
Abstract—
This work investigates in analytical capacities of arc atomic emission analysis (AAEA) in determination of As, Bi, Sb, Cu, and Te in rare earth metals (REM) and their oxides after preliminary group concentration using S,N-containing heterochain polymer sorbent. The investigations have been carried out using a Grand-Extra high-resolution spectrometer developed by VMK-Optoelektronika (Russia). Sorption kinetics and impurity extraction rate as a function of solution acidity have been studied for a selection of conditions of sorption concentration. Aiming at optimization of arc atomic emission detection of As, Bi, Sb, Cu, and Te, various flowcharts of their sorption concentration and subsequent treatment of obtained concentrated product have been considered with addition of a collector at various sorption stages. Powdered graphite has been used in this work as a collector in analysis of rare earth oxides, which is attributed to the relative simplicity of its emission spectrum and versatility. Analytical conditions and spectrometer parameters affecting the analytical signal (sample weight and composition, shape and size of electrodes, current intensity and operating mode of generator, interelectrode distance, wavelengths of analytical lines) have been selected. The evaporation curves of detected impurities have been analyzed, and the exposure time of As, Bi, Sb, Cu, and Te in the obtained sorption concentrate has been determined. Accuracy of the results has been evaluated using reference samples and by comparisons between methods. The results have been used for development of arc chemical atomic emission analysis of yttrium, gadolinium, neodymium, europium, scandium, and their oxides in the concentration range of n × (10–5–10–2) wt %.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Rare earth metals are called the vitamins for industry. Everyday all over the world, there appears new information about engineering innovations related to application of rare earth metals (REM) [1, 2]. Consumption is continuously expanding, and the possibilities are far from being exhausted; however, any use of REM assumes application of substances with strictly defined levels of chemical purity. In this regard, a critical issue is provision of quality control of REM and materials on their basis at various stages of their production and consumption.
Modern fields of science and engineering require constant expansion of the list of analyzed substances and associated impurities. The requirements on impurities contained in pure and highly pure substances are becoming more and more stringent. Not long ago, there existed demand for substances with purity in terms of a modest group of impurities. Nowadays, some fields of science require substances certified in terms of nearly all possible impurities. In addition, for various applications of the same substance, it is required to monitor the content of various impurities with low detection thresholds [3]. In many cases, instrumental sensitivity is insufficient for some elements, but it can be improved by preliminary separation and concentration of impurities.
This work is aimed at development of arc chemical spectral determination of As, Bi, Sb, Cu, and Te in rare earth metals after group sorption concentration of impurities.
The investigations were carried out using a Grand-Extra high-resolution spectrometer (VMK-Optoelektronika, Russia). The spectrometer is based on a new threaded nonclassic concave grating with ruling density of 2400 grooves/mm. The spectrum is recorded by two MAES analyzers, each containing a multichip assembly, units of electronic recording, and power supply. The entrance slit, concave grating, and radiation sensitive surfaces of two multichip assemblies are placed on a Rowland circle with the diameter of 1 m. The assembly with 12 strips records a spectral range of 190–350 nm and that with 8 strips records a range of 385–470 nm [4].
The instrumental capabilities of modern arc atomic emission analysis (AAEA) make it possible to detect non-rare-earth impurities in REM and their oxides at the level of n × 10–6–n × 10–5 wt %; however, direct determination of certain elements, arsenic, in particular, faces certain difficulties. Arsenic is among the most volatile elements in a carbon arc, positioned at the beginning of the series after phosphorus and mercury and before less volatile cadmium, zinc, and antimony. A few As spectral lines are positioned mainly in the short-wave region; here, additional processing of the spectrum and consideration for fractional evaporation with carrier prevent detection of arsenic at the required level of n × 10–4–n × 10–5 wt %: the lower limit of determinable contents in the best case is at the level of n × 10–3 wt %. In this regard, in order to achieve the required sensitivity of arsenic detection in REM and their oxides, preliminary concentration should be carried out.
Arsenic has a high affinity for sulfur: sulfuric compounds of arsenic is the form suitable for its extraction and concentration; hence, it was proposed to use S,N-containing sorbent based on primary ethylenediamine obtained by thiomethylation of polyamines (EDM).
