
Abstract
In this paper, the changes in the frequency and amplitude of action potentials (AP) were investigated depending on the depolarization caused by the Ca2+ channels activity during the spontaneous synchronous activity (SSA) of hippocampal neurons in culture. It is known that the pacemaker neuronal electrical activity can be both tonic and bursting. Using the image analysis to measure [Са2+]i and patch-clamp to register the membrane potential we show that depolarization caused by the GABA(A) receptor inhibitor results in a pattern of SSA in which tonic APs frequency of 2−3 Hz are generated by a neuron without any changes in cytosolic free Ca2+ concentration, ([Ca2+]i). The tonic mode is interrupted by bursts activity, which is accompanied by slow depolarization and calcium pulses. The inhibitor of T-type calcium channels, ML218, suppresses this process. The frequency and amplitude of the AP are regulated by slow depolarization pulses as follows: on the depolarization front, the APs frequency increases. At the same time, the amplitude decreases due to Na+ channels inactivation. The higher is the depolarization rate, the higher is the APs frequency. If slow depolarization amplitude exceeds Na+ channels reactivation potential, the neuronal impulse activity stops. As the [Ca2+]i increases and Ca2+-dependent K+ channels are activated, depolarization amplitude decreases slowly, and Na+ channels are reactivated, which leads to a gradual increase in the amplitude of APs against the background of depolarization decrease. The frequency of APs on the backside of depolarization pulse slows down to 3–4 Hz due to the occurrence of the faster Ca2+-potential oscillations (micro bursts of AP). Under these conditions, only one AP can be generated due to rapid depolarization at the leading edge. After that, APs generation is suppressed due to Na+ channel inactivation. The frequency of APs in this case coincides with the Ca2+ channel activation frequency (3−4 Hz). The burst firing is terminated due to [Ca2+]i increase, Ca2+-dependent K+ channels activation, and voltage-gated Ca2+ channels (VGCC) inactivation. As a result, the membrane is hyperpolarized even more (10 mV lower than the critical potential), that suppress AP generation, activate HCN-like channels and reactivate Na+ and VGCC. The activity of HCN-like channels increases, the membrane slowly depolarizes and reaches the threshold potential. The generation of tonic APs begins, and then, the Ca2+ channels opens, and Ca2+ potential and Ca2+ signal induced again. Thus, the Ca2+-channels, is determining the pulse of slow depolarization, control the frequency and the amplitude of APs during SSA, regulating the activation and inactivation conditions of Na+ channels. The T-type channels inhibitors reduce the duration of burst firing and Ca2+ impulse in neurons in vitro, stimulating their survival during hyperexcitation and ischemia. Thus, reducing the Ca2+ pulse duration caused by the inhibitors of T-type Ca2+-channels, can be one of the reasons for the known neuroprotective effect of these compounds.
Keywords: spontaneous synchronous activity of neurons, calcium signal, T-type calcium channels, voltage-gated calcium channels, action potential, genesis of bursting activity, action potential bursts, depolarization shift, critical potential
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Burst firing genesis. The primary neuronal function is to generate APs. The periodic electrical activity of neurons can be divided into background generation of APs, or tonic, and burst firing activity [1]. It is believed that tonic activity of APs occurs at certain depolarization, when Ca2+ channels are still inactivated, and Na+ channels have been already reactivated. However, there is currently no single criterion for stack of APs definition. In different areas of electrophysiology under the firing activity of the neural network are implied different phenomena [2]. In the present work, we will designate a burst firing of APs as the sequence of APs, followed by the Ca2+ pulse (or Ca2+depolarizing current) during the spontaneous synchronous activity (SSA) of neurons.
It has been shown earlier that different neurons during the SSA generate Ca2+ pulses of different shapes and amplitudes [3]. Differences in the form of calcium signals imply differences in the slow depolarization pulses. Currently it is not clear how these differences define the form of the burst activity, and how slow depolarization pulses influence on the structure of APs in the burst. Transition from tonic to burst firing activity involves the shift of resting potential towards depolarization (depolarization shift) to the values exceeding the critical potential (the threshold potential at which AP is initiated). Depolarization shift of the membrane in a certain neurons in the brain, is accompanied by hyperexcitability of these neurons in the form of induced and spontaneous epileptiform spiking discharges [4]. Phenomenon of hyperexcitation is also observed during depolarization at the initial stage of ischemia and it is suppressed by the inhibitors of T-type Ca2+ channels [5, 6]. In fact, this depolarization shift may result not only from the calcium channels opening, but also from the chloride or potassium channels closing. The range of depolarization pulse duration is wide enough, depending on the nature of depolarizing channels. For example, VGCC are capable of generating fast depolarization pulses, and potassium channels, regulated by calcium or G-proteins, are able to generate very slow depolarization pulses.
T-type Ca2+ channels play an important role in genesis of the burst firing activity. Many publications are devoted to a long history of the T-type Ca2+ channels discovery and study (see the review [7]). Depolarization caused by these slow, low-threshold, Ca2+ channels of low conductivity (8 pS) initiates AP, lowering the excitation threshold [8, 9]. Depolarization amplitude regulates the APs frequency and Na+ channel inactivation [10]. Low-threshold channels are activated by weak depolarization (at the potentials more positive than –70 mV) with further quick (t ≈ 20–50 ms) inactivation [8, 11]. It is believed that only the T-type channels can support Ca2+ entry and neuron pacemaker activity with such negative potentials [12]. For comparison, L‑type dihydropyridine-sensitive Ca2+ channels are activated at the potentials above 10 mV, characterized by the higher conductivity (25 pS) and very slow inactivation kinetics (t ≈ 500–2000 ms) [12]. In addition, they are regulated by G-proteins, which can provide the longer-term depolarization [13].
