
Abstract—Variants of miniplasminogen with an altered primary structure have been designed to study previously described changes in tryptophan fluorescence during plasminogen activation by urokinase. Miniplasminogens that contain the Trp141, Trp215, and Leu217 substitutions (the position of the amino-acid residue is specified by the Chymotrypsin nomenclature) have been tested. The activation of mutants containing a single tryptophan substitution induced changes in the fluorescence spectrum. No changes in the fluorescence spectrum were observed in the case of miniplasminogen containing a double tryptophan substitution. This indicates that both Trp141 and 215 contribute to the fluorescence spectrum shift; however, as suggested previously, the contribution of changes in the Trp215 microenvironment is greater. The Trp141, Trp215, and Leu217 amino-acid substitutions in miniplasminogen have no effect on the rate of plasminogen activation by urokinase. The replacement of the conserved Trp215 residue in serine proteases led to a significant reduction but not loss of the amidolytic activity of plasmin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Serine protease plasmin circulates in blood in the form of an inactive plasma enzyme plasminogen. Activation of plasminogen is a key stage of the process of dissolving blood clots (fibrinolysis). Moreover, both plasmin and plasminogen participate in a number of other physiological and pathological processes in the body, e.g., degradation of the extracellular matrix, angiogenesis, inflammation, and migration of tumor cells [1–6].
Plasmin is formed by urokinase or tissue plasminogen activators by the hydrolysis of the peptide bonds between the Arg and Val residues with coordinates 15 and 16, which leads to the reorganization of the spatial structure of the protease domain of the protein. (Hereinafter, the coordinates are given according to the chymotrypsin nomenclature. The Trp141 and Trp215 that are discussed in the text have coordinates of 685 and 761, respectively, when using Glu–Plg-numbering.)
In our previous work, we showed that the activation of plasminogen by urokinase is accompanied by significant changes in the fluorescence spectrum of the activated protein, i.e., the spectrum and, as a consequence, the fluorescence maximum of tryptophan for miniplasmin (mPln) is shifted by 6 nm in the long-wave region and the fluorescence intensity increases compared with miniplasminogen (mPlg). The time-dependence curve of the changes in the fluorescence spectrum of tryptophan during its activation correlates well with the kinetic curves of the activation, which have been obtained from both the measurement of the amidolytic activity of the resulting plasmin and the analysis of plasminogen cleavage products [7].
Comparison of the tertiary structures of plasmin and plasminogen allowed us to distinguish two tryptophan residues, Trp 215 and Trp141. During activation, the microenvironment of these residues changes significantly, which can cause differences in the fluorescence spectra of zimogen and the active enzyme (plasmin and plasminogen).
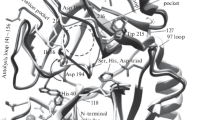
Previously the assumption was made that the Trp215 is responsible for the observed difference in the spectra caused by the significant change in the microenvironment during the conversion of plasminogen to plasmin [7]. When activated, Trp215 rotates around the axis passing through C-alpha 214 and C-alpha 216. In plasminogen, Trp215 is located in the substrate-recognizing pocket and completely blocks the access of the Arg side chain of the substrate to this zone (position Z, Fig. 1a). During rotation, the indole ring leaves the substrate-recognizing pocket (position Z) and moves to position A, which is between the 170–175 loop and beta chains F2 (226–231) and C2 (179–184). The benzene ring changes the polar environment from the negatively charged amino-acid residues (Asp194 and Glu143) to positively charged Arg175 and hydrophobic Leu217.

The microenvironment of Trp215 in plasminogen and plasmin.
The difference in the fluorescence spectra of zimogen and the active enzyme can depend on the change in the microenvironment of Trp 141 either. Trp141 itself does not change its position; however, the drift of Asp 194 and Val 16 during the plasminogen activation (Fig. 2) may affect the fluorescence of this tryptophan. The cleavage of the bond between Arg15 and Val16 leads to a radical change in the position of the Lys156, Glu143, and Asp194 residues relative to Trp141 (Fig. 2).

