
Abstract—
Using a model solution, we have demonstrated the feasibility of using titanium(IV) oxyhydroxyphosphate-based ion-exchange materials for extraction of Cs+ and Sr2+ cations from multicomponent high-salt bottom residues. Conditions for the use of the ion exchangers have been optimized experimentally: liquid : solid = 50, t = 25°C. It has been shown that Cs+ is effectively extracted at pH 2, whereas Sr2+, at pH 8. The zirconium-modified sorbent has been shown to have a higher affinity for metal cations. The modified sorbent is especially selective for cesium in the acid pH region owing to the higher mobility of the protons of its hydrogen phosphate groups in comparison with the unmodified sorbent composition.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
The operation of power reactors in nuclear power plants is accompanied by the formation of multicomponent liquid radioactive waste (LRW) [1, 2], which should be processed without delay. The most serious problem in handling LRW is posed by the bottom residue (BR), in which the total salt content can be as high as several hundred grams per liter. The prevalent salts are sodium and potassium nitrates. There are smaller amounts of chlorides and sulfates of these alkali metals [1]. A major contribution to the total specific activity of the LRW in question is made by radionuclides, such as 134,137Cs and 90Sr, whose half-lives are 2.06, 30, and 29.1 years, respectively [1–3]. For removing 134,137Cs and 90Sr radionuclides from high-salt multicomponent LRW, ion-exchange approaches are of interest [4]. To date, for extracting these radionuclides, researchers have proposed ion-exchange materials based on transition metal ferrocyanides, which are only selective to cesium, and those based on manganese dioxide [5], which are only selective to strontium and cannot ensure reliable radionuclide immobilization during storage. For resolving this technological issue, ion exchangers based on titanium(IV) phosphate compounds of various compositions can be of interest [6–8], in particular, amorphous hydrated titanium(IV) oxyhydroxyphosphates, which offer excellent sorption characteristics and ensure reliable radionuclide immobilization during prolonged storage [9, 10]. However, the feasibility of utilizing these ion-exchange materials in high-salt solutions has not yet been studied.
The objectives of this work were to test titanium(IV) oxyhydroxyphosphate-based ion exchangers for removing cesium and strontium cations from solutions modeling the composition of bottom residue solutions and optimize conditions for effective application of them.
EXPERIMENTAL
As shown earlier, the addition of dopants differing in acid–base properties from Ti(IV), in particular, the addition of Zr(IV), leads to polarization of functional hydrogen phosphate groups, considerably increasing the mobility of hydrogen ions.
Unmodified and Zr(IV)-modified ion-exchange materials were prepared stepwise, using a procedure proposed by Ivanenko et al. [10]. In the first step, hydrous titanium(IV) oxyhydroxide and mixed titanium(IV) zirconium(IV) oxyhydroxide precursors were prepared via precipitation from Ti-containing and Zr/Ti-containing reagents, followed by separation from the mother liquor and washing. In the second step, OH– groups were replaced by functional \({\text{HPO}}_{4}^{{2 - }}\) groups via treatment with orthophosphoric acid, separation, and drying at room temperature. Titanium was determined by atomic absorption and photocolorimetrically (with hydrogen peroxide), zirconium was determined volumetrically using Trilon B in the presence of Xylenol Orange as an indicator, and phosphorus was determined photocolorimetrically with ammonium molybdate (Leki-1107 photoelectrocolorimeter). The amounts of water and hydrogen phosphate groups were inferred from chemical analysis, differential thermal analysis (DTA), and thermogravimetry (TG) data. For DTA, we used an NTR-70 low-frequency thermoanalytical recorder in combination with a PRT-1000M programmed heater. Calcined Al2O3 was used as a reference substance. Thermogravimetric analysis was carried out with a VT-1000 tension balance. The temperature was measured by platinum/platinum–rhodium thermocouples in combination with a PP-63 potentiometer. The heating rate was 10°C/min. The specific surface area (S) of the sorbents was determined using temperature-programmed nitrogen desorption measurements with a Micromeritics TriStar II 3020 electronic surface area analyzer. The particle size distribution (PSD) was found using laser diffraction with a Shimadzu SALD-201 analyzer. Cs+ and Sr2+ concentrations in solutions was determined using a PerkinElmer ELAN 9000 DRC-e dynamic reaction cell inductively coupled plasma mass spectrometry (ICP-MS) system. The uncertainty in our measurements did not exceed 4%. The protons of the \({\text{HPO}}_{4}^{{2 - }}\) groups in our samples were ion-exchanged with metal cations under static conditions at a varied liquid : solid ratio, equilibrium pH value, and temperature. The working temperature chosen was maintained using an LOIP LB-140 thermostat. The equilibrium pH was adjusted with concentrated sodium hydroxide and nitric acid solutions. The solution pH was monitored with an Anion 7000 pH meter. An ESL-63-07 proton-selective electrode was used as an indicator electrode, and an EVL-1M3 Ag/AgCl silver/silver chloride electrode was used as an auxiliary electrode. After the sorption process, the liquid and solid phases were separated by filtration in a Buchner funnel through blue ribbon filter paper using an NIRA NVM 13 vacuum pump. The degree of metal cation extraction from solutions (R, %) was calculated using the relation R = (Vх/V0) × 100%, where Vx is the amount of metal cations extracted by the sorbent and V0 is the initial amount of metal cations in the aliquot. Distribution coefficients (Kd, mL/g)) were calculated using the relation Кd = Аα/(100 – А), where A is the percentage of sorbed metal cations and α is the ratio of the volume of the liquid phase to the weight of the sorbent.