VGCC not only depolarize the cells, but also increase [Ca2+]i [14], which triggers neurotransmitters’ secretion, closes the channel itself with further it inactivation. Hyperpolarization up to –70...–85 mV is required to reactivate T-type Ca2+ channels [15]. Hyperpolarization, needed for channels reactivation and termination of the burst firing activity, may occur due to activation of the calcium-activated K+ channels [16] during Ca2+ transport into the cells via VGСC. In some cells, the T-type Ca2+ channels are associated with BK [17] and SK channels [18]. Therefore, the K+ channels regulators determine the frequency of the burst firing activity and Ca2+ pulses [19]. The K+ channels are closed later at the intracellular calcium ions concentration decrease.
In addition to potential, several other endogenous mechanisms for regulating T-type calcium channels are known. CaV3.2 channels are activated САМКII [20], and stimulated by proteinkinase A and proteinkinase C phosphorylation [21]. Endocannabinoid [22, 23] and arachidonic acid inhibit the channels [24, 25] as well. βγ subunit of G-proteins selectively inhibits CaV3.2 [26], causing hyperpolarization shift of inactivation potential [27]. Activation of muscarinic acetylcholine receptors associated with Gaq/11 selectively inhibits CaV3.3 [28].
T-type channels are shown to participate in regulation of the epileptic rhythms [29], pain [30], Parkinson disease [31], and ischemia [5]. The channel blockers are used in the treatment of these diseases [31]. Neuroprotective effect of the channel blockers is also shown in vitro at the cellular level [16, 27, 30]. In the study of signaling pathways, regulating the activity of the channels, an attention is focused to inhibition of the T-type channels, the receptors associated with different Gi-proteins [14, 29], and the cGMP signaling pathway activators [32].
In the development central nervous system neurons form numerous synaptic contacts, as well as a neural network, where SSA of APs and Ca2+ pulses (Bursting) are observed. Violation of the SSA is seen in many neurological disorders and neurodegenerative diseases [33]. The main cause of the violations is depolarization (as many of the channels determining the excitability are voltage-gated). If close-to-threshold depolarization increases the frequency of discharges, excessive depolarization may suppress the activity due to inactivation of the Na+ channels. Transition from tonic APs to burst firing activity has been described for dopaminergic pacemaker neurons [34]. It is believed that rhythmic pacemaker activity of dopaminergic neurons in the substantia nigra pars compacta is necessary to maintain the basal level of dopamine in the striatum [35, 36] and at the same time is one of the important reasons for high vulnerability [37–39] associated with Ca2+ impulse duration. Rhythmic pacemaker activity is due to the properties of the pore-forming subunit of CaV1.3 of the L-type Ca2+ channels. However, general theory of the burst firing genesis and the rhythm control mechanisms is not yet available. In the present work we studied the dependence of the frequency and the amplitude of APs on the slow depolarization gene-rated by Ca2+ channels and on [Ca2+]i in the cell. Furthermore it has been shown how the slow depolarization pulses can encodings information transmitted AP.
METHODS
Membrane potential measurements. To analyze changes in membrane potential and ion channels activity, we used electrophysiological method (patch-clamp) in whole-cell configuration. The system of electrophysiological measurements (Molecular Devices) is built into the fluorescence station Axio Observer Imager Z1 (Carl Zeiss, Germany) equipped with a high-speed camera Hamamatsu ORCA-Flas, which allowed to study optical and electrophysiological parameters of the living cells simultaneously.
For currents registration an Axopatch 200B amplifier (Molecular Devices), ADC, Digidata 1440A and pClamp10 2 software. (Molecular Devices) were used. Changes in membrane potential were recorded in I-clamp mode. Electrophysiological data were analyzed using Clampfit 10.2.
[Ca2+]i measurements. To register quick changes of [Ca2+]i we used the Carl Zeiss Cell Observer on the basis of the inverted motorized microscope, Leica DMI6000, with a high-speed monochrome CCD-camera Hamamatsu C9100 and a high-speed light filter replacing system, Leica’s Ultra-Fast Filter Wheels. (switching time 10–30 ms). In the experiments we used an objective lens Leica HC PL APO 20×/0.7 IMM and Leica EL6000 illuminator with high-pressure mercury lamp HBO 103 W/2 as a source of fluorescence excitation. The [Ca2+]i was evaluated using a double-wave probe Fura-2, (Molecular probes, USA), in accordance with the known method [40, 41]. For cells staining Fura-2AM ether was used in a final concentration of 4 µm in a Hanks solution containing (in mM): 156 NaCl, 3 KCl, 1 MgSO4, 1.25 КН2РО4, 2 CaCl2, 10 glucose and 10 HEPES, рН 7.4. 200 µL of freshly prepared dye solution was added to each glass with the cell culture and incubated in the thermostat for 40 min at 37oC. Then, the cells were washed out with Hanks solution supplied with 0.2 mM L-arginine and incubated for 10–15 min to complete dye de-esterification.
