
Abstract
Nitrogen-doped carbon nanotubes (N-CNTs) with different degrees of nitrogen doping were synthesized. The effects of nitrogen in the N-CNTs on the hydrophilic–hydrophobic properties of carbon nanotubes and their activity in the decomposition of Н2О2 were studied. Various approaches to the surface modification of N-CNTs by alkyl groups were studied, and the high resistance of N-CNTs to alkylation was found. Heterogeneous catalysts based on the polyoxometallate [PO4{WO(O2)2}4]3– and N-CNTs with a low degree of nitrogen doping (≤1.8 at %) were successfully synthesized. The high efficiency of the catalysts in the liquid-phase reactions of selective oxidation of alkenes using H2O2 as a green oxidizer was found, and the heterogeneous nature of catalysis was confirmed.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Epoxides are widely used as building blocks for basic and fine organic synthesis due to the special reactivity of an oxirane ring [1]. The oxidation of alkenes using various substances containing a peroxide group is one of the main methods for the preparation of epoxides [2]. The majority of synthetic approaches are based on the use of peracids [2] and alkyl hydroperoxides [3] as oxidants. However, the use of aqueous hydrogen peroxide as a green oxidizing agent is of significant economic and environmental interest [4–6]. On the homolytic activation of hydrogen peroxide, radicals are formed to direct the reaction along the allylic oxidation route. As a result, the selective epoxidation of alkenes with hydrogen peroxide can be carried out only in the presence of a catalyst that heterolytically activates hydrogen peroxide [4, 5].
Metal-oxygen clusters or polyoxometallates (POMs) are of great interest as catalysts for oxidative processes because they are thermodynamically stable to oxidation, structurally various, and capable of activating various oxidizing agents [6]. The tetranuclear phosphotungstate [PO4{WO(O2)2}4]3– (PW4), which is also known as the Venturello complex, has long been used as an active and selective homogeneous catalyst for the epoxidation of alkenes with hydrogen peroxide [7, 8]. However, the effective use of this active component requires its immobilization on the surface of an inert support, which exhibits high stability under conditions of an aggressive liquid reaction medium.
It is well known that carbon nanomaterials (CNMs) are promising in the design of hybrid inorganic composites for electronics, energy conversion and storage, molecular sensors, and catalysis [9]. Composites based on POMs and CNMs are actively studied as electrocatalysts for water splitting or redox reactions in fuel cells [10–13]. Conventional adsorption [12] and impregnation [14] are the main methods of POM immobilization on the surface of CNMs; however, more complicated methods, such as electrostatic bonding through functional groups previously introduced onto the surface of CNMs [10] or covalent bonding through organically modified POMs [13], are also used.
The doping of CNM structures with nitrogen atoms (N-CNM) provides additional possibilities for the immobilization of active centers. Nitrogen in CNMs is incorporated into pyridine-like (NPy), pyrrole (NPyr), graphite-like (NQ), and oxidized (NOx) structural positions [15–17]. The morphology of CNMs, the amount of nitrogen, and a ratio between its forms are the main factors influencing the functional properties of composites based on N-CNMs. Numerous works have demonstrated an increase in the catalytic activity of catalysts supported onto nitrogen-containing carbon nanofibers [15, 18, 19], carbon nanotubes (N-CNTs) [20, 21], mesoporous carbon [22, 23], etc. Recently, Evtushok et al. [24] reported the possibility of stable immobilization of POMs on N-CNTs. According to the experimental data, the conversion and selectivity of the synthesized catalyst were comparable to those of a homogeneous catalyst in the selective oxidation reaction of alkyl phenol to a corresponding alkyl p-benzoquinone with the use of hydrogen peroxide as an oxidizing agent.
The aim of this work was to study N-CNTs with different degrees of doping with nitrogen and to obtain an effective heterogeneous catalyst based on POMs and N-CNTs for the selective oxidation of various alkenes.