The microenvironment of Trp141 in plasminogen and plasmin.
To clarify the contribution of these tryptophan residues and their environment in fluorescence of mPln and mPlg, we designed variants of miniplasminogen with an altered primary structure. The introduced mutations affected both the Trp141 and Trp215 residues and the neighboring amino-acid residues. In this work, we studied the properties of the resulting proteins.
MATERIALS AND METHODS
Reactants. We used reactants of analytical grade from Sigma (United States) and chromogenic substrate S-2251 (Chromogenix, Italy).
Recombinant proteins.We designed and used a producer of human miniplasminogen as the initial material. Recombinant miniplasminogen is the truncated variant of the native plasminogen composed of the catalytic domain and one (fifth) kringle domain. The structure of the protein and producer were described in [8–10]. Mutations were introduced by the standard methods of genetic engineering and verified by sequencing. The resulting recombinant proteins were purified as previously described [7].
А producer of the urokinase (urokinase type plasminogen activator (single-chain pro-form, sc_uPA)) was also designed in our laboratory. The two-chain activated form of urokinase-type plasminogen activator (tc_uPA) was prepared by incubation of the single-chain pro-form sc_uPA with plasmin [11].
The purification of the enzymes was evaluated by electrophoresis, followed by the digitization of the electrophoregrams using the TotalLab or GelAnalyzer2010a programs. The purity of the tc_uPA and mPlg variants was 80 and 95%, respectively.
The fluorescence spectra of activated miniplasminogen and its variants were recorded on a Cary Eclipse Fluorescence Spectrophotometer (Agilent, United States) at 24°C in standard cuvettes with an optical path of 1 cm. The signal accumulation time in the kinetic measurements was 0.2–0.5 s. Both slits had an optical resolution of 5 nm. The scanning rate was 100 nm/min. The ratio of fluorescence intensities at 320 nm and 360 nm (I320/I360) with the excitation at 279 nm was taken as the relative value of fluorescence [12]. The fluorescence spectra of the mPlg solution were recorded, followed by the addition of tc_uPA. After stirring the mixture, the fluorescence spectra were recorded at regular intervals at 320 and 360 nm. The data were recorded from the moment of the tc_uPA addition to the mPlg solution until reaching a plateau of the fluorescence intensity curve. Upon completion of the reaction, the fluorescence spectrum of the resulting product was recorded.
Preliminary experiments showed the molar ratio mPlg and tc_uPA, which provided a slow activation of mPlg W141W215 with reaching the plateau for 20–30 min. In further experiments, we used 1000 nM and 25 nM concentrations for the mPlg variants and tc_uPA, respectively.
At these concentrations, the absorption of the protein solutions at 280 nm was 0.06 o. u., which did not require an absorption correction in fluorescence measurements.
The results were processed using the MS and Office 2003 Cary Eclipse software packages.
Enzymatic activity of miniplasminogen and its variants towards chromogenic substrate S-2251 was evaluated by the previously described method [7].
RESULTS AND DISCUSSION
The miniplasminogen used in this work as a starting variant for the introduction of mutations was described in detail previously [8–10]. It is a truncated version (a catalytic domain and one (the fifth) Kringle domain) of the natural human plasminogen. Mutations in the coding part of the protein were introduced by the genetic engineering methods. The list of the analyzed mPlg variants is shown in Fig. 3.

Variants of miniplasminogen. Replacements of amino-acid residues in the studied variants of miniplasminogen are shown in the upper part of the figure. The designations of the variants are shown on the left (W141W215 (plasminogen) is the initial variant). Alignments of the primary structures of the catalytic domain fragments of plasminogen (P00747), trypsinogen (P07477), thrombin (P00734), and human urokinase (P00749) are shown in the lower part. Coordinates of amino-acid residues are given according to chymotrypsin (top line) and Glu−Plg (bottom line) nomenclatures.
The fluorescence spectra of the mPlg variants with Trp141 and Trp215 substitutions before and after activation are shown in Fig. 4. As can be seen from the figure, the activation of variants containing single substitutions is accompanied by the red shift in the spectrum, whereas the double replacement does not lead to any changes in the spectrum during the mPlg activation. Therefore, both tryptophan residues (141 and 215) contribute to the shift of the spectrum. The change in microenvironment of Тгр215 has the most significant influence on the shift (Figs. 4b and 4c). It is noteworthy that the signal intensities in the fluorescence maximum of the activated mPlg variants with the single tryptophan replacements change differently, i.e., the fluorescence intensities of the active forms of the Trp215- and Trp141-containing variants are lower and higher, respectively, compared to the corresponding zimogen.

The fluorescence spectra of mPlg variants before and after activation by urokinase. W141W215 (a), F141W215 (b), W141Y215 (c), F141F215 (d): (1) before activation, (2) after activation.
The time-dependence curves of fluorescence intensities at 320 and 360 nm and the shift values (I360/I320) during the activation of zymogen by urokinase are shown in Fig. 5. The uroknase activation of the mPlg variant containing no W141 did not change the fluorescence intensity at 320 nm, whereas this value increased at 360 nm (Figs. 4b and 5a). The activation of the mPlg variants that contain the Ala, Tyr, or Phe amino-acid residues instead of Trp215 led to a decrease in the fluorescence intensity in the short-wave region (320 nm) of the spectrum, and no changes were observed in the long-wave region (360 nm) (Figs. 4a, 5b, and 6).

