
Abstract
Series of high-quality spherical poly(methyl methacrylate) particles with a polydispersity less than 5% are prepared by chain-growth emulsifier-free controlled radical emulsion polymerization of methyl methacrylate in water. The average diameters in the series varied from 237 nm to 447 nm. The physico-chemical properties of obtained submicrospheres can be varied to make them more stable and stronger by a modified classical synthesis technology whereby 4-10 vol.% of dispersion medium is replaced by acetone. 2D and 3D photonic crystal structures, opals, are prepared from poly(methyl methacrylate) submicrospheres. The crystal structure of the opals is studied by IR spectroscopy and scanning electron microscopy. According to the spectroscopic data, the poly(methyl methacrylate) particles contain significant amounts of water whose evaporation leads to the shrinkage of the spheres. The stereoregularity of the synthesized polymer is studied, the glass-transition temperatures of obtained samples are determined.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Photonic crystal structures (PhCSs) currently attract the interest of researchers and engineers in various fields of modern technologies due to their unique optical properties resulting from the commensurability between the periodicity parameter of the photonic crystal (PhC) and the radiation wavelength. Opals and opal-like structures, which are 3D periodic colloidal crystals (due to 3D ordering of colloidal particles), hold a special place in the PhC hierarchy, particularly, because of the fact that they can be prepared using relatively simple and inexpensive production technologies [1-3].
The most common way of preparing opal-like structures is self-assembly [4] of monodisperse polymer submicrospheres. Poly(methyl methacrylate) (PMMA), a well known hard amorphous synthetic polymer, is one widely used material in this field. PMMA is more known as acrylic glass due to its high optical transparency. This polymer possesses high weather resistance and good chemical, physico-mechanical, and electrical insulating properties and is widely used as a replacement for inorganic glass since it has high toughness, low weight, high resistance to destruction, and can be easily processed by mechanical and thermal methods [5]. Understanding key properties of PMMA may largely enhance the efficiency of technological and laboratory production, chemical and physical conversion, and to expand the application area of this polymer. Polymers containing specific chemical groups underlying their new properties in physical, chemical, biological, and pharmaceutical applications are referred to as functional polymers [6]. It was shown that PMMA is a promising polymer for practical applications in biomedical [7-9], sensor [10-12], electrochemical, optical [13, 14], and electrically conductive devices [15], analytical separators [16], solar cell production technologies [17-22], nanotechnologies [23-26], etc.
Scanning electron microscopy (SEM) and optical spectroscopy are currently the main most commonly used methods to study the structural features of opal [27, 28]. The present work is aimed at investigating the possibilities of Fourier transform infrared spectroscopy (FTIR) and scanning electron microscopy to study the crystal structure parameters of 3D high-order opal consisting of monodisperse poly(methyl methacrylate) submicrospheres prepared by a modified synthesis of monodisperse polymer spherical PMMA particles and a hybrid method [29] for the preparation of opal-like PhCSs.
EXPERIMENTAL
The properties of classical PMMA spheres have been well studied. However, the polymerization technique can be modified using a more complex dispersion medium [30] to prepare submicroparticles with variable properties [31]. The chosen modified dispersion medium was a water-acetone mixture.
The PMMA submicrospheres with a polydispersity smaller than 5% [29] were prepared according to known procedure [27, 32] in a water dispersion medium (submicrospheres B) to be compared with the particles prepared in a water-acetone dispersion medium (submicrospheres A). The following reagents were used without additional purification: methyl methacrylate (MMA) (99.8%) (VitaReaktiv, Dzerzhinsk, Russia), gaseous nitrogen (99.6%) (Fakel, Krasnoyarsk, Russia), 2,2′-azobis(2-methylpropionamidine)dihydrochloride (97%) (SigmaAldrich, Saint Louis, Missouri, USA) along with distilled water and acetone (Omsky kauchuk, Omsk, Russia) that was purified by distillation.
Series of high-quality PMMA submicrospheres with a very narrow polydispersity were prepared by chain-growth emulsifier-free controlled radical emulsion polymerization of MMA in water [2, 33]. The size of the PMMA spheres obtained by this method strongly depends on the composition of the synthesis mixture, stirring rate, and reaction temperature. The morphological features of the samples are investigated on a high-resolution field emission scanning electron microscope (FE-SEM) S-5500 (Hitachi, Japan) at an accelerating voltage of 3 kV and a probe current of 10 µA. The average diameters in the series varied from 237 nm to 447 nm. The chain-growth emulsifier-free controlled radical emulsion polymerization of MMA was described in detail in [2, 27, 29, 32, 34].