EXPERIMENTAL
Materials
Cyclooctene (98%) and cyclohexene (98%) were obtained from Fluka and passed through neutral alumina before use. Acetonitrile (Panreac, HPLC grade) was dried and stored with activated molecular sieves 4 Å. All other reagents and solvents were of high purity grade, and they were used without further purification. The concentration of H2O2 (about 30 wt % in water) was determined by iodometry before use.
POM Synthesis
The tetrahexylammonium salt of polyoxotungstate ((C7H15)4N)3[PO4{WO(O2)2}4] was obtained according to a published procedure [25]. The purity of the compound was confirmed by IR spectroscopy and 31P NMR spectroscopy.
CNT and N-CNT Synthesis
Carbon nanotubes (CNTs) and nitrogen-containing carbon nanotubes (N-CNTs) were synthesized by the catalytic pyrolysis of ethylene or an ethylene–ammonia mixture on a Fe–Ni–Al2O3 catalyst at 700°C [26, 27]. The concentration of ammonia in the mixture was varied from 10 to 60 vol %. To remove the catalyst from CNTs and N-CNTs, the materials were treated with concentrated hydrochloric acid for seven days with the subsequent boiling in 2 M HCl for 6 h. The samples were washed to remove chloride ions by repeated boiling in distilled water until the disappearance of a positive test for Cl–. The catalyst content decreased to 1–2 wt %. High-resolution microscopy showed that the remaining catalyst particles were in an encapsulated state and, accordingly, the washed carbon nanotubes were inert in the H2O2 decomposition reaction. The washed samples were dried in air and then calcined in Ar at 170°C for 2 h. Before POM immobilization, the N-CNTs were additionally dried in a vacuum at 100°C.
N-CNT Alkylation with CH3I
A 200-mg weighed portion of 1.8 at % N-CNTs (1.8% N-CNTs) was placed in a high-pressure glass vessel with a screw cap; 50 μL of CH3I was added, and the contents were left under stirring for 48 h at 25°C. Then, the temperature was raised to 50°C, and the contents were stirred for another 8 h. The carbon nanotubes were filtered off, washed with a small amount of diethyl ether, and dried in a flow of air.
N-CNT Alkylation with C4H9Br
A 200-mg weighed portion of 4.8% at % N-CNTs (4.8% N-CNTs) was placed in a high-pressure glass vessel with a screw cap; 3 mL of toluene and 1 mL of C4H9Br were added, and the mixture was stirred for 24 h at 120°C. Then, the sample was filtered off, washed with a small amount of diethyl ether, and dried in a flow of air.
N-CNT Alkylation with C4H9Li
A 200-mg weighed portion of 4.8% at % N-CNTs was placed in a flask with 5 mL of a concentrated solution of KOH in ethanol; then, the N-CNTs were filtered off and washed with ethanol. After rapid air drying, the sample was dried in a vacuum at 100°C and the flask was filled with argon. In a flow of argon, 3 mL of hexane and 400 μL of a 2.5 M solution of C4H9Li in hexane were added to the N-CNTs at room temperature with stirring, and the mixture was allowed to stand for 24 h. Then, 100 μL of C4H9Br was added to the mixture, and it was kept for another 48 h. The reaction mixture was quenched with ethanol, and the N-CNTs were filtered off, washed with a small amount of diethyl ether, and dried in air.
POM Immobilization
The adsorption of PW4 on CNTs and N-CNTs from a MeCN solution was carried out at room temperature. To increase the adsorption capacity, HClO4 (1 equiv with respect to PW4) was added on the adsorption onto N-CNTs; the adsorption onto CNTs was performed without adding HClO4. The completion of the adsorption process was monitored using UV–visible spectroscopy. The resulting solid material was separated by filtration, washed three times with MeCN, and dried in a vacuum at 60°C.
