
Abstract
Two new ternary copper(II) complexes [Cu(2-phenoxyacetate)2(temed)] (1) and [Cu(4-chlorobenzoate)2 (temed)] (2) are synthesized by the addition of temed (N,N,N′,N′-tetramethylethylenediamine) to Cu(II) 2-phenoxyacetate/4-chlorobenzoate in a water:methanol (1:4, V/V) mixture. The newly synthesized complexes are characterized by elemental analyses, spectroscopic techniques (UV-Vis, FTIR), magnetic moment, molar conductance and single crystal X-ray diffraction. In both crystals, the asymmetric unit consists of one half of the neutral complex in which the central metal atom lies on a glide plane in 1 and on a twofold axis in 2. Both ternary complexes are neutral and consist of six coordinated CuN2O4 chromophores. In both complexes, the Cu(II) metal centre is coordinated by two nitrogen atoms of chelating temed and four oxygen atoms from two bidentate 2-phenoxyacetate/4-chlorobenzoate ligands. Detailed packing analyses of complexes 1 and 2 reveal that the C–H…π interaction along with weak intermolecular C–H…O hydrogen bonding interactions provide the stability and robustness of the crystal lattice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
In recent years, there is a tremendous advancement in the field of crystal engineering based on organic and/or inorganic building blocks for the successful synthesis and design of molecular structures and materials of wide applications due to the cooperative utilization of covalent, non-covalent, and coordinate bonding [1-4]. Non-covalent interactions have been exploited as one of the primary tools to establish the probable connection between organic and/or inorganic molecular building blocks [5, 6]. Although prodigious advances have been made in the past to decipher the complimentary traits responsible for the stability of molecules in the crystalline state, up to date, no reliable method exists which can design the anticipated crystalline supramolecular architecture. Indeed, an extremely trivial variation in the reaction condition, stoichiometry, organic building blocks, supporting ligands, and the coordination ability of the metal centre can change the nature and composition of the product [6-9].
However, a coherent or judicious selection of the metal center and the ligand to some extent can be employed as an effective approach for the construction of extended supramolecular architectures with preferred properties and functionalities [10-13]. In crystal engineering, carboxylic acids are generally chosen as building blocks because of their ability to form robust and directional hydrogen bonds with predictable properties [14, 15]. Aryl/alkyl carboxylates are flexible strong coordinating ligands that exhibit a variety of coordination modes (Fig. 1). A subtle variation in the position and nature of substituents in the ligands plays a decisive role in altering the coordination behaviour of the ligand towards the metal centre. For this purpose, strongly coordinating alky/aryl carboxylate i.e., 2-phenoxyacetic acid/4-chlorobenzoic acid and temed are taken into consideration (Fig. 2). The presence of electronegative oxygen and a chlorine group in aromatic acids enhances the probability of weak intermolecular hydrogen bonding interactions which sew the building blocks into an extended supramolecular framework. 2-Phenoxyacetic acid is also used in manufacturing pharmaceuticals, pesticides, herbicides, fungicides, and dyes whereas 4-chlorobenzoic acid is a precursor to a variety of drugs, food additives, and dyes [17-19].
Copper is an essential element for the sustenance of life. Copper(II) carboxylate complexes are among the most extensively studied transition metal compounds due to their wide array of applications [20-23]. Copper(II) 4-chlorobenzoate-based complexes can exist as mononuclear ionic complexes ([Cu(en)2(H2O)2](4-ClC6H4COO)2, [Cu(Cyclam)2(H2O)2](4-ClC6H4COO)2, [Cu(4-hexadecyloxypyridine)2(cyclam)2](4-ClC6H4COO)2), neutral monomeric complexes ([Cu(4-ClC6H4COO)2(4-hexadecyloxypyridine)2(H2O)], [Cu(pyr)2(4-ClC6H4COO)2(H2O)], [Cu(bpy)(4-ClC6H4COO)2(H2O)], [Cu(bzdh)2(4-ClC6H4COO)2]), and neutral dimeric complexes ([Cu2(4-ClC6H4COO)2(EtOH)2], [Cu2(4-ClC6H4COO)2(iPrOH)2]) in presence of nitrogen/oxygen donor ligands [24-26]. Metal carboxylates in the presence of temed can form both monomeric as well as polymeric complexes. Metal benzoate has gained rational interest not only for its magnetic properties, structural versatility, biological importance but also as a predecessor for designing new metal-organic frameworks [27-29]. Previously, our groups have successfully utilized a strong chelating ligand en (ethylenediamine) to study second sphere interactions between the cationic [Cu(en)2(H2O)2]2+ and anionic \(\text{RCOO}^{-}/\text{SO}_{3}^{2-}\) moieties for the creation and successful isolation of new entities in the solid state [30-32]. N,N,N′,N′-tetramethylethylenediamine (temed) is a derivative of en with increased basicity due to +I effect of the substituted methyl group and as en exhibits a considerable trait of molecular conformability and flexibility when coordinated to the metal centre to form a stable five-membered ring.