RESULTS AND DISCUSSION
We have determined the chemical composition of the synthesized materials in the form of oxide components. Comparing chemical analysis, DTA, and TG data, we inferred the formulas of the sorbents (Table 1). Prior to characterization, the sorbents were ground and size-classified by sieving to separate particles less than 0.04 mm in size. Next, their surface properties were studied (Table 1).
The physical parameters obtained can be favorable for a high mass transfer rate in ion-exchange processes owing to the accessibility of the functional groups for metal cations. The Zr(IV) cations in the modified material increase the rate of olation and oxolation of titanium(IV) oxyhydroxy complexes during synthesis, resulting in a partial increase in particle size and a decrease in the specific surface area of the final product [10] (Table 1).
Conditions for the effective use of the titanium phosphate sorbents for extracting Cs+ and Sr2+ cations from solution were optimized using a sample with the composition TiOНPO4·4.16Н2О. The cations were extracted from a high-salt multicomponent solution modeling technological BRs forming during the operation of water–water power reactors, with the following composition (g/L): Na+, 5.8; K+, 5; \({\text{NO}}_{3}^{ - }\), 150; Cl–, 7; \({\text{SO}}_{4}^{{2 - }}\), 6.4; \({{{\text{B}}}_{{\text{4}}}}{\text{O}}_{7}^{{2 - }}\), 25.1; Cs+, 2.42 × 10—3; Sr2+, 21.2 × 10–3 (total salt concentration, 269.3 g/L; pH 7) [1].
We examined the effect of sorbent consumption (liquid : solid ratio) on Cs+ and Sr2+ extraction from solution (Table 2). The addition of one of the sorbents to a solution reduced its pH from 7 to 5, an equilibrium value, as a consequence of the substitution of metal cations for protons of \({\text{HPO}}_{4}^{{2 - }}\) groups.
An increase in liquid : solid ratio was found to considerably reduce the R of metal cations at a given pH value. This was probably caused by the decrease in the number of functional groups per unit volume of the suspension formed. The optimal phase ratio was liquid : solid = 50, which ensured maximum extraction of Cs+ and Sr2+ cations (Table 2).
We examined the effect of the equilibrium pH value of the sorption process on Cs+ and Sr2+ extraction. For this purpose, a required amount of an acid or alkali was first added to a model solution. Next, one of the sorbents was added at a constant liquid : solid ratio and temperature and the solution was stirred for 1 h (Table 3).
It was found that pH 2 ensured extraction of Cs+ cations (over 80%), whereas Sr2+ cations were not sorbed. In this pH range, the ion-exchange process involved strong competition between hydrogen ions and metal cations, which appreciably suppressed sorption of cations with a large effective radius, that is, of Sr2+ [9]. Raising the equilibrium pH value reduced Cs+ extraction and increased Sr2+ extraction, which reached a maximum value at pH 8, as evidenced by the R and Kd values in Table 3. This is probably due to the reduction in the competition of hydrogen ions with Sr2+ and the increase in the mobility of the protons in the \({\text{HPO}}_{4}^{{2 - }}\) groups of the sorbent. As a result, the selectivity of the sorption for cesium cations, which have a considerably smaller effective radius than do other metal cations [11], considerably decreases. This is accompanied by an increase in the extraction of metal cations with larger effective radii, in agreement with previously reported data [11]. Moreover, the material undergoes hydrolytic destruction, accompanied by the formation of TiO(OH)2, which is capable of sorbing strontium cations to form titanates [12, 13].