For routine [Ca2+]i measurements we used an image analysis system based on an inverted motorized microscope Axio Observer Z1 (Carl Zeiss, Germany), equipped with a high-speed monochrome CCD ca-mera, Hamamatsu orca-Flash 2.8. As a source of fluorescence excitation, Lambda DG-4 Plus illuminator (Sutter Instruments) was used. For Fura-2 excitation and registration, we used a 21HE filter set (Carl Zeiss, Germany) with excitation filters BP340/30 and BP387/15, a beam splitter FT-409 and an emission filter BP510/90, objective lens Plan-Neofluar 10×/0.3. The coverslip, containing the cells loaded with Fura-2, was further mounted in the experimental chamber. The medium volume in the chamber was 0.5 mL. The reagents were added and washed off by replacing medium in a tenfold volume with the system that provides perfusion at the rate of 15 mL/min. A series of images were obtained at 1–2 s intervals. The recording time of one two-channel frame did not exceed 400 ms. Neurons in the experiment were detected by SSA and by the rapid response to depolarization caused by short-term (20 s) application of 35 mM KCl.
The resulting two-channel (when excited Fura-2 340 and 380 nm) time series of images were processed in the program ImageJ with plugin “Time series analyzer”. During image processing, we measured calcium response amplitudes of Fura-2 loaded cells expressed as the ratio of fluorescence intensities of Fura-2 upon excitation at the wave lengths of 340 and 380 nm. Origin 9.1 software packages were used for data processing, graph creation and statistical analysis. All values were given as mean signal of N cells in field of view ± standard deviation (N ± SD), or as typical calcium responses of the most cells or as signal of individual neurons. The data were compared statistically using one-way ANOVA, followed by the post hoc Student-Newman-Keuls test or a paired t-test and considered significantly different at p ≤ 0.05. All evidences were obtained from at least 3 different glass coverslips and 2–3 independent cell isolations.
Materials. N-methyl-D-aspartate (NMDA), bicuculline, domoic acid, and L-arginine were purchased from Tocris Bioscience (UK); balanced salt Hank’s solution, neurobasal medium, supliment B-27, fetal bovine serum (Gibco), gentamicin – Dal’khimfarm (Russia); 0.1% polyethylenimine; L-glutamine, L‑glutamate (Sigma-Aldrich, USA); KCl (Khimmed, Russia); Fura-2AM (Molecular probes, USA). All animal studies were carried out in accordance with the requirements of the legislation and were approved by the Animal Ethics Committees of ITEB RAS and IBC RAS. The work was performed on mixed culture of hippocampal neurons and astrocytes of different period of cultivation (shown in figures). Cell viability was controlled by double-staining with propidium Iodide and Hoechst 33342 probes.
RESULTS
The transition to the burst activity induced by depolarization. By measuring [Ca2+]i with fluorescent probe Fura-2 and the membrane potential with the patch-clamp in the whole-cell configuration, we investigated the transition from tonic to burst activity in rat hippocampal neurons in the culture upon inhibition removal by the agonist of GABA(A) receptors, bicuculline. At the same time, we recorded regular changes in the frequency and the amplitude of APs during slow depolarization pulse.
Figure 1 shows that the inhibitor of GABA(A) receptors, bicuculline, depolarizing neurons due to the Cl− channels closure, induces the mode of SSA, in which tonic APs frequency of 2–3 Hz (upper curve) is generated for some time by the neuron without [Ca2+]i changing (Fig. 1a, lower curve). During this period, the AP-dependent depolarization does not open Ca2+channels, as the latter are inactivated. Posthyperpolarization potential, developed after AP in the presence of bicuculline, is assumed to be not enough for Ca2+ channels reactivation. The tonic activity of APs (2–3 Hz) is interrupted periodically by AP burst firing. At this time the neuron generates a slow depolarization pulse and the corresponding Ca2+ signal. The frequency of Ca2+oscillations is ≈ 0.01 Hz. The burst activity (see Figs. 1a, 1b, 1d) begins with the opening of low-threshold voltage-gated calcium channels, which occurs due to their reactivation on the background of slow depolarization pulse [42]. The mechanism of periodic activation of these channels is not entirely understood.

(a) Spontaneous tonic AP activity during SSA periodically is interrupted by spontaneous burst of APs accompanied Ca2+spike and slow depolarization pulse. Electrical activity (upper curve) and [Ca2+]i of neuron (lower curve) during SSA. The electrical activity of the neuron is accompanied by the Ca2+ spike generation only during the burst activity. (b) AP generation disappears at the maximum of depolarization and gradually recovers during repolarization. A burst of APs ends with a slow strong hyperpolarization (~10 mV below the critical excitation potential), during which the neuronal impulse activity stops. This is followed by a slow depolarization phase, and when the critical excitation potential is reached, APs generation begins again. Bicu-culline (10 µM) is present in the medium. Hippocampal culture at 15 DIV (days in vitro). (c) A burst of APs from Fig. 1b during Ca2+ pulse. Slow decay of depolarization accompanied by an increase in the amplitude of AP, suggesting the reactivation of the Na+ channel. The frequency of APs on the back of the depolarization decreases continuously from 10 to 3 Hz due to the depolarizing inward current decrease. (d) The beginning of APs burst from Fig. 1c. The high-frequency APs, attenuated by amplitude (frequency ~ 140 Hz) are generated on the front of the slow depolarization (due to Na+ channels inactivation). (e) Micro burst consisting of one AP on the back front of the slow depolarization pulse from Fig. 1c is shown. The depolarization amplitude in the micro burst is constant and coincides with the amplitude of the initial depolarization (in Figs. 1c, 1d). The duration of the micro burst is ~ 10 ms.