Pictorial representation of different coordination modes exhibited by the carboxylate ligand.

2-phenoxyacetic acid (a), 4-chlorobenzoic acid (b), and temed (c).
The present study was envisaged to examine: (i) whether the enhanced basicity of temed will affect the coordination behavior (monodentate or bidentate) of carboxylate around the central metal ion when two different carboxylates, i.e. alkyl/aryl carboxylates (2-phenoxyacetate/4-chlorobenzoate) are employed; (ii) the effect of the increased basicity on the Cu–N bond length; (iii) whether the second sphere interaction will still be viable when the hydrogen atom attached to nitrogen in en is substituted by CH3, i.e blocking the probability of the N–H…X hydrogen bonding site; (iv) cumulative weak interactions responsible for their stability, which may arise due to the presence of electronegative oxygen and the chlorine group in acids; (v) whether subtle variation in the position of the substituent (chloro) in the ligand, i.e. 4-chlorobenzoate as compared to the previously reported isomeric 2-chlorobenzoate ligand [33], will alter the coordination ability of carboxylate and the packing architecture. To address all these queries, the syntheses, characterization, X-ray structural determination, and packing analyses of [Cu(2-phenoxyacetate/4-chlorobenzoate)2(temed)] were carried out.
EXPERIMENTAL
Materials and methods. Analytical grade reagents were used without any further purification. An automatic PerkinElmer 2400 CHN elemental analyzer was used to analyze carbon, hydrogen, and nitrogen; copper was estimated by the gravimetrical method [34]. FTIR spectra were recorded on a Spectrum Two FTIR spectrometer (PerkinElmer). Electronic spectra were recorded in methanol using a Shimadzu UV-1800 spectrophotometer. The molar conductance was determined in methanol at 25 °C using a CNO4091201 Pico Conductivity Meter. Gouy′s method was used to calculate the magnetic susceptibility of the newly synthesized complexes. Magnetic moments of the complexes were calculated according to the equation μ = 2.83(χMT)1/2.
Synthesis of [Cu(2-phenoxyacetate)2(temed)] (1). Sodium salt of 2-phenoxyacetic acid was prepared by dissolving 1.2 g (7.9 mmol) of 2-phenoxyacetic acid and 0.32 g (8 mmol) of sodium hydroxide in 10 mL of water. 1.0 g (4 mmol) of CuSO4·5H2O was dissolved in a minimum amount of distilled water. Both solutions were mixed and the blue-colored parent complex copper(II) 2-phenoxyacetate precipitated immediately. The obtained product was filtered through Whatman′s filter paper, washed with double distilled water, and dried in air. Temed was added to the blue-colored copper(II) complex suspended in water:methanol (1:4, V/V) until a clear dark blue solution was obtained. On evaporation at room temperature, after 35 days deep-blue crystals appeared which were separated from the mother liquor and dried at room temperature. Complex 1 is soluble in methanol, chloroform and insoluble in water. Complex 1 melts at 178-180 °C. Anal. calc. for C22H30CuN2O6 (%): C 54.77, H 6.22, N 5.80, Cu 13.17. Found (%): C 54.70, H 5.99, N 5.76, Cu 13.10. IR, cm–1 (neat): 3270 w, 1595 s, 1495 m, 1401 m, 1330 w, 1294 w, 1260 m, 1220 s, 1176 w, 1083 w, 1060 w, 1021 w, 789 w, 750 s 691 s, 601 w, 505 w; UV-Vis (H2O, λmax, nm: 233, 287, and 655 (εmax = 48.86 L·mol–1·cm–1).