As extraction rate as a function of solution acidity. The extraction rate of As was determined at various values of pH of the reference solution which together with As contained some other impurities, since application of EDM sorbent demonstrated its high efficiency as applied to a wide range of elements dictated by intermediate coordination of sorbed elements via nitrogen.
Oxides of the considered REM, pure in terms of impurities, with the weight of 1 g were placed into a 100 mL glass and wetted with water, 20 mL of hydrochloric acid was added, and the glass was heated at 60–70°C to complete dissolution. The solution was placed into a 500 mL beaker and solutions of impurities were added (Al, As, Ba, Be, Bi, B, Cd, Ca, Ce, Cs, Cr, Co, Cu, Dy, Er, Eu, Gd, Ga, Ho, In, Fe, La, Pb, Lu, Mg, Mn, Nd, Ni, Pr, Sm, Sc, Se, Tb, Tm, V, Yb, Zn, Sb, Ge, Hf, Mo, Nb, Si, Ag, Ta, Te, Sn, Ti, W, Zn) with the concentration of 100 µg/mL.
Sorption was performed from hydrochloric and sulfuric acid media in the static mode. The time of phase contact was 60 min at 100°C. Solution acidity was varied from 0.1 to 3 M HCl or H2SO4. The ratio of solution volume to sorbent weight was 1 : 1 (100 mL : 100 mg). Figure 1 illustrates the data on completeness of As extraction from solutions of the considered REM of different acidity.

Extraction rate of arsenic as a function of solution acidity: (a) HCl; (b) H2SO4.
If follows from the data that, with the increase in molarity of acids, the As extraction rate decreases. High extraction rates (92–95%) are observed upon the use of both 0.1 M HCl and 0.1 M H2SO4.
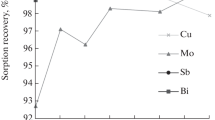
High extraction rates from 0.1 M HCl are also observed for Bi (99%), Te (96%), Cu (92%), and Sb (98%); partial extraction takes place in the case of Nb (65%) and Sn (79%); at the same time, from 0.1 M H2SO4, these elements are extracted somewhat worse (Fig. 2). Rare earth metals do not react with active centers of the sorbent, which facilitates separation of sorbed elements from REM base.

Extraction rate of impurities as a function of solution acidity: (a) HCl; (b) H2SO4.
Therefore, for As sorption, 0.1 M solution of HCl was selected; simultaneously, under the same conditions, Bi, Cu, Sb, and Te are quantitatively extracted to the obtained sorption concentrate. This group of elements was studied further. Simultaneous group concentration of these elements makes it possible to examine the accuracy of direct arc atomic emission analysis.
Extraction kinetics of impurities. The extraction kinetics of impurities from 0.1 M HCl was studied by their determination in the sorbate every 15 min. The obtained kinetic curves of extraction of analyzed substances by the S,N-containing EDM sorbent are illustrated in Fig. 3.

Kinetic curves of extraction of impurities by S,N-containing EDM sorbent.
The sorption rate depends on the time of contact between the sorbent and sample solution. Without heating and without agitation, the sorption is rather slow. Upon heating to 100°C and continuous agitation, the sorption rate of all analyzed substances in the initial 15 min reaches 40–50%, and in 60 min, complete sorption takes place. Therefore, the conditions of sorption concentration of impurities from REM and their compounds were experimentally determined; the flowchart is shown in Fig. 4.

Production of sorption concentrated product.
In AAEA, the obtained concentrate of impurities after sorption should be in the form suitable for direct excitation of the spectrum of the determined elements; that is, the impurities should be concentrated in a collector of low weight or localized on a moderate surface area. Powdered graphite was used as the collector in analysis of rare earth oxides, which was due to relative simplicity of its emission spectrum and versatility.
In order to optimize AAEA of the concentrate of impurities, various flowcharts of its production and subsequent processing were considered.
Flowchart 1.Direct analysis of sorption concentrate. The sorption concentrate obtained under the selected conditions was filtered, washed with diluted acid solution, and dried in air. Then spectral analysis of the sorbate was carried out.