During the in situ synthesis, multiple frustrated total internal reflection (MFTIR) spectra of the emulsion were recorded every 5 min by a FT-801 FTIR spectrometer (Simex, Novosibirsk, Russia) to control at least two polymerization features. The first one is related to the termination of the polymerization process: abrupt temperature rise at deep reaction stages due to a strong gel effect typical of MMA polymerization [2, 27]). The second one is establishing the absence of the monomer (i.e., the fact that MMA has reacted completely) using the lines characteristic of MMA [2]. The prepared PMMA dispersions were poured in glass vessels for further use.
The following equipment was used: a SU3500 scanning electronic microscope (Hitachi, Japan) to visualize large areas (at small magnifications) of PMMA opal films; a K575XD sputter coater (Emitech, United Kingdom) to cover the PMMA opal surface with a thin platinum film before SEM studied and to make them electrically conductive. The samples were dried at 350 °C in a SIBLAB 30L laboratory oven with a digital thermometer (DION, Novosibirsk, Russia). The MFTIR spectra were recorded with a Vertex 70 FTIR spectrometer (Bruker, Germany). The glass-transition temperatures were studied by conducting calorimetry experiments with a premium class Phoenix 204 F-1 differential scanning calorimeter (DSC) (NETZSCH, Germany).
2D and 3D colloidal structures based on PMMA spheres were prepared by different methods (vertical and horizontal deposition on cover glasses, meniscus, and by the hybrid method) [3, 29]. To clean the surface, the cover glass substrates were first sonicated for 10 min in acetone and then washed with distilled water. The ability of the particles to form ordered structures was estimated by instantly wetting a vertical degreased cover glass in obtained undiluted (13-15 vol.% of PMMA submicrospheres) suspensions and placing it on a horizontal surface in a Petri dish until the film dried completely. The laboratory conditions were maintained with a humidity of 60% and a temperature of 23 °C. The purpose of this stage was to study various ordering defects rather than to prepare a perfect single-crystal film.
Polymer particles can to shrink, melt, and even explode in the electron microscope chamber under the electron beam action. Therefore the sizes of individual PMMA particles were estimated at small magnifications (yet making them visible), an accelerating voltage of 3 kV, and an emission current of 10 µA [2], but with the maximum resolution (2560×1920 pixels) and the lowest possible scan speed to minimize the noise and ensure the possibility of subsequent digital image processing. Then the spheres sizes were estimated using open source programs ImageJ and GIMP (GNU Image Manipulation Program) for scientific analysis, editing and processing of raster graphics [29].
The electron microscopic experiments revealed that submicrospheres A and B shrink and decrease in diameter under the electron beam action. The shrinkage degree was estimated by choosing a particle on the aluminum substrate that was further processed with a method described in detail in [3]. Table 1 lists the average diameters before the shrinkage, shrinkage degrees, average initial dispersion viscosities, and synthesis parameters.
RESULTS AND DISCUSSION
The 3D colloidal crystal with a high-quality surface was prepared by the hybrid method from submicrospheres B (sample 7). The whole sample was divided into three fragments for further research: central region (sample H1, opal-substrate interface), meniscus region (sample H2, opal-air interface), and central region (sample H3, opal-air interface).
Only short-range order was revealed by SEM in sample H1, which had a mainly disordered structure (Fig. 1a). The inset shows a 2D Fourier transform of PhC coordinates demonstrating the amorphous structure. The sample H2 has a multidomain structure in which the long-range order is sometimes replaced by disordered domains, domain walls vacancies, and other defects (Fig. 1b). The inset depicts the Fourier transform demonstrating the polycrystalline structure of the surface. This is the classical picture of opal growth in the meniscus [35, 36]. In general, the ordering is quite high. Finally, sample H3 showed the most defect-free structure (Fig. 1c). The inset shows the Fourier transform demonstrating the fcc crystal structure of the surface.