Synthesis of [Cu(4-chlorobenzoate)2(temed)] (2). Sodium salt of 4-chlorobenzoic acid was synthesized by dissolving 1.25 g (8 mmol) of 4-chlorobenzoic acid and 0.32 g (8 mmol) of sodium hydroxide in 10 mL of water. 1.0 g (4 mmol) of CuSO4·5H2O was dissolved in a minimum amount of distilled water. On mixing both solutions the blue-colored product of parent copper(II) 4-chlorobenzoate was obtained immediately. The precipitated product was filtered through Whatman′s filter paper, washed with double distilled water, and dried in air. The above blue copper(II) complex was suspended in water:methanol (1:4, V/V) and temed was added. The addition was carried out until a clear dark blue solution was obtained. On evaporation at room temperature, after 40 days deep-blue crystals appeared which were separated from the mother liquor and dried at room temperature. Complex 2 is soluble in methanol, chloroform and insoluble in water. Complex 2 melts at 198-200 °C. Anal. calc. for C20H24Cl2CuN2O4 (%): C 48.97, H 4.89, N 5.70, Cu 12.93. Found (%): C 48.90, H 4.69, N 5.61, Cu 12.56. IR, cm–1 (neat): 3064 w, 2887 s, 2854 w, 1599 s, 1562 s, 1468 m, 1377 s, 1362 s,1291 w, 1171 w, 1135 s, 1082 s, 951 s, 866 m, 847 s, 805 w, 770 s, 731 w, 691 w, 629 w, 570 sh, 561 m, 520 s, 472 w; UV-Vis (H2O, λmax, nm: 237, 271, and 650 (εmax = 45.55 L·mol–1·cm–1).
The synthetic route for complexes 1 and 2 is shown in Scheme 1.

Schematic route for the synthesis of copper(II) complexes.
Single crystal X-ray diffraction. Diffraction data for complexes 1 and 2 were collected on a Nonius KAPPA diffractometer equipped with a CCD detector with graphite-monochromatized MoKα radiation (λ = 0.71069 Å) with φ scanning followed by ω scanning to fill the sphere. Data sets were integrated with Denzo-SMN [35]. Intensities were corrected for Lorentz, polarization, and absorption effects [36]. The structures were solved by direct methods with the SIR97 suite of programs [37] and refinements were performed on F2 by full-matrix least-squares methods with all non-hydrogen atoms in the anisotropic approximation. In both structures, hydrogen atoms were included in calculated positions, riding on their carrier atoms. All calculations were performed using SHELXL-2014/7 [38] implemented in the WINGX system of programs [39]. The crystal data are reported in Table 1. The drawings were made with Diamond [40] and ORTEPIII [41].
RESULTS AND DISCUSSION
Complexes 1 and 2 have been characterized by spectroscopic techniques (FTIR, UV-Vis), magnetic susceptibility, and conductivity measurements. The crystal structures of complexes 1 and 2 have been unequivocally established by single crystal X-ray diffraction.
Molar conductivity measurements. Conductivity measurements for complexes 1 and 2 were carried out in methanol at 25 °C. At infinite dilution, the limiting molar conductance was measured by plotting the molar conductance (Λ) against the square root of concentration (C1/2) [42]. On extrapolating the concentration to zero, it gave a small/negligible Λo value, indicating the formation of neutral complexes (Fig. 3).
Magnetic susceptibility measurements. Copper(II) compounds possessing ionic or weak covalent bonds exhibit a magnetic moment between 1.9-2.2 μB, whereas complexes possessing strong covalent bonds show a magnetic moment in the range of 1.73-1.82 μB [43]. The magnetic susceptibility of the present complexes measured at room temperature is 1.73 μB for 1 and 1.74 μB for 2, indicating the formation of strong covalent bonds.

Graph between the square root of the concentra-tion against the molar conductance of complexes 1 and 2.