Here, satisfactory results according to this flowchart were not obtained owing to uncontrollable emissions from the sample during exposure, which leads to low repeatability of the results even upon variation of the conditions of analysis and spectrometer parameters (shape and size of electrode, current intensity, and generator operating mode). Therefore, in order to improve the stability of supply of the sorption concentrate during the entire exposure, it would be reasonable to dilute it with powdered graphite.
Flowchart 2.Direct analysis of sorption concentrate with addition of powdered graphite. Mixing of the sorption concentrate with powdered graphite favorably affects the analytical signal of the considered impurities. The sorption concentrate was mixed with powdered graphite in the following ratios: 5 : 1, 2 : 1, 1 : 1, 1 : 2, 1 : 3. The stability of supply of the sample and intensity of spectral lines increase with content of powdered graphite; at the ratio of 1 : 1, a stable supply of the elements of the sample is fed to the arc discharge plasm. A further increase in the content of powdered graphite does not lead to an increase in the analytical signal, but can be a source of additional errors related to dilution of the sample upon determination of low contents of the impurities.
This flowchart is simple and does not require additional preparation stages; its execution takes a minimum amount of time.
Flowchart 3.Addition of powdered graphite during solution of sorbate. The obtained sorption concentrate was placed into a quartz cup, 2–5 mL of diluted nitric acid was added, and the cup was heated to complete dissolution. During heating, the matrix components C, N, and S are oxidized to oxides and extracted from the solution, and the considered impurities remain in the solution in the form of complex compounds. The substance from the cup was heated in a water bath until evolution of SO3 vapor, and 1–2 mL of a mixture of HCl and HNO3 and 100 mg of powdered graphite were added and evaporated to dryness.
A disadvantage of this flowchart is that the concentrate with powdered graphite should be evaporated to dryness, which can lead to loss of easily volatile compounds and distortion of experimental results.
Flowchart 4.Addition of powdered graphite during sorption. In this case, the sorption concentrate was diluted with powdered graphite directly during sorption. For this purpose, 100 mg of powdered graphite was added to 100 mg of sorbent, and sorption was carried out under previously selected conditions. Addition of powdered graphite complicates filtration of the obtained sorption concentrate; however, it provides a uniform distribution of its constituents, thus facilitating direct execution of AAEA. Hence, the stage of thorough mixing of sorption concentrate with powdered graphite, as proposed in Flowchart 2, is eliminated.
The aforementioned preparation procedures of the sorption concentrate for subsequent AAEA demonstrate that Flowcharts 2 and 4 are the simplest and provide complete sorption of the considered analyzed substances. However, Flowchart 2 has the additional stage of mechanical mixing of the sorption concentrate with powdered graphite, which can result in uncontrollable contamination of the concentrate; hence, in this work, Flowchart 4 was selected.
Selection of conditions of arc atomic emission analysis of sorption concentrate. The analytical lines of elements are located mainly in the short-wave UV band of the spectrum. Sorbent EDM was completely selective to REM; thus, upon analysis of the sorption concentrate, no matrix effects were revealed. In selection of spectral lines, only the requirements on sensitivity of determination of impurities were considered.
The selected most sensitive analytical lines of impurities are shown below:
Detected element | Analytical line, nm |
As | 234.98, 278.02, 286.04 |
Bi | 223.06, 293.87, 306.77 |
Cu | 324.75, 327.39 |
Sb | 231.15, 276.99, 287.79 |
Te | 214.28, 238.57, 238.6 |
Since the sorbate is composed mainly of sulfur, an easily volatile substance in carbon arc, and As, Bi, Te, Sb, Cu have comparatively high excitation potentials of spectral lines, then it would be reasonable to use a fairly deep electrode without a polished portion.
In this work, the electrodes with the diameter of 4 mm and different crater depth (4.6 and 9 mm) were applied.
On the basis of the weight of the obtained sorption concentrate, it would be reasonable to use a sample with the weight of 30 mg; in the case of the crater depth of 4 mm, it is required to compact the sample thoroughly since the crater is not filled completely. This results in emission of 20–30% of the sample during the initial seconds of arcing even at decreased current (5–8 A). Using an electrode with the crater depth of 6 mm, it is possible to achieve a uniform supply of components to the arc discharge plasma, which provides good convergence of results. In an electrode with the crater depth of 9 mm, the sample is positioned at the depth of about 4 mm from the edge, which leads to nonuniform heating of the sample and, hence, its supply to the plasma. In this regard, an electrode with the diameter of 4 mm and the crater depth of 6 mm was selected.