H2O2 Decomposition
A carbon support (10 mg) was placed in a glass reactor containing 5 mL of MeCN, and then 50 μL of 11 M H2O2 was added. The reaction mixture was vigorously stirred at 50°C, and the samples of the solution (300 μL) were taking at regular intervals with a syringe. The H2O2 concentration was determined by titration with a 0.002 M solution of KMnO4. The initial rates of decomposition of H2O2 W0 were found from the slopes of the initial sections of kinetic curves.
Catalytic Oxidation and Product Analysis
Catalytic experiments were carried out in thermostatically controlled glass vessels with vigorous stirring (500 rpm). Typical conditions for oxidation reactions were the following: 0.1 M alkene, 0.1–0.2 M H2O2, 10 mg of 15 wt % PW4/N-CNT, 1 mL of MeCN, and 50°C. The reaction conditions were chosen based on the results of earlier studies of heterogeneous catalysts based on PW4 [24, 25]. Reactions started with the addition of H2O2. The reaction products were identified by gas chromatography–mass spectrometry (GC–MS) and 1H NMR spectroscopy and quantitatively determined on a gas chromatograph using biphenyl as an internal standard.
Physicochemical Research Methods
The reaction mixtures were analyzed using a Khromos GKh-1000 gas chromatograph (Khromos Engineering, Russia) equipped with a flame ionization detector and a BPX5 quartz capillary column (30 m × 0.25 mm). The 31P NMR spectra were recorded on an AVANCE-400 spectrometer (Bruker, Germany) at 161.67 MHz. The chemical shift of P was determined relative to 85% H3PO4. The UV–visible spectra were recorded on a Cary 60 spectrophotometer (Varian, the United States). The X-ray photoelectron spectroscopy (XPS) spectra were measured on an ES-300 photoelectron spectrometer (KRATOS Analytical, the United Kingdom) using AlKα radiation (hν = 1486.6 eV). The energy scale of the spectrometer was calibrated using the Au 4f7/2 and Cu 2p3/2 spectra of gold and copper the binding energies of which were taken 84.0 and 932.7 eV, respectively. The nitrogen content of N‑CNTs was determined as the ratio N/C (at %), which was calculated from the intensities of the corresponding spectral lines (N1s and C1s) taking into account corrections for the atomic sensitivity factors of either of the elements. The texture characteristics were studied using the adsorption of nitrogen at 77 K on an ASAP-2400 specific surface area analyzer (Micrometrics, the United States) with preliminary training of the samples in a vacuum at 200°C. The moisture capacity of carbon nanotubes was found by a standard gravimetric method. For this purpose, a solvent (water or acetone) was added to a certain weighed portion of the sample with stirring until wet, and the resulting sample was weighed. The added amount of liquid referred to the sample weight was recorded as the moisture capacity of the sample.
The thermal analysis (TPO) of the samples was performed using an STA 449 С Jupiter synchronous thermoanalyzer (NETZSCH, Germany). The sample was placed in a corundum crucible without a lid in an atmosphere of air. The air flow rate into the sample chamber was 30 mL/min. Helium was supplied to the weighing block at a flow rate of 20 mL/min. The sample was first heated at a rate of 2 K/min from room temperature to 50°C and kept at this temperature for 30 min; then, a temperature-programmed heating to 1000°C was carried out at a rate of 5 K/min.
RESULTS AND DISCUSSION
Physicochemical Properties of N-CNTs
The concentrations of nitrogen and the fractions of various nitrogen species in N-CNTs were determined using XPS. Table 1 indicates that an increase in the concentration of ammonia in the reaction mixture from 10 to 60 vol % was accompanied by an increase in the nitrogen content of N-CNTs from 0.8 to 4.8 at %. In all cases, nitrogen was incorporated into standard structural positions (NPy, NPyr, and NQ), and the relative contribution of NQ decreased from 40 to 25% as the degree of doping was increased.
The study of the textural characteristics of carbon nanotubes showed that N-CNTs, like CNTs, were mesoporous materials, as indicated by the presence of a capillary-condensation hysteresis in the adsorption isotherms (Fig. 1). The CNT/N-CNT specific surface areas, average pore sizes, and pore volumes were 150–200 m2/g, 12–18 nm, and 0.5–0.9 cm3/g, respectively.