Spectroscopic characterization. The FTIR spectra of synthesized complexes 1 and 2 have been recorded in the range of 4000-400 cm–1 (Fig. 4a, b). Based on the literature values, tentative assignments have been made [44]. In both complexes 1 and 2, the bands between 3270-3064 cm–1 may be attributed to aromatic υ(=C–H) stretching vibrations. The bands between 2927-2854 cm–1 may be attributed to CH3 asymmetric stretching, υas(C–H) and asymmetric as well as symmetric stretchings, υas(C–H) and υs(C–H), of CH2 groups present in the temed ligand. The bands at 1595 cm–1, 1401 cm–1 in 1 and 1599 cm–1, 1362 cm–1 in 2 may be attributed to υas(COO)– and υs(COO)–, respectively. The magnitudes of Δυ(COO) = υas(COO)– – υs(COO)– of 194 cm–1 and 237 cm–1 in 1 and 2, respectively, are less than those in their sodium salt, indicating the bidentate mode of coordination. The bands between 1586-1562 cm–1 may be assigned to υ(C–C) stretching vibrations. The υ(C–N) absorption band occurs at 1176 cm–1 and 1171 cm–1 in 1 and 2, respectively. The sharp band at 750 cm–1 in 2 may be attributed to υ(C–Cl) stretching vibrations. The bands at 601 cm–1 in 1 and 520 cm–1 in 2 may correspond to υ(Cu–N) stretching whereas the bands at 505 cm–1 and 472 cm–1 in 1 and 2, respectively, may be attributed to the υ(Cu–O) stretching vibration in both complexes.

FTIR spectra of 1 (a) and 2 (b).
UV-Vis spectra of both complexes in methanol exhibited three bands. The bands at 233 nm in 1 and 222 nm in 2 are assigned to π…π* transitions whereas the bands at 250 nm in 1 and 269 nm in 2 are attributed to intraligand n…π* transitions/LMCT charge transfer. A broad band in the range of 500-800 nm corresponds to the d–d transition in octahedral copper(II) complexes [45]. Broad low-intensity absorption around 655 nm (εmax = 48.86 L·mol–1·cm–1) and 650 nm (εmax = 45.55 L·mol–1·cm–1) for 1 and 2, respectively, is due 2Eg → 2T2g, d–d transitions (Fig. 5). These λmax values observed for the title complexes are comparable with the reported values [33, 46].
The similarity in the FTIR and UV spectra of both complexes indicates a similar coordination environment around the copper(II) metal centre (CuN2O4 chromophore), i.e. identical modes or the coordination behavior of the carboxylate moieties and temed, which is later confirmed by X-ray crystallography.
CRYSTAL STRUCTURE
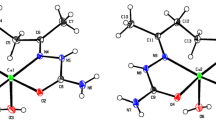
Coordination geometry and bonding. The structures of copper(II) complexes were unambiguously established by single crystal X-ray diffraction. The ORTEP diagram (Figs. 6, 7) and bond angles are given in Table 2. In both crystals, the asymmetric unit consists of one half of the neutral complex in which the central metal atom lies on a glide plane in 1 and on a twofold axis in 2. Both ternary complexes are neutral and consist of six-coordinated CuN2O4 chromophores. In both complexes, the Cu(II) metal center is coordinated by two nitrogen atoms of chelating temed and four oxygen atoms from two bidentate 2-phenoxyacetate/4-chlorobenzoate ligands, as anticipated by IR and UV-Vis spectroscopy. The oxygen atoms of the carboxylate group coordinate to the metal center in an uneven way, in which the Cu–O1 bond distances can in fact be regarded as short contacts (Table 2). This situation is quite common for copper(II) carboxylate complexes, as inferred by the analysis of the bond distance distribution that is clearly bimodal with two maxima at ca. 1.96 Å and 2.63 Å (948 hits in the Cambridge Crystallography Data Center). The bond distances between the copper metal center and the O2 oxygen atom are 1.968(2) Å and 1.961(2) Å for 1 and 2, respectively; they are shorter than the Cu–N bond distances. The similarity of these complexes to en complexes implies that there is no significant change in the Cu–N bong length with increasing basicity of the nitrogen donor ligand [24, 30-32]. The other Cu–O1 bond distances are quite long, being 2.645(2) Å and 2.618(2) Å in 1 and 2, respectively. In 1, the Cu–O1 bond distance is slightly more than that in 2 because the O1 atom is involved in bifurcated C–H…O1 hydrogen bonding interactions (Table 3), which decreases the electron density on the oxygen atom, resulting in an increase in the Cu–O1 bond distance. Consequently, the final coordination geometry for both structures can be better described as slightly distorted square planar. However, given that, according to the bond valence model [47], the valence of an atom is equal to the sum of valences of all the bonds it forms. In both complexes, each Cu atom reaches the valence of (II) only when the Cu–O1 contacts are considered.