The amplitude and the frequency of APs change on the background of slow depolarization pulse. Effect of calcium concentration.Figures 1c-1e shows the fragments of recording action potential trains (Fig. 1b) duration of several seconds (Fig. 1c), front (Fig. 1d) and back burst firing front (Fig. 1e). Against the background of slow depolarization front (Fig. 1d) burst firing of high-frequency APs activity is observed. The following sequence of events is assumed: the first AP always has maximum amplitude, since Na+ channels are fully reactivated by this time (Figs. 1c, 1d). Due to the high rate of slow depolarization increase, the frequency of APs in this area increases and tends to theoretical maximum (150 Hz in this case, Fig. 1d). The amplitude of each subsequent AP decreases with increasing depolarization (Fig. 1d), that indicates the potential-dependent inactivation of Na+ channel. Then, the amplitude of slow depolarization reaches the value at which all Na+ channels are inactivated and AP generation does not occur, since a certain degree of hyperpolarization is required for reactivation of the Na+ channel [40]. Further, several processes develop regulating activity of the repolarization phase channels: against the background of increasing [Ca2+]i calcium potential decreases, Ca2+ channels close, and Ca2+-dependent potassium channels are activated and slowly hyperpolarize the membrane, which leads to gradual Na+ channels reactivation and of AP generation recovery. Firing continues until the T-type channels inactivate in parallel with the activation of calcium-dependent potassium channels, which repola-rize the membrane back below the range of T-type channel activation [43]. Against the background of the back front reducing depolarization, the amplitude of every subsequent AP increases due to reactivation of the increasing number of Na+ channels (Fig. 1c). Since depolarization level exceeds the critical potential for APs generation, the channel stay time in inactivated state determines the APs generation frequency.
However, during each AP pulse against slow depolarization, the faster depolarization pulse (0.15 s) is observed. The amplitude of this pulse is constant and equal to the amplitude of slow depolarization (Figs. 1c, 1d, 1e), that indirectly indicates participation of the same channel in depolarization. Periodic activation of the Ca2+ channel indicates the absence of potential-dependent inactivation of the channel during this period. That is why we observe pulse activation of slow Ca2+ channel and inactivation of Na+ channel during each AP (Fig. 1e). Generation of such periodic depolarization pulses is a reason why the frequency of APs on the back of slow pulse depolarization is greatly reduced, coinciding with the frequency of Ca2+ channel activation (3–4 Hz).
Applying rapid registration of [Ca2+]i [44], we tried to detect rapid changes in the [Ca2+]i, corresponding to the Ca2+ channels activity fluctuations in micro burst (Figs. 1c, 1e). It has been found similar oscillations only at kainite receptors activation with domoic acid and only in certain populations of the neurons (Fig. 2). The figure shows that the domoic acid causes an increase in the basal [Ca2+]i level, an increase in the frequency of SSA and the generation of high-frequency calcium oscillations in individual neuronal populations in the culture with initially synchronous spontaneous activity (Fig. 2b). The oscillation frequency in this experiment was 2.5 Hz that corresponded to Ca2+ potential oscillation frequencies in Figs. 1c, 1e. Thus, rapid fluctuations of depolarization in Figs. 1c–1e may be a consequence of fluctuations in the Ca2+ channels activity in micro-burst and cause rapid changes in the Ca2+ concentration recorded in Fig. 2.

Changes in [Ca2+]i in seven neurons during SSA in hippocampal culture at 14 DIV. (a) Synchronous Ca2+ oscillations in neurons recorded after application of GABA(A) receptors inhibitor, bicuculline (10 µМ), and Ca2+ responses to kainite receptors agonist, domoic acid (100 nM) (DA). At the end of the experiment 35 mm KCl was added for standard cell depolarization. In the presence of DA, the frequency of SSA increased to 0.25 Hz. The initial oscillation frequency was 0.01–0.0025 Hz. Slow-responding cells (S) do not oscillate and is used as a control (gray curves). Quick recording taken during a spontaneous spike, during the second DA addition and during the Ca2+-response to KCl. (b) Quick recording (100 fps) of calcium-dependent fluorescence switched during DA-induced Ca2+ spike in neurons (from Fig. 2a). In some cell populations the high-frequency synchronous calcium oscillations were recorded (with a frequency of about 2.5 Hz). There are no oscillations in cells slow-responding on DA (gray curves). The main recording was carried out at 1 frame per 2 s.