Isotherms of N2 adsorption on (a) CNTs and (b) 4.8% N-CNTs at 77 K.
The ability of carbon nanotubes to adsorb various solvents was analyzed on the basis of data on the moisture holding capacity of the materials for water and acetone. As can be seen in Fig. 2, the moisture capacity of N-CNTs for both water and acetone increased with the nitrogen content to reach a maximum value of 6 mL/g. The found moisture capacity values are much higher than the pore volumes of carbon nanotubes determined based on nitrogen adsorption. It is most likely that nitrogen centers increase the hydrophilicity of a carbon surface to result in the volumetric filling of the inner channels of tubes [29, 30]. An increase in moisture capacity with the nitrogen content of N-CNTs can have a positive effect on the POM amount adsorbed on the surface.

Dependence of the moisture capacity of N-CNTs for water and acetone on the nitrogen content.
Thermal analysis showed that CNTs and 4.8% N-CNTs contained no amorphous carbon impurities because the DTA curves had only one maximum in the region of 500–600°С. The observed shift of a maximum toward low temperatures by 140°C (Fig. 3) indicates a decrease in the thermal stability of N-CNTs, as compared to that of CNTs, due to their greater defectiveness [27].

(a) TG and (b) DTA curves for CNTs and 4.8% N-CNTs.
In the framework of this work, we studied a possibility of the alkylation of nitrogen-containing carbon nanotubes with various reagents. The surface modification of N-CNTs with alkyl groups can increase the surface hydrophobicity; in turn, this can affect the selectivity of catalytic epoxidation reactions of alkenes with hydrogen peroxide in the presence of POM/N-CNT catalysts. Because the selectivity of epoxidation on POM/N-CNTs mainly depends on the rate of a side reaction of epoxy ring opening, surface hydrophobization will decrease the surface concentration of water and decrease the rate of hydrolytic ring opening thus increasing the yield of epoxides. In the case of N-CNTs, the above modification can be carried out by the alkylation of N groups on the surface of the N-CNTs using various alkylating agents, for example, CH3I and C4H9Br. It is assumed that pyridine-like nitrogen groups will be the most reactive in this process due to their high nucleophilicity. By analogy with pyrrole, pyrrole groups can also be accessible for alkylation, but after treatment with a base and deprotonation.
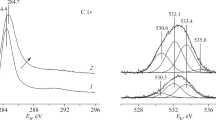
To modify the surface of 1.8% N-CNTs with methyl groups, methyl iodide CH3I was chosen as a methylating reagent, which can almost quantitatively methylate pyridine under mild conditions, because the fraction of pyridine-like N among the N groups in the N-CNTs was approximately 20% (Table 1). However, according to the XPS data (Fig. 4), the reaction of N-CNTs with a twofold excess of CH3I did not lead to surface modification with alkyl groups. The signal intensity of NPy (398.4 eV) did not change after the reaction. In addition, each pyridinium N+–Me group formed should correspond to the counterion I–, which was detected only in trace amounts according to the XPS data (Table 2).

N1s spectra of the 1.8% N-CNTs sample (a) before and (b) after methylation.
The next step was an attempt to use more severe conditions for the alkylation of N-CNTs, but the low boiling point of CH3I makes it difficult to carry out the process at higher temperatures. On this basis, 1-bromobutane C4H9Br was chosen to modify the surface of 4.8% N-CNTs [31]. The alkylation reaction of N‑CNTs was carried out in a toluene/bromobutane mixture with a tenfold excess of bromobutane with respect to the nitrogen of N-CNTs. TPO was used to analyze the surface state because the elimination of butyl groups in a temperature range of 100–250°C with a corresponding weight loss of ~10% should be observed upon the heating of N-CNTs modified with these groups. However, the thermogram of N-CNTs after the reaction did not exhibit a step of weight loss at these temperatures, and the DTA curve also did not have a corresponding maximum (Fig. 5).