Visible spectra of 1 and 2.

ORTEPIII view and atom numbering scheme for 1. Thermal ellipsoids are drawn at the 40% probability level. The close Cu–O1 contact is drawn as a dashed bond.

ORTEPIII view and atom numbering scheme for 2. Thermal ellipsoids are drawn at the 40% probability level. The close Cu–O1 contact is drawn as a dashed bond.
In both cases, temed has a gauche conformation with dihedral angles of 69.90° and 70.90° in complexes 1 and 2, respectively, and forms a five-membered ring through coordination with the copper(II) metal center with N–Cu–N#1 bite angles of 86.77(10) (#1 –x, –y, z ) in 1 and 85.56(8) (#1 1–x, y, –1/2–z) in 2.
The slightly larger Cu–O1 distance in [Cu(temed)(4-chlorobenzoate)2], as compared to that in the reported isomeric complex [Cu(temed)(2-chlorobenzoate)2], is due to the ortho effect in 2-chlorobenzoate. Resonance stabilization of carboxylate in the reported 2-chlorobenzoate complex is more than that in 4-chlorobenzoate, i.e. in 4-chlorobenzoate one carboxylate oxygen atom is more likely to have a negative charge than another, hence, the elongation of the Cu–O1 bond takes place in complex 2. Moreover, Cu–O1 participated in the hydrogen bonding, which may also enhance the probability of the increased bond length.
Packing. Our previous studies demonstrated that in the presence of the strongly chelating en ligand, copper(II) carboxylate was isolated in the crystalline solid state as ionic species with [Cu(en)2(H2O)2]2+ as a cationic moiety and the RCOO– anionic moiety. The second sphere interaction played an important role in stabilizing the complex. In the present study, both hydrogen atoms present on nitrogen were replaced with the methyl group. As a result, the probability of N–H…X interactions reduced to nil. A detailed packing analysis revealed the formation of a monomeric neutral complex, which confirmed the absence of any second sphere interaction. An almost similar packing arrangement is exhibited in both complexes with carboxylate organic moieties flanked outwardly towards each other (Fig. 8).

Packing analyses of complexes: 1 viewed along the a axis (a) and 2 viewed along the c axis (b).
In the packing architecture in complex 1, the adjacent units are mainly connected through weak intermolecular hydrogen bonding interactions C1–H…O1, 2.37 Å, 168° (1/2–x, y, z+1/2, between the carboxylate oxygen atom and the hydrogen atom of temed CH2) and C10H…O1, 2.64 Å, 149° (1/2–x, y, z–1/2, between the carboxylate oxygen atom and the hydrogen atom of 2-phenoxyacetate CH2, Table 3). Since the CH2 group in 2-phenoxyacetate is sandwiched between two electron-withdrawing groups, i.e. phenoxy and carboxylate, therefore, it participates in the hydrogen bonding (Fig. 9a). Moreover, CH…π interactions between C6–H…Cg also occur, where Cg is C4–C9, 2.89 Å, 173° in 1 (Fig. 9b), which arises between the H atom of one benzene ring and the π electron cloud of another aromatic moiety. It is interesting to note that the H atom from temed CH3 participates neither in hydrogen bonding interactions nor in CH…π interactions.
