribution method [21] based on a decrease in the substrate concentration in a gas phase at [MeCN] \( \gg \) [H2O2] \( \gg \) [Et2S]. Working solutions were prepared immediately before kinetic measurements. A mixture of the vapors of Et2S and toluene, which was stable under the reaction conditions and used as an internal standard, was introduced into the gas (air) phase of a thermostatically controlled shaken catalytic duck reactor (the total volume V = 62.3 cm3). At certain time intervals, gas-phase samples (0.1 cm3) were taken with a glass syringe through a hole in the reactor plug closed with rubber and Teflon septa. Changes in the substrate concentration in the gas phase were monitored by GLC using an LKhM-80 chromatograph (Russia), a flame-ionization detector, and a 2-m column with a 5% SE-30 stationary phase on a Chromaton N-AW support.
The high frequency of reactor shaking (500 min–1) makes it possible to exclude diffusion complications associated with the mass transfer of Et2S from the gas phase to the solution [21].
RESULTS AND DISCUSSION
Observed Kinetics of Diethyl Sulfide Oxidation
According to Lobachev et al. [6], the oxidation reaction of diethyl sulfide with hydrogen peroxide in water has the first order with respect to both Et2S and H2O2 in a range of pH 9 to 12. In H2O/MeCN solutions, the reaction order with respect to the substrate (n) determined by a differential method [22] depends on pH and varies from the first at pH 8.06 to nearly zero at pH 11.02 (Table 1).
The rate of consumption of Et2S from the gas phase in the Н2О/MeСN system at a constant pH value linearly increased with the concentration of Н2О2 (Table 2); in this case, the order of the reaction with respect to the substrate remained almost unchanged within the measurement accuracy. The rate of the reaction also linearly increased with the concentration of MeСN, and the reaction order with respect to Et2S did not depend on [MeСN] (Table 3).
Therefore, we used the initial rates of the reaction in order to analyze kinetic data obtained in Н2О/MeСN solutions under various conditions and compare them with data on the oxidation of Et2S with hydrogen peroxide in water.
Calculation of the Rates of Liquid-Phase Oxidation Reactions of Et2S in Н2О/MeСN Solutions
The kinetic distribution method (KDM) was proposed, widely studied, and applied in various versions to study the reaction kinetics of volatile low-soluble substrates, such as alkanes, in aqueous or sulfuric acid solutions [21]. The intense mixing of phases, which makes it possible to exclude complications related to diffusion and interphase mass transfer, is of great importance in the KDM.
In a simple KDM model [21], it is assumed that the substrate S is distributed in equilibrium between the gas (g) and liquid phases (l) of a closed reactor, but reaction with the reagent X proceeds only in solution. At [S] \( \ll \) [X], the kinetics of substrate loss from the gas phase corresponds to the equation
where kλ is the observed constant of substrate consumption from the gas phase at a given ratio between gas Vg and solution Vl volumes in the reactor, λ = Vg/Vl; k2 is the true liquid-phase second-order rate constant; and α = [S]g/[S]l is the equilibrium coefficient of substrate distribution between the gas and the solution. The method makes it possible to determine the values of k2 and α from the dependence of 1/kλ on λ by varying the values of λ.
Let us consider the KDM model for the rate of substrate consumption (W). We introduce the following designations: \(N_{{\text{g}}}^{{\text{S}}}\) and [S]g = \(N_{{\text{g}}}^{{\text{S}}}\)/Vg are the amount and concentration of the substrate S in the gas, respectively, and \(N_{{\text{l}}}^{{\text{S}}}\) and [S]l = \(N_{{\text{l}}}^{{\text{S}}}\)/Vl are the above quantities in the solution. Then, the total amount of the substance S (NS) in a closed system is
If the reaction proceeds only in solution, the rate of substrate consumption in the system is
where τ is the reaction time.