The kinetics of fluorescence change during activation of mPlg variants by urokinase. Variants F141W215 (a) (1) I360; (2) I320; (3) I320/I360) and F141F215 (4) I360, (5) I320; (6) I320/I360); variants W141W215 (b) ((1) I360; (2) I320; (3) I320/I360) and W141F215 (b) ((4) I360; (5) I320; (6) I320/I360).

The time dependence of fluorescence shift during urokinase activation of mPlg variants containing W215 replacements.
The kinetic curves for mPlg F141W215 can be approximated by an exponent (or hyperbola) with a characteristic time t1/2 = 12 min (Fig. 5a, Table 1). The activation of the variants that contain the replacement of Trp at position 215 occurred with similar characteristic times. In this case, the kinetic curves were the same for the W141F215 and W141Y215 variants, whereas the W141A215 variant led to the more pronounced shift in the fluorescence spectrum (Fig. 6, Table 1).
The analysis of the tertiary structure of serine proteases indicates that Trp215 in the activated enzyme closely contacts with the amino-acid residue located at position 217 (an example is shown in Fig. 1). We analyzed the mPlg variants containing leucine (L) replacements at this position by glutamic acid (E) or arginine (R). The choice of amino-acid residues for the replacement is due to the fact that glutamic acid and arginine are in a similar position in urokinase and thrombin, respectively (see Fig. 3). As expected, the change in the W215 microenvironment in the active enzyme (cf. Figs. 1 and 2) led to the change in the kinetic curves (Fig. 7).