(1) TG and (2) DTA curves for the 4.8% N-CNTs sample treated with bromobutane.
According to TPO data, the alkylation of pyrrole nitrogen groups after deprotonation with a strong base and the use of C4H9Li to modify the N-CNT surface due to nucleophilic substitution also did not lead to the expected result. This may be due to the accessibility of nitrogen centers to the reagent. Thus, regardless of the approach used, N-CNTs exhibited high resistance to alkylation, and further research is required to develop an effective method for the alkylation of the N groups of N-CNTs.
Activity of the 15 wt % PW4/N-CNTs Catalyst
Earlier, Evtushok et al. [24] found that nitrogen-containing tubes with the lowest nitrogen content are the most effective supports in the series 1.8% N‑CNTs–4.8% N-CNTs. It was found that the rate of the unproductive reaction of Н2О2 decomposition increased with the degree of doping with nitrogen. To confirm the higher efficiency of N-CNTs with low nitrogen content, we used 0.8% N-CNTs as a support for PW4 in this work. As can be seen in Fig. 6, the rate of decomposition of H2O2 decreased by a factor of more than 2 as the nitrogen content was decreased from 1.8 to 0.8%.

Effect of the nitrogen content of N-CNTs on the initial rate of H2O2 decomposition. Reaction conditions: 10 mg of CNTs, 0.11 M H2O2, 5 mL of CH3CN, and 50°C.
The PW4/N-CNTs catalysts were prepared by the adsorption of PW4 from a solution in MeCN on N-CNTs in accordance with a previously described procedure [24]. In the course of the sample preparation of 15 wt % PW4/0.8% N-CNTs and, for comparison, 15 wt % PW4/1.8% N-CNTs, 1 equiv of HClO4 was added. Acid addition was not used to obtain the 5 wt % PW4/CNTs catalyst.
The catalytic performance of 15 wt % PW4/0.8% N-CNTs was studied in the epoxidation reactions of various alkenes with a 30% aqueous solution of H2O2, and Table 3 summarizes these characteristics in comparison with those of other catalysts. The 15 wt % PW4/0.8% N-CNTs catalyst was slightly inferior to the 15 wt % PW4/1.8% N-CNTs catalyst in terms of conversion and selectivity in the oxidation of cyclooctene (Table 3, Nos. 1 and 2). However, the former sample demonstrated a 15% higher efficiency of H2O2 utilization due to a smaller amount of nitrogen in it, as found upon the determination of H2O2 amounts in the reaction mixtures at equal reaction times. This fact confirms the assumption that N-CNTs with a low degree of doping are optimal supports for the PW4 catalyst.
In the oxidation of cyclohexene and methyl oleate, the 15 wt % PW4/0.8% N-CNTs catalyst demonstrated moderate selectivity (43–80%) and conversion (55–80%) (Table 3, Nos. 3 and 5), and it was inferior to the acid-free 5 wt % PW4/CNTs catalyst in selectivity (Table 3, Nos. 4 and 6). The analysis of reaction products showed that the lower selectivity of the 15 wt % PW4/0.8% N-CNTs catalyst for epoxide in the oxidation of cyclohexene was related to the formation of a large amount of the corresponding diol. Previously, it was found that the activity of PW4/CNTs heterogeneous catalysts in epoxidation reactions increases with the amount of an HClO4 acid used for the immobilization of PW4, whereas the selectivity in the epoxidation of alkenes, whose epoxides are sensitive to acid-catalyzed epoxy ring opening, decreases [25].