The termination of the burst firing seems to be due to the hyperpolarization caused by the Ca2+-dependent K+ channel opening, as well as to the closure and inactivation of the VGCC. Figure 1B shows that the resting potential at this time is restored, and the membrane is further hyperpolarized (by 10 mV) due to the of Ca2+-dependent K+-channels activity, since [Ca2+]i is still high (Fig. 1b). This hyperpolarization is believed to be necessary for Ca2+ and HCN channels reactivation. Ca2+ is pumped out of the cell (Fig. 1b). Sodium channels are reactivated, however negative potential is higher than the critical one, and APs generation is absent. The activity of HCN-like channels increases, the membrane slowly depolarizes and reaches the critical excitation potential, APs generation begins, but Ca2+ channels are opened later. The mechanisms of Ca2+ channel-dependent activation of slow depolarization remain debatable and will be discussed below.
Thus, Ca2+ channels determining the pulse of slow depolarization form the burst activity of two types: macro and micro-bursts of APs, which are accompanied by Ca2+ pulses of low and high frequencies, respectively. Against increasing depolarization, the APs frequency increases strongly (100-fold), and the amplitude decreases due to Na+ channels inactivation. While keeping depolarization potential above the reactivation one for Na+ channel, APs generation is terminated.
During repolarization due to [Ca2+]i increase and Ca2+-dependent K+ channels activation, gradual inactivation of Na+ channels occurs, and APs generation is restored, APs frequency decreases and amplitude increases (Fig. 1b). The [Ca2+]i increase during slow depolarization terminates the ongoing APs burst, causing hyperpolarization and APs termination. Thus, the slow depolarization pulses generated by Ca2+ channels regulate the AP frequency and the amplitude by monitoring the Na+ channels inactivation/reactivation.
The dependence of the AP frequency and the amplitude on depolarization are simulated in the experiments with evoked potentials in the mode of current fixation (Fig. 3). By changing the value of the inward current, a number of dependencies of the burst parameters activity on the depolarizing potential can be simulated. Figure 3a shows that depolarization shift above the critical potential causes the APs burst. The APs frequency grows with depolarization increase by increasing the inward current (HCN channel current analogue). If depolarization is small and constant, the AP amplitude during burst does not change (Fig. 3a), since the number of inactivated Na+ channels remains unchanged. At high inward current, when the maximum depolarization amplitude exceeds the inactivation potential of the Na+ channel, the growth of high-frequency APs occurs at the front of depolarization, the amplitude of which decreases with increasing depolarization, and APs is attenuates due to Na+ channels inactivation (Fig. 3b). Hence, experiments with the potentials caused by depolarizing steps DC, demonstrate the mechanisms of APs frequency and amplitude regulation by the depolarization magnitude described above for the bursting activity while SSA.

Frequency and amplitude of evoked APs with increasing depolarizing current. (a) The increase of evoked APs frequency during the depolarization amplitude increase by increasing the inward current. The curves given registered in 40 pA. (b) The depolarization kinetic determines the frequency and amplitude of APs. At low depolarization the APs amplitude are constant. (black curve). At large depolarization the APs damped at the forefront of depolarization due to the inactivation of Na+ channels (gray curve). The curves given registered at 50 and 450 pA.
The amplitude shift of slow depolarization by GABA(A) and glutamate receptors. As shown above, SSA is accompanied by Ca2+ increase in neurons, which causes the secretion such neurotransmitters as glutamate and GABA. The change of the corresponding receptors activity can change the slow depolarization amplitude in the SSA.
Figure 4 show that manipulations with the potential-regulating channels activity (GABA(A) and AMPA receptors) control the APs frequency and amplitude in the bursts. Figure 4a shows the initial APs fragments in the bursts in the control and in the presence of the GABA(A) receptors inhibitor, bicu-culline. Bicuculline increases the slow depolarization amplitude by reducing the inward (hyperpolarizing) Cl--currents. The depolarization amplitude increase in the presence of bicuculline above the Na+ channel inactivation potential leads to a temporary APs damping in the bursts.

Changes in neuronal membrane potential during SSA in the presence of GABA(A) and AMPA receptors inhibitors. (a) The initial part of the burst in the control (black curve) and in the presence of 10 µM bicuculline (gray curve). Decrease of hyperpolarizing Cl− channel activity increases the amplitude of slow depolarization (from –42 mV to –18 mV) above the Na+ channel inactivation potential, which leads to AP amplitude damping on the front of slow depolarization pulse. (b) AMPA channel inhibitor (500 nM NBQX) reduces the slow depolarization amplitude to –25 mV. In this case Na+ channels are not fully activated. The average APs frequency on anterior and back front of slow depolarization is changed in accordance with a depolarization rates decrease (compare with Fig. 1c).
Figure 4b shows that the inhibitor of AMPA receptors, NBQX, in small concentrations reduces the depolarization amplitude below the Na+ channel inactivation potential. In this case, the full Na+ channel inactivation in the maximum of slow depolarization does not occur, and when the anterior and back front of depolarization slows down, the number of APs in the bursts increases, the average frequency decreases and the duration of the AP bursts increases. Thus, adjustment of slow depolarization parameters continuously changes the APs frequency and amplitude.