The time dependence of fluorescence shift during urokinase activation of mPlg variants containing replacements at position 217. W141W215 is an initial variant of plasminogen (Leu at position 217).
In our previous work, we studied the dependences of the fluorescence spectra of mPlg and mPln on temperature and pH [7]. For all analyzed mutants, both temperature and pH dependencies did not differ from those for the wild-type protein in the state of both the pro-enzyme and activated enzyme (data not shown).
Enzyme activity. The amidolytic activities of the mPln variants are presented in Table 1. It should be emphasized that none of the studied variants had detectable enzymatic activity prior to activation of urokinase. The data show that the introduced mutations led to a decrease in the activity with the exception of L217E. It is noteworthy that, despite the conservation of tryptophan at position 215 in serine proteases [13, 14] (Fig. 3), its replacement leads to a significant decrease but not to a complete loss of the mPln enzymatic activity.
It is interesting to note that the value of the electric induction vector projection on the vector drawn from the center of the benzene ring to the center of the pyrrole ring of Trp215 calculated for plasmin [7] is almost the same for either trypsin or all hemostasis serine proteases, those do not require additional factors for their enzymatic activity. For the calculation, we used the 1lmw (for tcuPA), 1rtf (tPA), 1bui (plasmin), 1eb1 (alpha-thrombin), and 4cr9 (FXIa factor) structures. This field is primarily created by the negatively charged Asp102 amino-acid residue localized near the pyrrole ring of Trp215 and the positively charged amino-acid residues in the 170–175 loop (or Arg217 for tcuPA) located near the benzene ring of indole in Trp215 (see Fig. 1).
The coagulation factors FVIIa, FIXa, and FXa require binding to the activator factors TF (tissue factor), FVIIIa, and FVa, respectively, in addition to cleavage of the peptide bond between the amino-acid residues 15 and 16 [15]. When binding, cofactors stabilize (make them less mobile) both the position of the indole ring of Trp215 and the position of the 170–175 loop. This loop (like the autolysis loop) often is so mobile that it becomes invisible during the X-ray analysis [16, 17].
It is noteworthy that the replacement of the 170 loop in factor FVIIa by a shortened fragment (similar to the region of trypsin 170) makes the activity of FVIIa almost independent of the binding to the tissue factor [18].
We believe that any weakening of van der Waals contacts of the indole ring of Trp215 or the replacement of tryptophan, which enhances the mobility of the 215 amino-acid residue, should lead to a noticeable decrease in the enzyme activity because of the disorientation of the carbonyl group of the peptide bond between serine 214 and amino acid 215. The field of positively charged amino acids near the benzene ring of Trp215 leads to polarization of the indole ring of Trp215. Apparently, this polarization not only plays an important role in the fluorescence spectrum of tryptophan but also enhances the proton ability to be a donor in the formation of the hydrogen bond with the nearest carboxyl (as in plasmin) or carbonyl group or the water molecule (due to a decrease in the partial electron charge on the pyrrole ring) [19, 20].
The calculations of the structure of the environment of amino acid 215, which are beyond the limits of this publication, show that Trp215 in plasminogen can be replaced only by His without a significant loss of activity.
It should be mentioned that we did not investigate possible changes in substrate specificity in the mutant mPln variants. In the case of thrombin, the replacement of Trp215 with another amino acid leads to significant changes in the rates of hydrolysis of different substrates [21].
CONCLUSIONS
It was shown in this work that:
(1) Changes in the fluorescence spectrum during plasminogen activation by urokinase are caused by changes in the microenvironment of Trp141 and Trp215.
(2) Replacement of Trp141, Trp215, and Leu217 in mPlg does not affect the rate of activation of plasminogen by two-chain urokinase.
(3) Replacement of conservative Trp in serine proteases at position 215 does not lead to the complete loss of the amidolytic activity.
REFERENCES
R. B. Aisina, L. I. Mukhametova, D. V. Tyupa, et al., Russ. J. Bioorg. Chem. 40 (5), 516 (2014).
J. C. Chapin and K. A. Hajjar, Blood Rev. 29, 17 (2015).
D. A. Ayon-Nunez, G. Fragoso, R. J. Bobes, and J. P. Laclette, Biosci. Rep. 38, BSR20180705 (2018).
E. V. Kugaevskaya, T. A. Gureeva, O. S. Timoshenko, and N. I. Solov’eva, Biomed. Khim. 64, 472 (2018).
E. I. Deryugina and J. P. Quigley, J. Biomed. Biotechnol. 2012, 564259 (2012).
C. Oh, J. Hoover-Plow, and E. F. Plow, J. Thromb. Haemost. 1, 1683 (2003).
T. I. Belyanko, Ya. G. Gursky, N. I. Dobrynina, et al., Biophysics (Moscow) 63, 683 (2018).
R. Sh. Bibilashvili, A. A. Belogurov, Ya. G. Gursky, et al., RF Patent No. 2 432 396 (November 24, 2009).
R. Sh. Bibilashvili, A. A. Belogurov, Ya. G. Gursky, et al., RF Patent No. 2 432 397 (November 24, 2009).
Ya. G. Gurskii, M. M. Minashkin, E. S. Feoktistova, et al., Appl. Biochem. Microbiol. 46, 776 (2010).
A. Ya. Shevelev, T. N. Barshevskaya, A. A. Belogurov, et al., Mol. Biol. (Moscow) 20, 778 (1986).
E. A. Burstein, N. S. Vedenkina, and M. N. Ivkova, Photochem. Photobiol. 18, 263 (1973).
G. M. Yousef, M. B. Elliott, A. D. Kopolovic, et al., Biochim. Biophys. Acta – Proteins Proteomics 1698, 77 (2004).
A. Jendroszek, J. B. Madsen, A. Chana-Munoz, et al., J. Biol. Chem. 294 (10), 3794 (2019).
H. H. Versteeg, J. W. M. Heemskerk, M. Levi, and P. H. Reitsma, Physiol. Rev. 93, 327 (2013).
R. B. Peacock, J. R. Davis, P. R. L. Markwick, and E. A. Komives, Biochemistry 57, 2694 (2018).
J. J. Madsen, E. Persson, and O. H. Olsen, J. Thromb. Haemost. 13, 262 (2015).
A. B. Sorensen, J. J. Madsen, L. A. Svensson, et al., J. Biol. Chem. 291, 4671 (2016).
L. J. Juszczak and A. S. Eisenberg, J. Am. Chem. Soc. 139, 8302 (2017).
A. S. Eisenberg, M. Nathan, and L. J. Juszczak, J. Mol. Struct. 1118, 56 (2016).
D. Arosio, Y. M. Ayala, and E. Di Cera, Biochemistry 39, 8095 (2000).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Translated by A.S. Levina
Abbreviations: mPln, miniplasmin; mPlg, miniplasminogen; sc_uPA, single-chain pro-form of urokinase-type plasminogen activator; tc_uPA, two-chain activated form of urokinase-type plasminogen activator.
Rights and permissions
About this article

Cite this article
Belyanko, T.I., Feoktistova, E.S., Skrypina, N.A. et al. A Study of the Structure of Trypsin-Like Serine Proteinases. 2. A Study of Tryptophan Fluorescence in Variants of Miniplasminogen with an Altered Primary Structure. BIOPHYSICS 64, 331–338 (2019). https://doi.org/10.1134/S0006350919030035
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006350919030035