A similar type of weak intermolecular hydrogen bonding interactions, C2–H…O2, 2.61 Å, 153° (x, –y, z–1/2, between the 4-chlorobenzoate oxygen atom and the hydrogen atom adjacent to chlorine present in 4-chlorobenzoate), C9–H…O1 2.41 Å, 162° (x, 1–y, z+1/2, between the 4-chlorobenzoate oxygen atom and the hydrogen atom of the temed methyl group, Table 3), and C3–H…π where Cg is C5–C10, 3.07 Å, 151° (x, y–1, z, between the methyl H atom and the π electron cloud of the aromatic moiety) intermolecular interactions are observed in 2 (Fig. 10a, b). It is interesting to note that in the present complex, no hydrogen bonding interaction exists between the electronegative moieties present on carboxylate and H atoms from the temed methyl group, i.e. C–H…Cl interactions in complex 2, which were present in its reported isomeric complex, i.e. [Cu(temed)(2-chlorobenzoate)2], since the temed hydrogen atom in complex 2 is involved in the interaction with the π electron cloud of the aromatic moiety of the neighbouring molecule (C3H…π) and with the carboxylate oxygen atom (C9H…O1, Fig. 10b). No significant π…π interactions have been observed in the packing analyses of both complexes.

Depiction of non-covalent interactions in 1: hydrogen bonding interactions (a) and C–H…π interactions (b).

Depiction of non-covalent interactions in 2: hydrogen bonding interactions (a) and C–H…π interactions (b).
CONCLUSIONS
The reaction of hydrated copper(II) sulphate with a sodium salt of 2-phenoxyacetic acid/4-chlorobenzoic acid and temed resulted in the formation of two novel ternary complexes. The molar conductivity and magnetic susceptibility measurement indicated the formation of neutral and monomeric formulations in both complexes. FTIR spectra suggested the asymmetric bidentate coordination mode of the carboxylate moiety with the Cu(II) metal center, whereas the electronic spectra indicated the presence of the CuN2O4 chromophore in both complexes. The X-ray structural analysis of both complexes confirmed the presence of the bidentate asymmetric mode of the carboxylate moiety and a five-membered ring of the symmetric bidentate temed ligand around the copper(II) metal center. Detailed packing analyses revealed the presence of weak intermolecular hydrogen bonding interactions in both complexes. In complex 1, only one oxygen atom (O1) of the carboxylate moiety is involved in bifurcated hydrogen bonding interactions with the temed CH2 group as well as the aromatic moiety. In complex 2, both oxygen atoms present in the carboxylate moiety are involved in hydrogen bonding interactions with the hydrogen atom adjacent to the chloro group in carboxylate and the hydrogen atom from temed CH3. The CH…Cl interaction observed in its reported isomeric complex was absent. Apart from this, in both complexes the CH…π interaction provides the robustness and stability to the crystal lattice.
ADDITIONAL INFORMATION
Appendix A. Supplementary data. CCDC 1957495 and 1958748 contain the supplementary crystallographic data for complexes 1 and 2, respectively. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
REFERENCES
D. Braga. J. Chem. Soc., Dalton Trans., 2000, (21), 3705–3713. https://doi.org/10.1039/B007602N
G. Desiraju. Angew. Chem., Int. Ed., 2007, 46(44), 8342–8356. https://doi.org/10.1002/anie.200700534
J. W. Steed and P. A. Gale. Supramolecular Chemistry: from Molecules to Nanomaterials, Vol. 8. Wiley, 2012.
L. J. Zhou, Y. Y. Wang, C. H. Zhou, C. J. Wang, Q. Z. Shi, and S. M. Peng. Cryst. Growth Des., 2007, 7(2), 300–306. https://doi.org/10.1021/cg060369l
A. Westcott, J. Fisher, L. P. Harding, P. Rizkallah, and M. J. Hardie. J. Am. Chem. Soc., 2008, 130(10), 2950–2951. https://doi.org/10.1021/ja8002149
J. W. Steed, D. R. Turner, and K. J. Wallace. Core Concepts in Supramolecular Chemistry and Nanochemistry. John Wiley & Sons: Chichester, 2007, 194.
