
Abstract
G protein-coupled receptors (GPCRs) are an important family of membrane proteins responsible for many physiological functions in human body. High resolution GPCR structures are required to understand their molecular mechanisms and perform rational drug design, as GPCRs play a crucial role in a variety of diseases. That is difficult to obtain for the wild-type proteins because of their low stability. In this review, we discuss how this problem can be solved by using protein design strategies developed to obtain homogeneous stabilized GPCR samples for crystallization and cryoelectron microscopy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
G protein-coupled receptors (GPCRs), which are one of the most important classes of membrane proteins in pharmacology and the most represented family of proteins in the human proteome, are characterized by the amphiphilic nature and low stability. GPCRs consist of seven transmembrane (TM) helices and are expressed on the cell plasma membrane. They recognize a broad range of extracellular ligands and transmit signals into the cells, thus triggering different types of cellular responses. GPCRs play a paramount role in many physiological functions of the human organism, such as vision, gustation, olfaction, regulation of activity of the nervous, immune, and cardiovascular systems, and maintenance of homeostasis and cell density in tissues. Dysfunctions in these processes lead to serious diseases that can be corrected by blocking or activating the respective receptors. Up to 40% of currently prescribed drugs target GPCRs [1, 2]. Knowing the high-resolution structures of GPCRs in different functional states is essential for understanding of the molecular mechanism underlying their action, as well as for the creation of highly efficient medicines with minimal side effects. The time and the cost of drug development can be significantly decreased by using the structure-based drug design [3]; hence the studies of GPCRs can benefit basic science, medicine, and pharmacology.
Currently used methods for obtaining high-resolution structures of proteins are X-ray crystallography [4], cryoelectron microscopy (cryo-EM) [5], microcrystal electron diffraction (microED) [6], and biomolecular nuclear magnetic resonance (NMR) spectroscopy [7].
Studying GPCR structure and functions requires homogeneous stabilized samples of these receptors. Each of the involved procedures (e.g., expression and purification of stable monomeric receptor) represents an independent scientific task requiring development of the corresponding protein engineering strategy.
In this article, we reviewed the strategies that proved to be the most successful in resolving the structures of GPCRs and their complexes, including rational design of genetic modifications (prediction of necessary point mutations, deletions; the use of tags and fusion proteins), selection of the optimal expression systems, development of strategies for protein purification and stabilization (including the use of various membrane-modeling media, antibodies and their fragments, and ligands), as well as the basic approaches to the characterization and quality control of the obtained protein samples.
GENETIC CONSTRUCTS
Introduction of point mutations. In most cases, point mutations leading to the increase in the receptor expression level, protein homogeneity, and higher thermostability and, in some cases, aimed to eliminate undesired posttranslational modifications, are introduced into receptor-encoding genetic constructs. For example, it has been shown that for some class A GPCRs, amino acid substitutions in the allosteric sodium-binding site upregulate protein expression [8] and decrease the conformational heterogeneity of the receptor via its stabilization in the states preferential for the binding of antagonists [9] or agonists, or in the intermediate states [10]. Similar modifications were used for the class B receptors [11].
Posttranslational modifications can reduce the homogeneity of protein sample during crystallization. To eliminate the negative effect of posttranslational modifications, some receptor residues were substituted with amino acids with different functional side groups. N-glycosylation is the most common posttranslational modification in GPCRs, so deleting the glycosylation sites has proven to be beneficial [12-15]. However, in most cases, posttranslational modifications are functionally important and can affect protein expression, folding, and in some cases, the ability to bind ligands; hence elimination of postranlational modifications can result in the decreased surface expression for many GPCRs [16].
Another approach is introduction of cysteine bridges into the protein molecule. It has been commonly recognized that disulfide bonds have a direct effect on the protein folding and stability [17-19]. For some GPCRs, introduction of point mutations leading to the formation of disulfide bridges between two closely spaced cysteine residues proved to be quite successful, for example, introduction of disulfide bond into the rhodopsin molecule between the N-terminus and the third extracellular loop (ECL3). This modification increased the thermostability of the receptor by 10°C [20], both as the opsin apoprotein [21] and in a complex with retinal [20] in a detergent solution. At the same time, it had no significant effect on the receptor function [20-22]. In another example, the position for the disulfide bond introduction into the LPA1 receptor was determined using a predictive algorithm [23]. The formation of a disulfide bond between the external sides of the TM domains 5 and 6 increased the thermostability of the LPA1 receptor by 5°C in the presence of a ligand [24]. The formation of a disulfide bond between the TM helices 5 and 6 in the GLP-1 receptor resulted in the stabilization of its inactive conformation [25].
Despite the existing concepts on the mechanisms of receptor stabilization, the choice of point mutations that would lead to the desired effects remains a difficult task. Below, we present some approaches aimed to solve this problem.
Alanine scanning and StaR™ technology. Alanine scanning is a method of sequential site-directed mutagenesis used to identify specific amino acid residues that influence the stability or function of a studied protein. Because alanine has only a beta-carbon atom in its side chain [26], it is a small amino acid that can replace almost any other residue without creating steric hindrances. At the same time, alanine preserves the polypeptide backbone conformation (unlike glycine and proline) [27]. Alanine residues tend to form alpha-helices, which are common for membane proteins [28]. Due to these properties, alanine is preferred as a substituting residue in the search for the functional amino acids in a receptor.