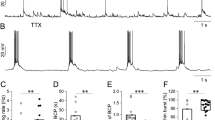
T-type Ca2+ channel inhibitors decrease the Ca2+ pulse duration at SSA. Since the T-type Ca2+ channels blockers cause hyperpolarization and decrease the Ca2+ concentration in the cells; they predictably should have neuroprotective effect during hyperexcitation caused by their activation. The protective effect of VGCC inhibitors can be due to both subsequent hyperpolarization and inhibition of toxic Ca2+-dependent processes. The potential and Ca2+ concentration interdependence is clearly demonstrated in pacemaker neurons, for which it is shown that Ca2+ pulse duration (determined by the burst duration) during the pacemaker activity is critical for Ca2+ ions accumulation in toxic concentrations in the cells [45]. As a rule, these neurons are characterized by a more intense and prolonged calcium signal due to the lack of channel desensitization, the absence of calcium-binding proteins in buffer concentrations and the absence of GABA-dependent inhibition [46, 47]. Therefore, to reduce toxic effect of Ca2+, it is necessary to reduce Ca2+ signal duration (bursting time), especially at high frequencies of SSA. Figure 5 shows that the T-type Ca2+ channels blocker, ML218, in concentration 500 нМ gradually reduces the neuronal depolarization amplitude during SSA. Slow dynamics of the ML218 action is explained by slow penetration rate into the cells. To show all phases of burst activity inhibition we selected neurons with initially high resting membrane potential. In these neurons the tonic activity of APs between bursts is absent. The ML218 in concentration 1 µM induces a gradual depolarization decrease and reduces the burst activity up to 1 AP. Then the slow depolarization amplitude is reduced to values below the threshold level and APs generation stops. However, slow potential fluctuations are continued for some time without APs generation. Therefore, the T-type of Ca2+ channels take part in initiation burst activity oscillations during SSA, periodically depolarize the neurons. The T-type Ca2+ channels modulate the frequency and amplitude of the APs. Increasing the depolarizing current increases the APs frequency and depolarization inactivates Na+ channels and reduces the AP amplitude. The T-type Ca2+ channel activity inhibitors reduce the Ca2+ pulse duration, which may explain their neuroprotective effect in is-chemia and hyperexcitation [5]. The mechanism of these channels periodic reactivation remains unknown

Changes in neuron membrane potential during SSA. Hippocampal cultures at 16 DIV. (a) Blocker of T-type channels, ML218, at concentration of 500 nM reduces slow depolarization amplitude and reduces the duration of every burst. (b) T-type channels blocker, ML 218, at a concentration of 1 µM reduces duration of every burst, suppresses the APs generation, hyperpolarizes the cell, and reduces the amplitude of the slow depolarization; (c, d, e) 1 µM ML218 decrease burst duration (d) suppresses the APs generation and reduces depolarization amplitude (d, e). The dotted lines show the location of AP bursts shown in Figs. 5c, 5d, 5e, during the SSA before and after ML218 application. Culture at 14 DIV.
DISCUSSION
SSA is observed throughout the brain, and plays a key role in processing neuronal information, brain development, neuronal plasticity and synaptogenesis [32, 33]. During SSA, tonic activity is intermittently interrupted by bursting of APs. Each burst is followed by a period of quiescence before the next burst occurs. Bursting is a very general phenomenon and is observed in many contexts in many neural systems [48]. This process also relies on calcium channels, which depolarize the neuron and increase an influx of calcium ions. In 1993, it was shown that the rising phase of each calcium spike was associated with a burst of APs, and that every APs burst was superimposed on the period of continuous membrane depolarization [49]. It has become apparent that voltage-gated calcium channels should also participate in the SSA. It is known that Ca2+ ions are important regulators of neuronal electrical activity. On the one hand, Ca2+ channels depolarize the cell and, thus, contribute to its excitation, regulating the state of Na+ channels inactivation. On the other hand, the Ca2+ concentration increase regulates the activity of the others channels, forming AP. Ca2+ spike generated by the VGCC through the secretion process involves neurotransmitters—GABA and glutamate, which regulate neuron depolarization through GABA(A), NMDA and AMPA channels. Synchronous changes in membrane potential and Ca2+ concentrations in large populations of neurons occurring during periodic changes in the activity of T-type Ca2+ channels activating synaptic transmission and genes expression are necessary for the development of neural network without any hormones and other external stimuli.
To induce synchronous bursts of spikes (SSA), we used depolarization shift, which occurs after inhibition removal in neural network by GABA receptors inhibitor, bicuculline [50]. Disinhibition of neurons with bicuculline induces the mode of neuronal spontaneous activity, in which tonic high-frequency APs are generated without [Ca2+]i changing, follow the period of the APs burst activity with simultaneous generation of slow depolarization and Ca2+ spikes in the neural network. Changes in the parameters of the AP are due to the fact that Ca2+ channels activation occurs quite slowly, on the one hand, providing several APs generations on the depolarization front line, but at the same time occurs quite intensively to provide high frequency of APs. The mechanism of APs frequency regulation is known to consist of time-separated voltage-dependent opening/closing and inactivation/reactivation of the ionic channels. Fine regulation of membrane potential provides tonic or bursting activity of the neurons. The slow depolarization amplitude determines not only the frequency of APs generation, but also the AP amplitude due to the potential-dependent Na+ channels inactivation. As shown in Figs. 1 and 4, the change of the depolarizing current is the main regulator of the AP frequency inside the burst. It has been shown earlier that despite synchronicity of the SSA, Ca2+ pulses in different neurons have different shape and amplitude [3]. This means that synchronous pulsating neurons generate APs bursts of different frequency and amplitude and, thus, transmit different signals. Thus, the mechanisms of burst activity formation and changes in the frequency and the amplitude of APs due to Na+ and Ca2+ channels voltage-inactivation and reactivation may be a mechanism for encoding electric signal, transmitted by the neurons. Slow depolarization, caused by the opening of VGCC, not only changes the APs frequency in a wide range, but also, forming the burst activity, in fact, converts the analog signal (potential) to the digital one (AP frequency), which is automatically modulated in the amplitude.