Electronic micrographs (top row) of colloidal structures based on PMMA submicrospheres and absolute reflectance spectra (bottom row) of prepared opals: disordered surface (111) of sample H1 (a); quasi-ordered multidomain surface (111) of sample H2 (b); perfectly ordered surface (111) of sample H3 (c); reflectance spectra of samples H1, H2, H3 for an angle of incidence of 12° (d); reflectance spectrum of sample H3 for an angle of incidence of 56° (multiple Bragg diffraction) (e); absolute reflectance as a function of the angle of incidence for sample H3 (f). The insets show a 2D Fourier transform of PhC coordinates.
The angular dependences of reflection spectra were recorded on a Vertex 70 FTIR Spectrometer within an angle range of 12-84° and a wavelength range of 400-1200 nm (Fig. 1d). Obtaining such spectra is an essential part of the spectroscopy of periodic colloidal structures such as opals. The reflectance of sample H1 was less than 4%, that of samples H2 and H3 was 30% and 86%, respectively. The maximum reflectance was reached at 12° and decreased with increasing angle. Full widths at half maximum were determined for each sample at 12°: FWHMH1 = 203 nm, FWHMH2 = 74 nm, FWHMH3 = 70 nm. From these data, the following quality factors λmax/Δλ were determined for the peaks at 14°: 4.1, 11.4, 12.3 for H1, H2, H3, respectively. λmax/Δλ at 12° for H3 was 12.4. All these data are summarized in Table 2 for the sake of easier comparison.
The modified Bragg– Snell law (1)
(where d111 is the interplanar distance; θ is the angle of incidence; neff is effective refractive index) along with the data of angle-resolved spectroscopy were used to calculate the following crystal lattice parameters: average diameter of submicrospheres B, packing density, and quality factor. The calculated data agree well with experimental SEM results (Table 2). Moreover, the spectral data obtained for sample H3 were used to estimate the intensity of maximum absolute reflectance λmax at normal incidence (Fig. 1f). As can be seen, the estimated absolute reflectance at 0° was 98%.
The reflectance spectrum at 56° measured for sample H3 showed a peak splitting at 706 nm and 725 nm (Fig. 1e). Small diffraction peaks were also observed at 481 nm, 524 nm, and 599 nm. These data indicate light diffraction on different planes of the 3D lattice of perfectly ordered spheres.
Electron microscopy revealed different shrinkage degrees of submicrospheres A and B. Thus, submicrospheres A (prepared with acetone) showed a smaller shrinkage under the electron beam action than submicrospheres B (prepared without acetone). Note also that submicrospheres B were damaged much more often during sample preparation than submicrospheres A.

SEM images of PMMA submicroparticles demonstrating shrinkage under the electron beam action: submicrosphere A (sample 5) (a); submicrosphere B (sample 4) (b). The outer boundary of the blurred halo around a clear-contoured bright spot is the boundary of the submicrosphere before the shrinkage. SEM images of PMMA submicroparticles that were damaged during sample preparation: submicrospheres A (sample 5) (c); submicrospheres B (sample 4) (d).
To estimate the degree of shrinkage, one of the particles on the substrate was chosen and measured by the method described in the experimental section. Fig. 2 shows the electron microscopy images of submicrospheres A and B before and after shrinkage. Fig. 2a shows shrinkage Δ = 7% for sample 5, Fig. 2b shows shrinkage Δ = 25% for sample 4. Fig. 2c and 2d show the damages of submicrospheres A and B, respectively. Submicrospheres B were found to be more plastic and damageable, whereas water-acetone submicrospheres A were more stable and stronger.
Below are the average shrinkages in the group (Fig. 3a) and particle sizes (Fig. 3b) plotted as functions of the acetone volume. Fig. 3c shows average shrinkage as a function of the average initial viscosity of dispersion. It is known that particle size depends on the monomer concentration (increases together with the monomer concentration). It is shown in the present work that the volume of particles depends also on the acetone concentration: the submicrosphere diameter increases together with the volume of added acetone. This statement is true for acetone concentrations in the dispersion up to 10%.
The experiments were carried out with the following assumptions. First, water-acetone medium provides higher concentrations of dissolved monomer than water medium. Second, the longer polymer chain, which precipitates into a solid phase from the solution, captures more of dispersion medium (water + acetone + MMA). Thus, higher acetone concentrations result in a larger number of MMA and oligomers captured by polymeric globules (nanoparticles) to form the polymer sphere [2]. As a result, nanoparticles A contain more MMA and growing oligomers than nanoparticles B by the time the gel effect starts resulting in larger and more dense particles in the acetone medium.