At the same time, according to Eq. (2),
From the equality
there follows a relation linking the measured (observed) rate of substrate consumption in the gas phase Wg with the reaction rate in solution Wl:
By varying λ, the liquid phase rates (Wl) and the distribution coefficient α can be calculated from the linear dependence
The same equation is valid for the initial rates of reaction. As an example, Fig. 1 shows a plot of relationship (7) for the oxidation reaction of Et2S with hydrogen peroxide at [H2O2]0 = 0.006 M, [MeCN] = 1 vol % (0.19 M), NS = 8.4 × 10–7 mol, and pH 10.0. The calculated values of the initial rate Wl and α were (1.4 ± 0.1) × 10–8 M s–1 and 0.06, respectively. The coefficient α in this system was almost two times lower than that in water (0.1) [6], and this fact indicated an increase in the solubility of diethyl sulfide on going from water to the Н2О/MeСN system (1 vol %). The same increase in the solubility of Et2S was found in aqueous alcohol solutions of H2O/ROH (70 : 30, vol %) [6], α = 0.04–0.08.

Plot of relationship (7) for the oxidation reaction of Et2S with hydrogen peroxide in Н2О/MeСN solutions at [Н2О2] = 0.006 M, [MeCN] = 1 vol % (0.19 M), NS = 8.4 × 10–7 mol, and pH 10.0.
The observed initial rates were determined from the time dependence of the measured substrate concentration in the gas phase \(({{N_{\tau }^{{\text{S}}}} \mathord{\left/ {\vphantom {{N_{\tau }^{{\text{S}}}} {{{V}_{{\text{g}}}}}}} \right. \kern-0em} {{{V}_{{\text{g}}}}}})\) in the region of a decrease in NS < 20%. The current substrate concentration in the gas phase \(({{N_{\tau }^{{\text{S}}}} \mathord{\left/ {\vphantom {{N_{\tau }^{{\text{S}}}} {{{V}_{{\text{g}}}}}}} \right. \kern-0em} {{{V}_{{\text{g}}}}}})\) was calculated using the equation
where \(({{N_{0}^{{\text{S}}}} \mathord{\left/ {\vphantom {{N_{0}^{{\text{S}}}} {{{V}_{{\text{g}}}}}}} \right. \kern-0em} {{{V}_{{\text{g}}}}}})\) is the initial concentration of diethyl sulfide; φτ is the measured ratio between the chromatographic peak heights (or areas) of the substrate (hS) and a reference standard (hst), proportional to the current concentration of Et2S; φ0 is the intercept of φτ as a function of the reaction time, which corresponds to the composition of the initial mixture.
Effect of Acidity on the Rate of Et2S Oxidation
According to Lobachev et al. [6], the oxidation of Et2S with hydrogen peroxide (PH) in aqueous solutions at pH 8–12 proceeds according to two parallel reaction paths with the participation of HOOH and HOO–. In this case, the dependence of the initial rate of consumption of diethyl sulfide (WРН) on acidity has the form
where [PH]0 and [Et2S]0 are the initial concentrations of hydrogen peroxide and diethyl sulfide in solution, respectively, and Kа is the acid ionization constant of Н2О2.
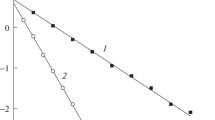
The dependence of the initial rates of Et2S oxidation in water (\({{W}^{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}\)), calculated from previously published data [6], was adequately described by Eq. (9) at the parameters kHOOH = 2.7 × 10–2 M–1 s–1, \({{k}^{{{\text{HO}}{{{\text{O}}}^{ - }}}}}\) = 0.41 × 10–2 M–1 s–1, and рKа = 11.5 (Fig. 2, curve 1).

In contrast to aqueous solutions, the rate of reaction in a Н2О/MeСN mixture (1 vol %) increased exponentially with a decrease in the acidity (Fig. 2, curve 2), and it was higher at pH 11 by a factor of more than 10 than the rate of oxidation in water (Table 1). These results indicate that the concentration of active species participating in the oxidation of Et2S in the above system increased with pH. Note that, at pH 8–9, the oxidation rate in Н2О/MeСN solutions weakly depended on the acidity, and it was close to the corresponding values in water.