For selection of the current intensity, the curves of burning off of impurities and intensity of analytical lines were studied. At 8 A, supply of As, Bi, Sb, Te, and Cu is observed during the first 10–15 s. Increase in current leads to decrease in exposure time of the considered elements in the arc discharge plasma, and the sensitivity of impurity detection is also impaired. On the basis of the curves of burning off of impurities and intensity of analytical lines, the following conditions of analysis were selected: the current—8 A; the interelectrode distance—3 mm; the exposure time required for complete burning off of all considered impurities—20 s.
In order to obtain reliable AAEA results, it is required that the composition of reference samples (RS) be as close as possible to that of the considered samples. With this aim, pure EDM sorbent was mixed with RS based on powdered graphite in the ratio of 1 : 1 (15 mg of sorbent : 15 mg of RS based on powdered graphite). Calibration dependences were plotted using RS based on graphite with the contents of As, Bi, Cu, Sb, and Te in the range of 3 × 10–5–1 × 10–1 wt %.
On the basis of the obtained experimental results, the following conditions of AAEA of the As, Bi, Cu, Sb, and Te sorption concentrate in the range of 3 × 10–5–1 × 10–1 wt % were selected: the sample weight—30 mg; the electrode with the diameter of 4 mm and the depth of 6 mm; the operating mode of generator—direct current; the current intensity—8 A; the analytical distance—3 mm; the exposure time—20 s.
The accuracy of the results was verified using reference samples and by cross-method comparisons. This is exemplified by the results of analysis of neodymium oxide after sorption by S,N-containing EDM sorbent.
As evidenced by the presented data, it can be seen that the results obtained by the AAEA and ICP-AES methods agree between themselves and certified values. The absence of significant differences in the results obtained by these methods upon analysis of samples from solutions or solid phase after group concentration of As, Bi, Sb, Cu, and Te using S,N-containing sorbents confirms the efficiency of AAEA for rare earth metals and materials on their basis.
On the basis of the performed study, a procedure of chemical atomic emission analysis of yttrium, gadolinium, neodymium, europium, scandium, and their oxides was developed including group concentration of As, Bi, Cu, Sb, and Te using S,N-containing complex-forming EDM sorbent and facilitating execution of arc atomic emission determination of impurities in the range of 1 × 10–5–1 × 10–2 wt %. The relative standard deviation of the results of analysis according to the developed procedure is 20–8%, respectively.
REFERENCES
Sarychev, G.A., Kosynkin, V.D., and Glebov, V.A., Location of rare earth elements in the Mendeleev’s Periodic Table, Tsvetn. Met., 2012, no. 3, pp. 24–31.
Kablov, E.N., Ospennikova, O.G., and Vershkov, A.V., Rare metals and rare earth elements are materials for modern and advanced technologies, Aviats. Mater. Tekhnol., 2013, suppl. 2, pp. 3–10.
Cherednichenko, I.V., Ospennikova, O.G., Piskorskii, V.P., et al., The economics aspects of manufacturing of permanents magnets (a review), Nov. Materialoved. Nauka Tekhn., 2016, no. 4, pp. 58–76.
Labusov, V.A., Put’makov, A.N., Zarubin, I.A., and Garanin, V.G., New multichannel optical spectrometers based on MAES analyzers, Zavod. Lab., Diagn. Mater., 2012, vol. 78, no. 1 (2), pp. 7–13.
Funding
This work was supported by state assignment of the Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, in the field of fundamental scientific research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by I. Moshkin
Rights and permissions
About this article

Cite this article
Koshel’, E.S., Baranovskaya, V.B. & Doronina, M.S. Arc Atomic Emission Analysis of Rare Earth Metals and Their Oxides with Preliminary Sorption Concentration of Impurities. Inorg Mater 55, 1403–1407 (2019). https://doi.org/10.1134/S0020168519140085
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0020168519140085