W. Xu, Z. X. Si, M. Xie, L. X. Zhou, and Y. Q. Zheng. Cryst. Growth Des., 2017, 17(4) 2147–2157. https://doi.org/10.1021/acs.cgd.7b00097
S. W. Jaros, M. F. C. G. D Silva, M. Florek, M. C. Oliveira, P. Smolenski, A. J. L. Pombeiro, and A. M. Kirillov. Cryst. Growth Des., 2014, 14, 5408–5417. https://doi.org/10.1021/cg500557r
P. C. Lemaire, D. T. Lee, J. Zhao, and G. N. Parsons. ACS Appl. Mater. Interfaces, 2017, 9(26), 22042–22054. https://doi.org/10.1021/acsami.7b05214
D. Imbert, M. Cantuel, J. C. G. Bunzli, G. Bernardinelli, and C. Piguet. J. Am. Chem. Soc., 2003, 125(51), 15698–15699. https://doi.org/10.1021/ja0386501
S. Zang, Y. Su, Y. Li, Z. Ni, and Q. Meng. Inorg. Chem., 2006, 45(1), 174–180. https://doi.org/10.1021/ic051502m
R. J. Hill, D. L. Long, N. R. Champness, P. Hubberstey, and M. Schroder. Acc. Chem. Res., 2005, 38(4), 335–348. https://doi.org/10.1021/ar040174b
Y.-Z. Zhang, S. Gao, H.-L. Sun, G. Su, Z.-M. Wang, and S.-W. Zhang. Chem. Commun., 2004, Iss. 17, 1906–1907. https://doi.org/10.1039/b405167j
M. Lackinger and W. M. Heckl. Langmuir, 2009, 25(19), 11307–11321. https://doi.org/10.1021/la900785f
J. Gu, M. Wen, X. Liang, Z. Shi, M. V. Kirillova, and A. M. Kirillov. Crystals, 2018, 8(2), 83. https://doi.org/10.3390/cryst8020083
R. P. Sharma, A. Saini, S. Kumar, P. Venugopalan, G. Yanan, J. Yu, and V. Ferretti. Inorg. Chim. Acta, 2016, 442, 37–45. https://doi.org/10.1016/j.ica.2015.11.020
E. Regulska, M. Samsonowicz, R. Wislocka, and W. Lewandowski. Int. J. Spectrosc., 2012, 27, 321–328. https://doi.org/10.1155/2012/498439
C. Timchalk. Toxicology, 2004, 200, 1–19. https://doi.org/10.1016/j.tox.2004.03.005
S. Psaltou, S. Stylianou, M. Mitrakas, and A. Zouboulis. Separations, 2018, 5(3), 42–54. https://doi.org/10.3390/separations5030042
and . Curr Biol., 2011, 21(21), R877–R883. https://doi.org/10.1016/j.cub.2011.09.040
M. C. Linder. The Biochemistry of Copper. Plenum Press: New York, 1991. https://doi.org/10.1007/978-1-4757-9432-8
M. M. O. Pena, J. Lee, and D. J. Thiele. J. Nutr., 1999, 129(7), 1251–1260. https://doi.org/10.1093/jn/129.7.1251
S. J. Jenniefer and P. T. Muthiah. Chem. Cent. J., 2013, 7(35), 1–15. https://doi.org/10.1186/1752-153X-7-35
J.-C. Lee, H. Takahashi, and Y. Matsui. Z. Kristallogr. – New Cryst. Struct., 2005, 220, 491–492. https://doi.org/10.1515/9783110938227.bm
A. A. Ajibola, F. Perveen, K. Jan, I. I. Anibijuwon, S. E. Shaibu, L. Siero, and W. Maniukiewicz. Crystals, 2020, 10(11), 991. https://doi.org/10.3390/cryst10110991
N. Abdullah, N. Sharmin, L. N. Ozair, A. R. Nordin, W. S. N. M. Nasir, and M. I. Mohamadin. J. Coord. Chem., 2015, 68(8), 1347–1360. https://doi.org/10.1080/00958972.2015.1016428
L. Findorakova, K. Gyoryova, M. Melnik, M. Koman, and F. A. N. El-Dien. J. Coord. Chem., 2010, 63(19), 3348–3355. https://doi.org/10.1080/00958972.2010.512083
E. K. Lim, S. G Teoh, and S. M. Goh. Polyhedron, 2009, 28(7), 1320–1330. https://doi.org/10.1016/j.poly.2009.02.006