The data on the orders of reaction and the dependence of the rate on the acidity of solution allowed us to assume that, at pH < 9, reaction paths with hydrogen peroxide, Eq. (9), made the main contribution to the rate of Et2S oxidation in the Н2О/MeСN system (WМеCN). This is evidenced by the orders of the reaction with respect to Et2S (n > 0.5), the constancy of the rates in the test range of pH, and their similarity to the corresponding values for the reaction in water.
An increase in the reaction rate at pH ≥ 10 indicates that, under these conditions, the route with an activated form of H2O2 (PI), peroxyimidic acid MeC(O2H)=NH2 or peroxyimidate MeC(O2H)=NH–, played the main role in the oxidation of Et2S; these active species are formed in the interaction of HOO– with MeСN [15–17, 23], Scheme 1:

Scheme 1 .
Here, kf and k–f are the rate constants of the forward and reverse reaction stages of the formation of PI; kS is the rate constant of the reaction of Et2S with PI; and kR is the rate constant of the reaction path of PI with hydrogen peroxide leading to the formation of acetylamide [23].
Separation of Et2S Oxidation Routes in the Н2О/MeСN System
The contribution of the WРН route to the oxidation of diethyl sulfide with hydrogen peroxide in Н2О/MeСN solutions was calculated according to Eq. (9) using the rate constants kНООН = 2.7 × 10–2 M–1 s–1 and \({{k}^{{{\text{HO}}{{{\text{O}}}^{ - }}}}}\) = 0.41 × 10–2 M–1 s–1, рKа = 11.5 [6]Footnote 1, and the concentration of diethyl sulfide in solution calculated at α = 0.06 and under the assumption that the rates of Et2S oxidation in this system and in water are similar. This can be evidencedFootnote 2 by the coincidence of the reaction rates in aqueous solutions and in the Н2О/MeСN system at pH < 9 (Table 1). The contribution of the route with peroxyimidate (W PI) (Scheme 1) was determined as the difference between the measured initial rate of Et2S oxidation in H2O/MeCN solutions (WMeCN) and the rate of oxidation with hydrogen peroxide (WPH).
The rate of the W PI route increased with pH and was a linear function of the concentration of HOO– (Fig. 3, curve 1). The concentrations of HOO– (Tables 1–3) were calculated using the equation
at pKa = 11.5.

Dependences of the initial rates of Et2S consumption in the reaction path with peroxyimidate in the Н2О/MeСN system on (1) the concentration of HOО– and (2) the concentration of MeСN. See Table 1 for the reaction conditions.
In a range of pH 9.08–10.02, where the contribution of the route with peroxyimidate became significant or major, the values of WPI/[HOО–][MeCN] were the same (1.2 ± 0.1) × 10–4 M–1 s–1 (Table 3). The values of WPI at constant pH also linearly depended on [HOО–] with varying the initial concentration of hydrogen peroxide (Table 2, Fig. 3) and the concentration of MeCN (Fig. 3, curve 2).
The found value of W PI/[HOО–][MeCN] = (1.2 ± 0.1) × 10–4 M–1 s–1 differs from the value of W МеCN/[HOО–][MeCN] = (7.8 ± 0.8) × 10–4 M–1 s–1 found by Gillitt et al. [17] for the oxidation of thioanisole with hydrogen peroxide in aqueous solutions of NaOH (0.01–0.02 M) in the presence of acetonitrile. This may be due to the fact that Gillitt et al. [17] used the measured rates WМеCN and did not take into account the contribution of the route with hydrogen peroxide.
On the Mechanism of Et2S Oxidation by Peroxyimidate
According to published data [16, 17], the oxidation of thioethers with peroxyimidates (or peroxyimidic acids) proceeds according to Scheme 1, which includes a slow stage of the formation of an active PI intermediate in the reaction of MeCN with HOO– and its rapid reaction with the substrate, as evidenced by the first orders of the reaction with respect to MeCN and HOO– and the zero order with respect to RSR'. The data obtained in this work (Tables 1–3) are also consistent with the mechanism proposed previously [16, 17].