We studied the effect of temperature on Cs+ and Sr2+ extraction from a solution under optimal conditions (liquid : solid ratio and pH). For this purpose, the solution was heated to a required temperature, its pH was adjusted, and one of the sorbents was immersed in the solution, following which it was reprecipitated over a period of 1 h (Table 4). Raising the sorption extraction temperature from 25 to 60°C was found to have a negligible effect on the R for Cs+ and Sr2+, and Kd increased by less than twofold. This was possibly due to the insignificant change in the effective radius of the metal cations as a consequence of their dehydration in such complex multicomponent systems.
Under the experimentally found optimal conditions for the use of titanium phosphate ion exchangers (as exemplified by TiOНPO4·4.16Н2О), we carried out Cs+ and Sr2+ sorption from a model solution for the modified sorbent composition. The process was run at a temperature of 25°C, liquid : solid ratio of 50, equilibrium pH of 2 for Cs+ and 8 for Sr2+, and reprecipitation time of 1 h. The residual Cs+ concentration in solution was 0.18 mg/L, with R = 93% and Kd = 622 mL/g, and the residual Sr2+ concentration was 0.79 mg/L, with R = 96% and Kd = 1292 mL/g. Because of the high mobility of the protons in the \({\text{HPO}}_{4}^{{2 - }}\) groups of the modified sorbent, Cs+ sorption was more effective at pH 2: R increased by 10%, and Kd, by more than a factor of 2.5. At equilibrium pH 8, Sr2+ was sorbed essentially in the same manner as in the case of the unmodified ion exchanger composition. Modified samples are more hydrolytically stable [10], so the contribution of TiO(OH)2 to Sr2+ extraction was extremely small, unlike in the case of the zirconium-free sample.
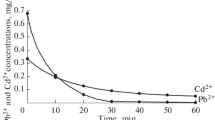
We examined the time variation of metal cation extraction for both sorbent compositions under the optimal conditions (Figs. 1, 2). For this purpose, we adjusted the pH of a model solution, added one of the sorbents, and then took an aliquot every 10 min for determining the residual metal cation concentration in the solution after separation of the sorbent. If necessary, the solution pH was again adjusted. It was found that, during the first 30 in of the ion-exchange process, most of both types of metal cations were sorbed by all of the sample compositions. In the acid region, the modified sorbent ensured a higher cesium extraction rate owing to the higher mobility of the protons of its functional groups, whereas in the alkaline region the strontium cation extraction curves for the two sorbent compositions were similar in shape. Running the ion-exchange process for ~40 min was shown to ensure complete chemical equilibrium.

Time variation of cesium cation extraction from solution by the sorbents with the compositions (1) Zr0.1(TiO)(OH)0.4(HPO4)⋅1.76H2O and (2) TiOНPO4·4.16Н2О at liquid : solid = 50, pH 2, and t = 25°C.

Time variation of strontium cation extraction from solution by the sorbents with the compositions (1) Zr0.1(TiO)(OH)0.4(HPO4)⋅1.76H2O and (2) TiOНPO4·4.16Н2О at liquid : solid = 50, pH 8, and t = 25°C.
Thus, we have tested titanium phosphate sorbents and demonstrated the feasibility of using them for extraction of cesium and strontium cations from solutions with complex chemical compositions modeling bottoms compositions.
CONCLUSIONS
We have studied the properties of titanium(IV) oxyhydroxyphosphate-based sorption materials for extraction of cesium and strontium cations from multicomponent high-salt solutions modeling the composition of the BR forming at nuclear power plants during the operation of nuclear power reactors. The conditions for the use of the sorption materials in such solutions, namely, the liquid : solid ratio, solution pH, and temperature, have been optimized. Experimental data demonstrate that cesium cations are well extracted in the acid region at pH 2, liquid : solid = 50, and t = 25°C, whereas strontium cations are well extracted at pH 8 and the same liquid : solid ratio and temperature. The zirconium-modified material is more effective in extracting cesium cations and ensures slightly better extraction of strontium cations in comparison with the unmodified sorbent. The particle size composition of the materials ensures a high rate of substitution of metal cations for protons of the \({\text{HPO}}_{4}^{{2 - }}\) groups.
REFERENCES
Ryabchikov, B.E., Ochistka zhidkikh radioaktivnykh otkhodov (Purification of Liquid Radioactive Waste), Moscow: DeLi Print, 2008.