J. Medvecka, J. Halaska, K. Jomova, and J. Moncol. Acta Chim. Slov., 2012, 5(1), 15–20. https://doi.org/10.2478/v10188-012-0003-5
R. P. Sharma, A. Saini, S Kumar, J. Kumar, R. S. Kumar, P. Venugopalan, and T Aree. Polyhedron, 2017, 123, 430–440. https://doi.org/10.1016/j.poly.2016.11.042
R. P. Sharma, A. Saini, P. Venugopalan, S. Khullar, and S. Mandal. Polyhedron, 2013, 56, 34–43. https://doi.org/10.1016/j.poly.2013.03.040
R. P. Sharma, A. Saini, S. Singh, A. Singh, P. Venugopalan, and V. Ferretti. J. Mol. Struct., 2010, 969(1–3), 155–162. https://doi.org/10.1016/j.molstruc.2010.01.063
S. S. Batool, S. R. Gilani, M. N. Tahir, W. T. A. Harrison, L. Mitu, and S. Mazharr. J. Coord. Chem., 2018, 71(16–18), 2569–2583. https://doi.org/10.1080/00958972.2018.1503653
M. A. Malati. Experimental Inorganic Chemistry. Harwood Publishing: Chichester, 1999.
Z. Otwinowski and Z. Minor. In: Methods in Enzymology / Eds. C. W. Carter and R. M. Sweet. Academic Press: London, 1977, Vol. 276, Part A, 307–326.
R. H. Blessing, Acta Crystallogr., Sect. A, 1995, 51, 33–38. https://doi.org/10.1107/S0108767394005726
A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. Moliterni, G. Polidori, and R. Spagna. J. Appl. Crystallogr., 1999, 32(1), 115–119. https://doi.org/10.1107/S0021889898007717
G. M Sheldrick. Acta Crystallogr., Sect. C, 2015, 71, 3–8.
L. J. Farrugia. J. Appl. Crystallogr., 1999, 32(4), 837–838. https://doi.org/10.1107/S0021889899006020
K. Brandenburg. DIAMOND, Version 2.0. Crystal Impact GbR: Bonn, Germany, 1998.
M. N. Burnett and C. K. Johnson. ORTEPIII, Report ORNL-6895. Oak Ridge, Tennessee: Oak Ridge National Laboratory, 1996.
W. J. Geary. Coord. Chem. Rev., 1971, 7(1), 81–122. https://doi.org/10.1016/S0010-8545(00)80009-0
M. Kato, H. B. Jonassen, and J. C. Fanning. Chem. Rev., 1964, 64(2), 99–128. https://doi.org/10.1021/cr60228a003
J. Bellamy. The Infrared Spectra of Complex Molecules, 2nd ed., Vol. 2. Chapman & Hall: London, 1980. https://doi.org/10.1007/978-94-011-6520-4
V. Zelenak, Z. Vargova, and K. Gyoryova. Spectrochim. Acta, Part A, 2007, 66(2), 262–272. https://doi.org/10.1016/j.saa.2006.02.050
S. S. Batool, S. R. Gilani, S. S. Zainab, M. N. Tahir, W. T. A. Harrison, M. S. Haider, Q. Syed, S. Mazhar, and M. Shoaib. Polyhedron, 2020, 178, 114346. https://doi.org/10.1016/j.poly.2020.114346
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interests.
Additional information
Text © The Author(s), 2021, published in Zhurnal Strukturnoi Khimii, 2021, Vol. 62, No. 9, pp. 1495-1507.https://doi.org/10.26902/JSC_id79926
Rights and permissions
About this article

Cite this article
Saini, A., Gupta, P., Bansal, P. et al. SYNTHESES, CHARACTERIZATION, X-RAY STRUCTURAL DETERMINATION, AND PACKING ANALYSES OF TERNARY COPPER(II) COMPLEXES: [Cu(2-PHENOXYACETATE/4-CHLOROBENZOATE)2(TEMED)]. J Struct Chem 62, 1398–1410 (2021). https://doi.org/10.1134/S0022476621090080
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022476621090080