There are no unambiguous published data on the nature of the first stage in Scheme 1 and the degree of protonation of the PI intermediate [17]. In the papers [16, 17] devoted to the study of ArSMe, it was hypothesized that this stage is slow (possibly, reversible) but nonequilibrium. At the same time, according to Laus [15], the equilibrium constants K = kf/k–f × 104 determined by spectrophotometry in a phosphate buffer solutions (pH 7) were 2.0, 3.3, and 5.2 M–1 at 20, 30, and 40°C, respectively.
For Scheme 1 with the rapid equilibrium formation of peroxyimidate, the rate of Et2S consumption should be described by the equation
According to Eq. (11), in the case when the formation of peroxyimidate is an equilibrium stage, the rate of diethyl sulfide consumption should be of the first order with respect to the substrate over the entire range of pH in contrast to the experimental data.
Let us consider a mechanism (Scheme 1) with the reversible but nonequilibrium formation of peroxyimidate. Under the assumption of the steady-state concentration of the PI intermediate, the observed initial rate of Et2S consumption is described by the equation
If we assume that k–f \( \ll \) kS[Et2S] \( \gg \) kR[PH]0, the expression for the initial rate of Et2S oxidation by peroxyimidate in the test range of pH has the form:
That is, the slow stage of the reaction is the interaction of acetonitrile with the hydroperoxide anion with the formation of peroxyimidate PI, and the rate of Et2S consumption corresponds to the zero order with respect to the substrate. The constancy of the values of W PI/[MeCN][HOО–] in the test range of pH (Tables 1–3) is consistent with this assumption and the data reported by Bethell et al. [16], who found that kS \( \gg \) kR \( \gg \) kf.
Reactivity of Peroxyimidates
The reactions of RSR′ with Н2О2, peroxyformic acid, peroxyacetic acid, peroxynitrous acid, the peroxosulfate anion [14], peroxocarbonate anion, and mono- and diperoxoborate anions [8] are of the first order with respect to sulfide. The rate constants of the second-order reactions of Et2S with В(ОН)3(OOH)–, \({\text{B}}{{\left( {{\text{OH}}} \right)}_{2}}\left( {{\text{OOH}}} \right)_{2}^{ - }\), and \({\text{HCO}}_{4}^{ - }\) are higher than by factors of 2.5, 100, and 97, respectively, that that of the reaction with Н2О2 [8].
Peroxyimidates react rapidly with sulfides, and the observed zero-order kinetics of the oxidation of Et2S and PhSMe [17] may indicate that RC(O2H)=NH2 (or RC(O2H)=NH–) are much more reactive than other typical peroxo acids, and the rate of substrate consumption is limited by the rate of a reaction between MeCN and HOO–. Another explanation of the zero order with respect to RSR′ is that the reaction of peroxyimidate formation is a nonequilibrium process, and it proceeds much more slowly than that in the case of the formation of other peroxo acids.
CONCLUSIONS
We found that the rate of Et2S oxidation by hydrogen peroxide in aqueous solutions of MeCN increases with the pH of the solution. The nearly zero order of reaction with respect to the substrate indicates that the reaction of HOO– with МеCN, which leads to the formation of active peroxyimidate PI, is a rate-limiting stage of the process; then, PI reacts with Et2S in a rapid stage, Scheme 1. At pH 10, the rate of Et2S consumption in the МеCN/Н2О system was higher by a factor of almost 15 than the rate of oxidation of diethyl sulfide by hydrogen peroxide in water. Hence, we can conclude that acetonitrile is one of the most effective activators of Н2О2 in the oxidation reactions of organic sulfides.
Notes
According to Gillitt et al. [17], we hypothesized that the additives of MeCN do not have a significant effect on Ka.
As demonstrated previously [6], an increase in the solubility of Et2S in aqueous alcohol solutions makes it possible to compensate increase in the reaction rate constants and provide a high rate (W = k[Et2S]l of its oxidation.
REFERENCES
Wagner, G.W. and Yang, Y.C., Ind. Eng. Chem. Res., 2002, vol. 41, no. 8, p. 1925.
Anisimov, A.V. and Tarakanova, A.V., Russ. J. Gen. Chem., 2008, vol. 79, p. 1264.
Fernandez, I. and Khiar, N., Chem. Rev., 2003, vol. 103, no. 9, p. 3651.