It is assumed, that slow depolarization currents convert single short-time AP spike into the longer lasting burst patterns APs of different frequency and amplitude, as a way to focusing on a new stimulus and activation of important ways processing this information in networks [51, 52]. AP coding in frequency and amplitude by slow depolarization spikes can represent a way to place a much larger amount of information in a single burst, which leads to a more reliable signal transmission. Due to the complexity of the Ca2+ signaling system in neurons, the mechanism of periodical bursts start during the SSA remains questionable and the detailed mechanisms of Ca2+ channel reactivation are largely unclear.
Neuroprotective properties of T-type Ca2+-channels. As shown in Fig. 5, the T-type calcium channels not only determine the AP structure in the burst, but also regulate the Ca2+ spike duration. It is known that, despite the quasi-synchronicity of Ca2+ oscillations in neurons during SSA, the Ca2+ spike duration in individual neurons varies [3]. The Ca2+ spike duration is determined by several factors: 1 – Ca2+-binding proteins deficiency, Ca2+ channels desensitization, lack of GABA(A) receptors, T-type calcium channels activity. A number of studies have shown that the Ca2+ spike duration is a critical parameter at high frequencies of SSA [54] and at neuron pacemaker activity [36]. Neurons damaged primarily during hyperexcitation are characterized by prolonged Ca2+ spike, rapid rise of [Ca2+]i during the burst firing, absence of desensitization, slow pumping of Ca2+ from the cytoplasm and the Ca2+-binding proteins deficiency [53, 54]. Figure 5 demonstrates that the known neuroprotective effect of T-type calcium channel inhibitors may be due to a decrease in Ca2+ spike duration, which can be a critical moment for pacemaker neurons and neurons containing Ca2+-permeable kainite and AMPA receptors and responding to glutamate by [Ca2+]i rise without desensitization [54].
The bursting activity regulation with participation of VGCC opens up wide possibilities in the transformation and coding of the electrical signal transmitted by the neuron. A major role in this transformation can play Ca2+-transporting systems and Ca2+-binding proteins that determine the different forms of Ca2+ pulse.
REFERENCES
S. M. Lu, W. Guido, and S. M. Sherman, J. Neurosci. 68 (6), 2185 (1992).
S. Anava, A. Greenbaum, E. Ben Jacob, et al., Biophys. J. 96 (4), 1661 (2009).
A. V. Kononov, N. V. Ball, and V. P. Zinchenko, Biochemistry (Moscow). Suppl. Ser. A: Membr. Cell Biol. 5 (2), 162 (2011).