The hot filtration test confirmed the heterogeneous nature of catalysis (Fig. 7). This conclusion is based on the observed zero conversion of a substrate after the removal of the catalyst from the reaction medium. The strong immobilization of the active component on the surface of N-CNTs was found in cycling experiments, in which a catalyst sample was reactivated by heating at 50°C in a vacuum between cycles. With the use of 15 wt % PW4/0.8% N-CNTs in four reaction cycles of cyclooctene epoxidation, no significant drops in conversion (90 vs. 87%) and selectivity (95 vs. 93%) were found (Fig. 8). It should be noted that the resulting mesoporous 15 wt % PW4/0.8% N-CNTs catalyst is able to participate in the oxidation of bulky alkenes, such as cyclooctene or cyclohexene, in contrast to a well-known microporous titanium silicate catalyst TS-1 for the selective oxidation of alkenes with hydrogen peroxide. It is well known that the use of TS-1 without diffusion hindrances is limited to small alkenes such as propene [32, 33]. In addition, in the case of mesoporous titanium silicates, rapid catalyst deactivation in three to four cycles with the use of aqueous H2O2 as an oxidizing agent is a problem, which is associated with Ti leaching in the course of the reaction, loss of the hierarchical structure, or the oligomerization of active centers [34]. In turn, the Ti-MMM-2 catalyst, which is stable in the epoxidation of alkenes with aqueous H2O2, is less active and selective in the epoxidation of cyclooctene than 15 wt % PW4/0.8% N-CNTs under comparable catalysis conditions. Thus, in the presence of Ti-MMM-2, the conversion of cyclooctene after a 24-h reaction was only 12% with 65% selectivity for epoxide formation [35]. Therefore, the above comparison shows that the use of N-CNTs as the effective support of a heterogeneous catalyst for selective oxidation of a wide range of different alkenes is promising.

Hot filtration test in the epoxidation reaction of cyclooctene in the presence of 15 wt % PW4/0.8% N-CNTs. Reaction conditions: 0.1 M cyclooctene, 10 mg of the catalyst, 0.1 M H2O2, 1 mL of CH3CN, and 50°C.

Activity and selectivity of the 15 wt % PW4/0.8% N-CNT catalyst in four cycles of the cyclooctene epoxidation reaction. Reaction conditions: 0.1 M cyclooctene, 10 mg of the catalyst, 0.2 M H2O2, 1 mL of CH3CN, 50°C, and 240 min.
CONCLUSIONS
The synthesized N-CNTs with various degrees of nitrogen doping were studied as supports of a heterogeneous catalyst for the selective oxidation of alkenes. We demonstrated that the affinity for various solvents increased with the nitrogen content of the N-CNTs and the rate of the unproductive decomposition of Н2О2 increased by a factor of 5. With the use of CH3I, 1-bromobutane (C4H9Br), and C4H9Li, we found a high resistance of N-CNTs to alkylation as a means of increasing the hydrophobicity of a carbon surface. The use of the most hydrophobic N-CNTs with a low degree of doping with nitrogen (≤1.8%) allowed us to perform strong immobilization of PW4 on the surface of carbon nanotubes. The resulting catalysts exhibited high activity and selectivity in the epoxidation reaction of cyclooctene, and they can be used repeatedly without a significant decrease in performance characteristics.
REFERENCES
Sienel, G., Rieth, R., and Rowbottom, K.T., Ullmann’s Encyclopedia of Industrial Chemistry, 2000, vol. 13, p. 139.
Swern, D., Organic Peroxides, Vol. II, New York: Wiley-Interscience, 1971, p. 963.
Fine Chemicals through Heterogeneous Catalysis, Sheldon, R.A. and van Bekkum, H., Eds., Wiley: Weinheim, 2001, p. 636.
Jones, C.W., Applications of Hydrogen Peroxide and Derivatives, Cambridge: Royal Society of Chemistry, 1999, p. 264.
Catalytic Oxidations with Hydrogen Peroxide as Oxidant, Strukul, G., Ed., Berlin: Springer, 2013, p. 286.
Hill, C.L. and Prosser-McCartha, C.M., Coord. Chem. Rev., 1995, vol. 143, p. 407.