Das, R. and Chakraborty, D., Tetrahedron Lett., 2010, vol. 51, no. 48, p. 6255.
Richardson, D.E., Yao, H., Frank, K.M., and Bennett, D.A., J. Am. Chem. Soc., 2000, vol. 122, no. 8, p. 1729.
Lobachev, V.L., Savelova, V.A., and Prokop’eva, T.M., Teor. Eksp. Khim., 2004, vol. 40, no. 3, p. 157.
Davies, D.M., Deary, M.E., Quill, K., and Smith, R.A., Chem. Eur. J., 2005, vol. 11, no. 12, p. 3552.
Lobachev, V.L., Zimtseva, G.P., Matvienko, Ya.V., and Rudakov, E.S., Teor. Eksp. Khim., 2007, vol. 43, no. 1, p. 38.
Durrant, M.C., Davies, D.M., and Deary, M.E., Org. Biomol. Chem., 2011, vol. 9, no. 20, p. 7249.
Lobachev, V.L., Dyatlenko, L.M., and Zimtseva, G.P., Teor. Eksp. Khim., 2012, vol. 48, no. 3, p. 168.
Chiarini, M., Gillitt, N.D., and Bunton, C.A., Langmuir, 2002, vol. 18, no. 10, p. 3836.
Lobachev, V.L., Dyatlenko, L.M., and Zimtseva, G.P., Teor. Eksp. Khim., 2012, vol. 48, no. 5, p. 322.
Lobachev, V.L., Zimtseva, G.P., and Rudakov, E.S., Teor. Eksp. Khim., 2005, vol. 41, no. 5, p. 290.
Amels, P., Elias, H., and Wannowius, K.-J., J. Chem. Soc., Faraday Trans., 1997, vol. 93, no. 15, p. 2537.
Laus, G., J. Chem. Soc., Perkin Trans. 2, 2001, p. 864.
Bethell, D., Graham, A.E., Heer, J.P., Markopoulou, O., Page, P.C.B., and Park, B.K., J. Chem. Soc., Perkin Trans. 2, 1993, p. 2161.
Gillitt, N.D., Domingos, J., and Bunton, C.A., J. Phys. Org. Chem., 2003, vol. 16, p. 603.
Payne, G.B., Deming, P.H., and Williams, P.H., J. Org. Chem., 1961, vol. 26, no. 3, p. 659.
Veigand-Khil’gentag, Metody eksperimenta v organicheskoi khimii (Methods of Experiment in Organic Chemistry), Moscow: Khimiya, 1969, p. 944.
Vaisberger, A., Proskauer, E., Riddik, J., and Tups, E., Organic Solvents, Moscow: Inostrannaya Literatura, 1958.
Rudakov, E.S., Reaktsii alkanov s okislitelyami, metallokompleksami i radikalami v rastvorakh (Reactions of Alcanes with Oxidizers, Metallocomplexes and Radicals in Solutions), Kiev: Naukova Dumka, 1985.
Semiokhin, I.A., Strakhov, B.V., and Osipov, A.I., Kinetika khimicheskikh reaktsii (Kinetics of Chemical Reactions), Moscow: MSU, 1995.
McLsaac, J.E., Jr., Ball, R.E., and Behrman, E.J., J. Org. Chem., 1971, vol. 36, no. 9, p. 3048.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by V. Makhlyarchuk
Abbreviations and designations: RSR', organic sulfides; MeCN, acetonitrile; RCN, nitriles; ArSMe, aryl methyl sulfides; Et2S, diethyl sulfide; PH, hydrogen peroxide; PI, activated form of Н2О2, peroxyimidic acid MeC(O2H)=NH2 or peroxyimidate MeC(O2H)=NH–; GLC, gas–liquid chromatography; KDM, kinetic distribution method.
Rights and permissions
About this article

Cite this article
Liubymova, A.K., Bezbozhnaya, T.V. & Lobachev, V.L. Activation of Hydrogen Peroxide by Acetonitrile in the Oxidation of Thioethers: Reaction Kinetics and Mechanism. Kinet Catal 62, 342–349 (2021). https://doi.org/10.1134/S002315842103006X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S002315842103006X