Abdel-Karima, A.M., Zaki, A.A., Elwana, W., El-Naggar, M.R., and Gouda, M.M., Experimental and modeling investigations of cesium and strontium adsorption onto clay of radioactive waste disposal, Appl. Clay Sci., 2016, nos. 132–133, pp. 391–401. https://doi.org/10.1016/j.clay.2016.07.005
Mansy, M.S., Hassana, R.S., Selim, Y.T., and Kenawy, S.H., Evaluation of synthetic aluminum silicate modified by magnesia for the removal of 137Cs, 60Co and 152+154Eu from low-level radioactive waste, Appl. Radiat. Isot., 2017, no. 130, pp. 198–205. https://doi.org/10.1016/j.apradiso.2017.09.042
Yaroslavtsev, A.B., Ion exchange on inorganic sorbents, Russ. Chem. Rev., 1997, vol. 66, no. 7, pp. 579–596.
Milyutin, V.V., Nekrasova, N.A., and Kozlitin, E.A., Selective inorganic sorbents in modern applied radiochemistry, Materialy II Vserossiiskoi nauchnoi konferentsii s mezhdunarodnym uchastiem “Issledovaniya i razrabotki v oblasti khimii i tekhnologii funktsional’nykh materialov” (Proc. II All-Russia/Int. Sci. Conf. Research and Development in the Field of the Chemistry and Technology of Functional Materials), Apatity: Kol’sk. Nauchn. Tsentr Ross. Akad. Nauk, 2015, pp. 418–421.
Elghniji, K., Saad, M.E.K., Araissi, M., Elaloui, E., and Moussaoui, Y., Chemical modification of TiO2 by \({{{\text{H}}}_{{\text{2}}}}{\text{PO}}_{4}^{ - }\)/\({\text{HPO}}_{4}^{{{\text{2}} - }}\) anions using the sol–gel route with controlled precipitation and hydrolysis: enhancing thermal stability, Mater. Sci.-Pol., 2014, vol. 32, no. 4, pp. 617–625. https://doi.org/10.2478/s13536-014-0237-610.2478/s13536-014-0237-6
Ortiz-Oliveros, H.B., Flores-Espinosa, R.M., Ordonez-Regil, E., and Fernandez-Valverde, S.M., Synthesis of α-Ti(HPO4)2·H2O and sorption of Eu(III), Chem. Eng. J., 2014, vol. 236, pp. 398–405. https://doi.org/10.1016/j.cej.2013.09.103
Garc’a-Glez, J., Trobajo, C., Khainakov, S.A., and Amghouz, Z., α-Titanium phosphate intercalated with propylamine: an alternative pathway for efficient europium(III) uptake into layered tetravalent metal phosphates, Arab. J. Chem., 2017, no. 10, pp. 885–894. https://doi.org/10.1016/j.arabjc.2016.07.013
Trublet, M., Maslova, M.V., Rusanova, D., and Antzutkin, O.N., Mild syntheses and surface characterization of amorphous TiO(OH)(H2PO4)·H2O ion-exchanger, Mater. Chem. Phys., 2016, vol. 183, pp. 467–475. https://doi.org/10.1016/j.matchemphys.2016.09.002
Ivanenko, V.I., Lokshin, E.P., Aksenova, S.V., Korneikov, R.I., and Kalinnikov, V.T., Thermodynamics of cationic substitutions on titanyl hydrogen phosphate, Russ. J. Inorg. Chem., 2008, vol. 53, no. 4, pp. 503–508.
Korneikov, R.I. and Ivanenko, V.I., Extraction of cesium and strontium cations from solutions by titanium(IV) phosphate-based ion exchangers, Inorg. Mater., 2020, vol. 56, no. 5, pp. 502–506. https://doi.org/10.1134/S0020168520050088
Korneikov, R.I., Synthesis and properties of sorption materials based on titanium(IV) oxyhydroxyphosphates, Extended Abstract of Cand. Sci. (Eng.) Dissertation, Apatity, 2009.
Ivanenko, V.I., Yakubovich, E.N., Vladimirova, S.V., and Lokshin, E.P., Synthesis of powders of solid solutions between barium, strontium, and lead titanates and zirconates, Inorg. Mater., 2015, vol. 51, no. 9, pp. 916–922, https://doi.org/10.1134/S0020168515090095
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by O. Tsarev
Rights and permissions
About this article

Cite this article
Korneikov, R.I., Tikhomirova, E.L., Aksenova, S.V. et al. Sorption of Cs+ and Sr2+ Cations by Titanium(IV) Phosphates from Solutions Modeling the Nuclear Power Plant Bottom Residue. Inorg Mater 59, 81–85 (2023). https://doi.org/10.1134/S0020168523010119
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0020168523010119