Venturello, C., D’Aloisio, R., Bart, J.C.J., and Ricci, M., J. Mol. Catal., 1985, vol. 32, no. 1, p. 107.
Panicheva, L.P., Meteleva, G.P., Berlina, O.V., and Panichev, S.A., Pet. Chem., 2006, vol. 46, no. 6, p. 422.
Eder, D., Chem. Rev., 2010, vol. 110, p. 1348.
Toma, F.M., Sartorel, A., Iurlo, M., Carraro, M., Parisse, P., Maccato, C., Rapino, S., Gonzalez, B.R., Amenitsch, H., Da, RosT., Casalis, L., Goldoni, A., Marcaccio, M., Scorrano, G., Scoles, G., Paolucci, F., Prato, M., and Bonchio, M., Nat. Chem., 2010, vol. 2, p. 826.
Guo, S.-X., Liu, Y., Lee, C.-Y., Bond, A., Zhang, M.J., Geletii, Y.V., and Hill, C.L., Energy Environ. Sci., 2013, vol. 6, p. 2654.
Kawasaki, N., Wang, H., Nakanishi, R., Hamanaka, S., Kitaura, R., Shinohara, H., Yokoyama, T., Yoshikawa, H., and Awaga, K., Angew. Chem., Int. Ed., 2010, vol. 50, p. 3471.
Ji, Y., Huang, L., Hu, J., Streb, C., and Song, Y.-F., Energy Environ. Sci., 2015, vol. 8, p. 776.
Wang, R., Yu, F., Zhang, G., and Zhao, H., Catal. Today, 2010, vol. 150, p. 37.
Podyacheva, O.Yu. and Ismagilov, Z.R., Catal. Today, 2015, vol. 249, p. 12.
Arrigo, R., Schuster, M.E., Xie, Z., Yi, Y., Wowsnick, G., Sun, L.L., Hermann, K.E., Friedrich, M., Kast, P., Hävecker, M., Knop-Gericke, A., and Schlögl, R., ACS Catal., 2015, vol. 5, p. 2740.
Inagaki, M., Toyoda, M., Soneda, Y., and Morishita, T., Carbon, 2018, vol. 132, p. 104.
Zacharska, M., Podyacheva, O.Y., Kibis, L.S., Boronin, A.I., Senkovskiy, B.V., Gerasimov, E.Y., Taran, O.P., Ayusheev, A.B., Parmon, V.N., Leahy, J.J., and Bulushev, D.A., ChemCatChem, 2015, vol. 7, no. 18, p. 2910.
Ayusheev, A.B., Taran, O.P., Seryak, I.A., Podyacheva, O.Y., Descorme, C., Besson, M., Kibis, L.S., Boronin, A.I., Romanenko, A.I., Ismagilov, Z.R., and Parmon, V.N., Appl. Catal., B, 2014, vol. 146, p. 177.
Arrigo, R., Schuster, M.E., Xie, Z.L., Yi, Y.M., Wowsnick, G., Sun, L.L., Hermann, K.E., Friedrich, M., Kast, P., Havecker, M., Knop-Gericke, A., and Schlogl, R., ACS Catal., 2015, vol. 5, no. 5, p. 2740.
Suslova, E.V., Savilov, S.V., Egorov, A.V., and Lunin, V.V., Kinet. Catal., 2019, vol. 60, p. 87.
Bulushev, D.A., Zacharska, M., Shlyakhova, E.V., Chuvilin, A.L., Guo, Y.N., Beloshapkin, S., Okotrub, A.V., and Bulusheva, L.G., ACS Catal., 2016, vol. 6, no. 2, p. 681.
He, L., Weniger, F., Neumann, H., and Beller, M., Angew. Chem., Int. Ed., 2016, vol. 55, no. 41, p. 12582.
Evtushok, V.Yu., Suboch, A.N., Podyacheva, O.Yu., Stonkus, O.A., Zaikovskii, V.I., Chesalov, Yu.A., Kibis, L.S., and Kholdeeva, O.A., ACS Catal., 2018, vol. 8, p. 1297.