It was hypothesized in [37] that this phenomenon is often observed in dispersion polymerization when the monomer and the initiator are well dissolved in an organic dispersion environment. At the early stage of dispersion polymerization, oligomers formed in the homogeneous phase (i.e., polymerization stage in the solution) reach the critical length of the chain which can no more remain dissolved in the medium and precipitates from the solution to form primary nanoglobules. As the medium solubility increases, a smaller number of primary particles are formed; since most of oligomers occur in the medium, larger final particles are produced.

Average shrinkage as a function of acetone volume in the group: averaged experimental data (dots); approximated experimental data (curve). Fitting equation is also shown (a). Average submicrosphere diameter as a function of acetone volume for various MMA contents (b). Average submicrosphere diameter as a function of the average initial viscosity of dispersion (c).
In the case of emulsifier-free emulsion polymerization, the reaction also begins in the solution phase to form surfactant oligomeric forms resulting from the initiator decomposition. The diffusion of originating primary particles can be described by the Stokes–Einstein equation:
where T is the absolute temperature; kB is the Boltzmann constant; η is the viscosity of the liquid; r is the particle radius.
Reducing the dispersion viscosity due to the addition of acetone affects the mobility of primary particles and the growth of submicron PMMA spheres.
As an indirect confirmation of above conclusions, the DSC data show that the glass-transition temperature Tg increases with increasing MMA and acetone volumes. The lowest glass-transition temperature (121 °C) was observed for sample 1 and the highest one (126 °C) for sample 6. The difference between these glass-transition temperatures is probably related to different polymer chain lengths [38-40]. As is known, the glass-transition temperature is a non-linear function of the chain lengths in polymers [38]. Therefore, the difference between the glass-transition temperatures is not so large. According to [41, 42], Tg for for syndiotactic PMMA is ≈125 °C. Thus, the DSC results confirmed that the stereoregularity of prepared PMMA is syndiotactic (Fig. 4).
The data on the PMMA glass-transition temperature allow estimating the annealing temperature required for the hardening of opal matrix using slight sintering of polymer particles.

DSC data for samples 4 (water), 5 (30 mL acetone), and 6 (70 mL acetone).

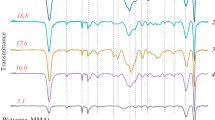
MFTIR spectra: PMMA dispersion (sample 4) (a); dried PMMA spheres (sample 6) (b).
Fig. 5 shows the obtained MFTIR spectra (final emulsion and PMMA submicrospheres dried at 100 °C for 3 h). The spectrum a freshly prepared emulsion was recorded on a Simex FT-801 spectrometer. The spectrum of dried spheres was recorded on a Bruker Vertex 70 spectrometer. The emulsion was to be dried since the spectrum of water overlapped with the polymer lines. As can be seen in Fig. 5a, no monomer lines are present in the reaction mixture, i.e. the monomer reacted completely.
Fig. 5b shows a distinct absorption band (from 1150 cm–1 to 1250 cm–1) which can be assigned to C–O–C stretchings. Two bands at 1388 cm–1 and 754 cm–1 are assigned to methyl group vibrations. The band at 987 cm–1 along with the bands at 1062 cm–1 and 843 cm–1 are characteristic vibrational absorptions of PMMA. The band at 1732 cm–1 shows the presence of the acrylate carboxyl group. The band at 1444 cm–1 can be assigned to C–H bendings of the –CH3 group. Two bands at 2997 cm–1 and 2952 cm–1 correspond to C–H stretchings of –CH3 and –CH2– groups, respectively. There are also two weak bands at 3437 cm–1 and 1641 cm–1 probably corresponding to the stretching of group –OH and to the bendings of molecules of physically absorbed water, respectively (Table 3).
Thus, it can be concluded that the obtained polymer is indeed a macromolecular syndiotactic PMMA, which is confirmed by works [43-45].
CONCLUSIONS
Series of high-quality spherical poly(methyl methacrylate) particles with a polydispersity less than 5% were prepared by a modified classical procedure of chain-growth emulsifier-free controlled radical emulsion polymerization of methyl methacrylate in water. Submicrospheres with an average diameter from 237 nm to 447 nm and variable physico-chemical properties were obtained. Replacing up to 10 vol.% of water on acetone in the dispersion medium during the synthesis yielded more stable and strong spherical particles. 2D and 3D opals based on the above particles were prepared. Dimensional parameters of obtained opals (particle diameters, degree of shrinkage under the electron beam action, crystal lattice cell parameters), stereoregularity of the synthesized polymer, and glass-transition temperatures of obtained samples were studied by scanning electron microscopy, infrared spectroscopy, and differential scanning calorimetry. It was established by electron microscopy and IR spectroscopy methods that the surface of the photonic crystal is highly ordered and exhibits high reflectance.