D. A. McCormick and D. Contreras, Annu. Rev. Physiol. 63, 815 (2001).
I. Nikonenko, M. Bancila, A. Bloc, et al., Mol. Pharmacol. 68 (1), 84 (2005).
B. J. Kopecky, R. Liang, and J. Bao, Pflügers Arch. 466 (4), 757 (2014).
B. Nilius, K. Tala, and A. Verkhratsky, Cell Calcium 40 (2), 81 (2006).
D. Kim, I. Song, S. Keum, et al., Neuron 31 (1), 35 (2001).
I. Song, D. Kim, S. Choi, et al., J. Neurosci. 24 (22), 5249 (2004).
S. Huc, A. Monteil, I. Bidaud, et al., Biochim. Biophys. Acta 1793 (6), 947 (2009).
E. Perez-Reyes, L. L. Cribbs, A. Daud, et al., Cell. Mol. Life Sci. 56 (7–8), 660 (1999).
M. Chevalier, P. Lory, C. Mironneau, et al., Eur. J. Neurosci. 23 (9), 2321 (2006).
J. Proft and N. G. Weiss, Mol. Pharmacol. 87 (6), 890 (2015).
E. Perez-Reyes, Mol. Pharmacol. 77 (2), 136 (2010).
J. L. Sánchez-Alonso, J. V. Halliwell, and A. Colino, Neurosci. Lett. 439 (3), 275 (2008).
R. Iyer, M. A. Ungless, and A. A. Faisa, Sci. Rep. 7 (1), 5248 (2017).
R. Rehak, T. M. Bartoletti, J. D. T. Engbers, et al., PloS One 8, e61844 (2013). doi 10.1371/journal.pone. 0061844
J. Wolfart and J. J. Roeper, Neuroscience 22 (9), 3404 (2002).
J. Xu and C. E. Clancy, PloS One 3 (4), e2056 (2008). doi 10.1371/journal.pone. 0002056
J. T. Wolfe, H. Wang, E. Perez-Reyes, and P. Q. Barrett, J. Physiol. 538 (2), 343 (2002).
J. Chemin, A. Mezghrani, I. Bidaud, et al., J. Biol. Chem. 282 (45), 32710 (2007).
J. Chemin, A. Monteil, E. Perez-Reyes, et al., EMBO J. 20 (24), 7033 (2001).
J. Chemin, J. Nargeot, and P. Lory, J. Biol. Chem. 282 (4), 2314 (2007).
K. Talavera, M. Staes, A. Janssens, et al., J. Gen. Physiol. 124 (3), 225. (2004).
Y. Zhang, L. L. Cribbs, and J. Satin, Am. J. Physiol. Heart Circ. Physiol. 278 (1), H184 (2000).
J. T. Wolfe, H. Wang, J. Howard, et al., Nature 424 (6945), 209 (2003).
J. Tao, M. E. Hildebrand, P. Liao, et al., Mol. Pharmacol. 73 (6), 1596 (2008).
M. E. Hildebrand, L. S. David, J. Hamid, et al., J. Biol. Chem. 282 (29), 21043 (2007).
M. T. Nelson, S. M. Todorovic, and E. Perez-Reyes, Curr. Pharmaceut. Design 12 (18), 2189 (2006).
S. M. Todorovic and V. Jevtovic-Todorovic, Pflügers Arch. 466 (4), 701 (2014).
B. J. Kopecky, R. Liang, and J. Bao, Eur. J. Physiol. 466 (4), 757 (2014).
P. Orestes, D. Bojadzic, R. M. Chow, and S. M. Todorovic, Mol. Pharmacol. 75 (3), 542 (2009).
N. C. Spitzer and E. X. Olson, J. Neurobiol. 26 (3), 316 (1995).
H. P. Robinson, M. Kawahara, et al., J. Neurophysiol. 70 (4), 1606 (1993).
J. N. Guzman, J. Sánchez-Padilla, C. S. Chan, and D. J. Surmeier, J. Neurosci. 29 (35), 11011 (2009).
D. J. Surmeier and P. T. Schumacker, J. Biol. Chem. 288 (15), 10736 (2013).
O. J. Lieberman, S. J. Choi, E. Kanter, et al., eNeuro. 4 (6), 0167 (2017).
D. J. Surmeier, J. A. Obeso, and G. M. Halliday, Nat. Rev. Neurosci. 18 (2), 101 (2017).
C. S. Chan, J.N. Guzman, E. Ilijic, et al., Nature 447 (7148), 1081 (2007).
G. Grynkiewicz, M. Poenie, and R. Y. Tsien, J. Biol. Chem. 260 (6), 3440 (1985).
H. Hayashi, H. Miyata, H. Terada, et al., Jpn. Heart J. 35 (5), 673 (1994).
L. Cozzi, P. D’Angelo, and V. Sanguineti, Biol. Cybern. 94 (5), 335 (2006).
S. M. Cain and T. P. Snutch, Biochim. Biophys. Acta 1828 (7), 1572 (2013).
M. V. Turovskaya, E. A. Turovsky, V. P. Zinchenko, et al., Neurosci. Lett. 516 (1), 151 (2012).
E. A. Turovsky, M. V. Turovskaya, S. G. Gaidin, and V. P. Zinchenko, Arch. Biochem. Biophys. 615, 35 (2017).
E. A. Turovsky, V. P. Zinchenko, S. G. Gaidin, and M. V. Turovskaya, Biochemistry (Moscow). Suppl. Ser. A: Membr. Cell Biol. 12, 74 (2018).
V. P. Zinchenko, M. V. Turovskaya, I. Yu. Teplov, et al., Biophysics (Moscow) 61 (1), 85 (2016).
E. M. Izhikevich, Int. J. Bifurcat. Chaos 10 (6), 1171 (2000).
H. P. Robinson, K. Torimitsu, Y. Jimbo, et al., Jpn. J. Physiol. 43 (1), 125 (1993).
V. V. Dynnik, A. V. Kononov, A. V. Sergeev, I. Yu. Tep-lov, V. P. Zinchenko, Plos One. 10 (7) e0134145. doi: 10.1371/journal.pone.0134145 (2015)
D. Cooper, Neurochem. Int. 41 (5), 333 (2002).
H. A. Swadlow, A. G. Gusev, and T. Bezdudnaya, J. Neurosci. 22 (17), 7766 (2002).
V. P. Zinchenko, E. A. Turovsky, M. V. Turovskaya, et al., Biochemistry (Moscow). Suppl. Ser. A: Membr. Cell Biol. 10 (2), 118 (2016).
V. P. Zinchenko, S. G. Gaidin, I. Y. Teplov, and A. M. Kosenkov, Biochemistry (Moscow). Suppl. Ser. A: Membr. Cell Biol. 11 (4), 261 (2017).
ACKNOWLEDGMENTS
This work was supported by the Committee of Science of MES RK, grant AP05133528.
Author information
Authors and Affiliations
Corresponding author
Additional information
1The article was translated by the authors.
Abbreviations: SSA – spontaneous synchronous activity, Ca2+ signal, T-type Ca2+ channels, VGCC – voltage-gated calcium channels, AP – action potential, genesis of bursting activity, action potential burst, depolarization shift, critical potential, [Ca2+]i – cytoplasmic Ca2+ concentration.
Rights and permissions
About this article

Cite this article
Teplov, I.Y., Tuleukhanov, S.T. & Zinchenko, V.P. Regulation of Action Potential Frequency and Amplitude by T-type Ca2+ Channel During Spontaneous Synchronous Activity of Hippocampal Neurons. BIOPHYSICS 63, 566–575 (2018). https://doi.org/10.1134/S0006350918040206
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006350918040206