Evtushok, V.Yu., Ivanchikova, I.D., Podyacheva, O.Yu., Stonkus, O.A., Suboch, A.N., Chesalov, Y.A., Zalomaeva, O.V., and Kholdeeva, O.A., Front. Chem., 2019, vol. 7, p. 858:1-14.
Suboch, A.N., Cherepanova, S.V., Kibis, L.S., Svintsitskiy, D.A., Stonkus, O.A., Chesnokov, V.V., Romanenko, A.I., Ismagilov, Z.R., and Podyacheva, O.Yu., Fullerenes, Nanotubes, Carbon Nanostruct., 2016, vol. 24, p. 520.
Podyacheva, O.Y., Cherepanova, S.V., Romanenko, A.I., Kibis, L.S., Svintsitskiy, D.A., Boronin, A.I., Stonkus, O.A., Suboch, A.N., Puzynin, A.V., and Ismagilov, Z.R., Carbon, 2017, vol. 122, p. 475.
de Correa, C.M., J. Mol. Catal. A: Chem., 2002, vol. 185, nos. 1−2, p. 269.
Chizari, K., Janowska, I., Houlle, M., Florea, I., Ersen, O., Romero, T., Bernhardt, P., Ledoux, M.J., and Pham-Huu, C., Appl. Catal., A, 2010, vol. 380, p. 72.
Kumar, K.V., Preuss, K., Guo, Z.X., and Titirici, M.M., J. Phys. Chem. C, 2016, vol. 120, p. 18167.
Xu, J., Wu, F., Jiang, Q., and Li, Y.-X., Catal. Sci. Technol., 2015, vol. 5, p. 447.
Clerici, M. G., Kinet. Catal., 2015, vol. 56, no. 4, p. 450.
Esipovich, A.L., Belousov, A.S., Kanakov, E.A., Mironova, V.Yu., Rogozhin, A.E., Danov, S.M., Vorotyntsev, A.V., and Makarov, D.A., Kinet. Catal., 2019, vol. 16, p. 62.
Kholdeeva, O.A. and Trukhan, N.N., Russ. Chem. Rev. 2006, vol. 75, no. 5, p. 411.
Bonon, A.J., Mandelli, D., Kholdeeva, O.A., Barmatova, M.V., Kozlov, Y.N., and Shul’pin, G.B., Appl. Catal., A, 2009, vol. 365, no. 1, p. 96.
ACKNOWLEDGMENTS
We are grateful to O.A. Nikolaeva for studying the samples by a TPO method.
Funding
This work was supported by the Russian Foundation for Basic Research (grant no. 18-33-00764) and the Ministry of Science and Higher Education (project no. AAAA-A17-117041710084-2). The studies were carried out using the equipment of the Center of Collective Use “National Center for Catalyst Research.”
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors have no conflicts of interest.
Additional information
Translated by V. Makhlyarchuk
Abbreviations and designations: CNTs, carbon nanotubes; N‑CNTs, nitrogen-containing carbon nanotubes; POMs, polyoxometallates; PW4, the tetranuclear phosphotungstate [PO4{WO(O2)2}4]3–; NPy, NPyr, NQ, and NOx, pyridine-like, pyrrole, graphite-like, and oxidized structural positions, respectively; TG, thermogravimetry; DTA, differential thermal analysis; DSC, differential scanning calorimetry; TPO, temperature-programmed oxidation; XPS, X-ray photoelectron spectroscopy.
Rights and permissions
About this article

Cite this article
Suboch, A.N., Evtushok, V.Y., Kibis, L.S. et al. Nitrogen-Doped Carbon Nanotubes as an Effective Support of Heterogeneous Catalysts for Selective Alkene Oxidation. Kinet Catal 62, 288–298 (2021). https://doi.org/10.1134/S0023158421020105
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0023158421020105