ACKNOWLEDGMENTS
The authors acknowledge the Krasnoyarsk Regional Center of Research Equipment of Federal Research Center “Krasnoyarsk Science Center SB RAS” for providing the equipment.
REFERENCES
M. A. Boles, M. Engel, and D. V. Talapin. Chem. Rev., 2016, 116, 11220.
O. V. Shabanova, M. A. Korshunov, I. V. Nemtsev, and A. V. Shabanov. Nanotechnol. Russ., 2016, 11, 633.
I. V. Nemtsev, O. V. Shabanova, N. P. Shestakov, A. V. Cherepakhin, and V. Y. Zyryanov. Appl. Phys., A, 2019, 125, 738.
K. Hou, W. Ali, J. Lv, J. Guo, L. Shi, B. Han, X. Wang, and Z. Tang. J. Am. Chem. Soc., 2018, 140, 16446.
N. Berrahou, A. Mokaddem, B. Doumi, S. Hiadsi, N. Beldjoudi, and A. Boutaous. Polym. Bull., 2016, 73, 3007.
F. Adams, P. Pahl, and B. Rieger. Chem. - Eur. J., 2018, 24, 509.
Y. Chen, L. Zhang, and G. Chen. Electrophoresis, 2008, 29, 1801.
C. Bouzigues, T. Gacoin, and A. Alexandrou. ACS Nano, 2011, 5, 8488.
S. Lazzari, D. Moscatelli, F. Codari, M. Salmona, M. Morbidelli, and L. Diomede. J. Nanopart. Res., 2012, 14, 920.
W.-K. Kuo, H.-P. Weng, J.-J. Hsu, and H. Yu. Appl. Sci., 2016, 6, 67.
S. Y. Lin, J. G. Fleming, D. L. Hetherington, B. K. Smith, R. Biswas, K. M. Ho, M. M. Sigalas, W. Zubrzycki, S. R. Kurtz, and J. Bur. Nature, 1998, 394, 251.
A. Bearzotti, A. MacAgnano, S. Pantalei, E. Zampetti, I. Venditti, I. Fratoddi, and M. Vittoria Russo. J. Phys. Condens. Matter, 2008, 20, 474207.
K. Gipson, K. Stevens, P. Brown, and J. Ballato. J. Spectrosc., 2015, 2015, 9.
I. Venditti. Materials, 2017, 10, 97.
H. Hashim, N. I. Adam, N. H. M. Zaki, Z. S. Mahmud, C. M. S. Said, M. Z. A. Yahya, and A. M. M. Ali. In: CSSR 2010-2010 International Conference on Science and Social Research: Conf. Proc., Kuala Lumpur, Malaysia, 2010. Piscataway, N.J.: IEEE, 2010, 485-488.
G. Bin Lee, S. H. Chen, G. R. Huang, W. C. Sung, and Y. H. Lin. Sens. Actuators, B, 2001, 75, 142.
L. Xu, C. Aumaitre, Y. Kervella, G. Lapertot, C. Rodríguez-Seco, E. Palomares, R. Demadrille, and P. Reiss. Adv. Funct. Mater., 2018, 28, 1706291.
X. Huang, S. Han, W. Huang, and X. Liu. Chem. Soc. Rev., 2013, 42, 173.
H.-Q. Wang, M. Batentschuk, A. Osvet, L. Pinna, and C. J. Brabec. Adv. Mater., 2011, 23, 2675.
B. Shao, Z. Yang, Y. Wang, J. Li, J. Yang, J. Qiu, and Z. Song. ACS Appl. Mater. Interfaces, 2015, 7, 25211.
D. Bi, C. Yi, J. Luo, J.D. Décoppet, F. Zhang, S.M. Zakeeruddin, X. Li, A. Hagfeldt, and M. Grätzel. Nat. Energy, 2016, 1, 16142.
M. K. Assadi, H. Hanaei, N. M. Mohamed, R. Saidur, S. Bakhoda, R. Bashiri, and M. Moayedfar. Appl. Phys. A: Mater. Sci. Process., 2016, 122, 821.
M. Perween, D. B. Parmar, G. R. Bhadu, and D. N. Srivastava. Analyst, 2014, 139, 5919.
J. H. Sung, H. S. Kim, H. J. Jin, H. J. Choi, and I. J. Chin. Macromolecules, 2004, 37, 9899.
L. Zhang, Y. Ren, S. Peng, D. Guo, S. Wen, J. Luo, and G. Xie. Nanoscale, 2019, 11, 8237.
X. Wang, P. Wang, Y. Jiang, Q. Su, and J. Zheng. Compos. Sci. Technol., 2014, 104, 1.
I. V. Nemtsev, O. V. Shabanova, and A. V. Shabanov. Sib. Zh. Nauki Tekhnol., 2012, 1(41), 126.
M. M. Gafurov and K. S. Rabadanov. J. Struct. Chem., 2009, 50(2), 246.
I. V. Nemtsev, I. A. Tambasov, A. A. Ivanenko, and V. Y. Zyryanov. Photonics Nanostruct. - Fundam. Appl., 2018, 28, 37.
R. D′Amato, I. Venditti, M. V. Russo, and M. Falconieri. J. Appl. Polym. Sci., 2006, 102, 4493.
R. De Angelis, I. Venditti, I. Fratoddi, F. De Matteis, P. Prosposito, I. Cacciotti, L. D′Amico, F. Nanni, A. Yadav, M. Casalboni, and M. V. Russo. J. Colloid Interface Sci., 2014, 414, 24.
O. V. Shabanova, A. V. Shabanov, and I. V. Nemtsev. Sib. Zh. Nauki Tekhnol., 2011, 4(37), 201.
N. Matsuura, S. Yang, P. Sun, and H. E. Ruda. Appl. Phys. A: Mater. Sci. Process., 2005, 81, 379.
E. V. Morozov, O. V. Shabanova, V. F. Pavlov, and O. V. Falaleev. Abstracts of Papers: 11th International Conference on Magnetic Resonance Microscopy (ICMRM), Beijing, China, Aug 14-18, 2011. China, 2011. NMR/MRI 66.
H. G. Campos, K. P. Furlan, D. E. Garcia, R. Blick, R. Zierold, M. Eich, D. Hotza, and R. Janssen. Int. J. Ceram. Eng. Sci., 2019, 1, 68.
K. T. Hufziger, A. B. Zrimsek, and S. A. Asher. ACS Appl. Nano Mater., 2018, 1, 7016.
Q. Ye, Z. Zhang, H. Jia, W. He, and X. Ge. J. Colloid Interface Sci., 2002, 253, 279.
K. O′Driscoll and R. A. Sanayei. Macromolecules, 1991, 24, 4479.
S. Ogden, L. Klintberg, G. Thornell, K. Hjort, and R. Bodén. Microfluid. Nanofluidics, 2014, 17, 53.
D. Marsh. Biochim. Biophys. Acta, Biomembr., 2010, 1798, 40.
L. Chang and E. M. Woo. Polym. Chem., 2010, 1, 198.
K. H. Liao, S. Kobayashi, H. Kim, A. A. Abdala, and C. W. Macosko. Macromolecules, 2014, 47, 7674.
L. Zeng, P. Bermel, Y. Yi, B. A. Alamariu, K. A. Broderick, J. Liu, C. Hong, X. Duan, J. Joannopoulos, and L. C. Kimerling. Appl. Phys. Lett., 2008, 93, 221105.
V. L. Furer. J. Struct. Chem., 1995, 36(2), 321.
K. Nakanishi. J. Pharm. Sci., 1963, 52, 716.
Funding
The research was carried out within the state assignment of the Ministry of Science and Higher Education of the Russian Federation (theme No. AAAA-A18-118041990041-8).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interests.
Additional information
Russian Text © The Author(s), 2021, published in Zhurnal Strukturnoi Khimii, 2021, Vol. 62, No. 4, pp. 684-693.https://doi.org/10.26902/JSC_id71171
Rights and permissions
About this article

Cite this article
Nemtsev, I.V., Shabanova, O.V., Tambasov, I.A. et al. METHODS TO DETERMINE CRYSTAL LATTICE PARAMETERS OF OPAL-LIKE STRUCTURES. J Struct Chem 62, 641–650 (2021). https://doi.org/10.1134/S0022476621040168
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022476621040168