In the course of alanine scanning, all amino acid residues are sequentially replaced by alanine, while native alanine residues are typically replaced by leucine [29, 30] and less often, by other amino acids [31]. The stability of the resulting mutant proteins is evaluated experimentally and the best mutations are selected. Alanine scanning has been used to design more stable receptors, as well as to identify amino acids involved in the signal transduction [32].
As a rule, alanine scanning is not performed for wild-type receptors. Usually, the starting point is a modified construct, e.g., protein with deleted flexible N- and C-terminal domains, since such deletions can promote protein expression and reduce proteolytic cleavage of the terminal receptor sequences [29, 30, 33].
Although using alanine and leucine significantly helps in the initial search for positions that can be mutated to stabilize the protein, using other amino acid as substitutions might result in better stabilization of the receptors [29, 30].
Stabilized Receptor™ (StaR™) technology involves a design of thermostable receptors by inserting point mutations into their structure. The thermostability increases due to the protein stabilization in any of its conformations existing in nature: from the ground state to the fully active G protein-bound state. The choice of the state is determined by the type of ligand (agonist, antagonist, inverse agonist) used in the experiments. Thus, when the agonist-binding conformation is chosen, the receptor will have a high affinity to other agonists, and vice versa. For example, the constructs with an enhanced affinity to agonists or antagonists have been designed for the adenosine A2A receptor (A2AR) [30].
In essence, the StaR™ technology is based on alanine scanning. All mutants with the single point substitutions are expressed and their ability to bind radioactively labeled ligand at increasing temperatures is assessed in comparison with that of the wild-type protein [34]. Next, point mutations promoting the thermostability of the ligand–receptor complex are sequentially combined with each other to achieve the maximum melting temperature [35].
The first crystal structure obtained with the help of the StaR™ technology was the structure of the turkey β1-adrenergic receptor (β1AR) [29]. The authors used the alanine scanning, as well as substitutions based on the amino acid sequence alignment with the sequence of bovine rhodopsin, the only GPCR with the known structure at that time.
Because of the need to test a large number of mutants (more than 300) for thermostability, designing a StaR™ construct might take months of laborious work. However, this technique still stays relevant for obtaining receptors for crystallization [20, 36-48].
Directed evolution. Directed evolution can assist in the search for and selection of functionally expressed receptors that cannot be produced in commonly used expression systems, such as insect and mammalian cells.
The directed evolution of GPCRs includes the following steps: (i) creation of a library of the receptor-encoding gene mutants; (ii) detection of receptor variants exposed to selection pressure, whose increased functional expression is confirmed by the binding of a specific ligand; and (iii) subsequent rounds of evolution/selection to find the most preferable variant [49]. It should be taken into account that all the procedures described in this section require the use of receptor-specific ligands.
Originally, the libraries of mutant GPCR genes were analyzed in the Escherichia coli cells as the most convenient and simplest object for genetic manipulations. Permeabilization of the E. coli outer membrane made the receptors expressed on the inner membrane accessible to fluorescently labeled ligands. Then the fluorescence-activated cell sortinf (FACS) was performed. Hence, a great number of receptor mutants could be screened simultaneously without the need to express each receptor separately. The use of several rounds of selection has made it possible to identify receptors with an enhanced functional expression not only in prokaryotic, but also in eukaryotic expression systems [50]. The CHESS technique (cellular high-throughput encapsulation, solubilization, and screening) developed later allowed to identify receptors with an increased resistance to detergents, which was possible due to the encapsulation of E. coli cells within a polysaccharide matrix, followed by the cell membrane disruption and solubilization of receptors with a detergent. Large cellular components, including the receptor itself and the plasmid DNA, remained within a capsule. Further incubation of the semipermeable capsules with a fluorescently labeled ligand and FACS made it possible to select the clones with the receptors staying functional under the given conditions. The selected genes were exposed to further rounds of mutagenesis and selection [51]. The methods of directed evolution in E. coli cells [51, 52] together with further modifications allowed to produce the neurotensin receptor 1 (NTSR1) applicable for structural studies [47].
The drawback of the above-mentioned techniques is the need to express functional receptors in E. coli cells, which is not always possible for the native eukaryotic membrane proteins.
The SaBRE (Saccharomyces cerevisiae-based receptor evolution) method, which appeared later [53], is based on the directed protein evolution in yeast cells and allows expression of a large variety of GPCRs due to the existence of posttranslational mechanisms in yeast. Only two rounds of directed evolution in S. cerevisiae cells were needed to obtain receptors with an enhanced expression and capable of ligand binding. Mutations selected by the SaBRE method ensured an increased expression in insect cells of receptors with a significantly elevated thermostability. The method was used to create the constructs for crystallization of the oxytocin receptor [54], parathyroid hormone receptor PTH1R [55], and opiod receptor δOP with eight introduced point mutations that had been identified earlier by the SaBRE method for the related κOP receptor [56].
Recently, researchers at the Andreas Plückthun lab have combined several techniques of directed evolution for the purpose of high-throughput CHESS screening for the oxytocin receptor, which is originally toxic for E. coli cells. The mutants selected by the SaBRE method were used in subsequent rounds of evolution in E. coli cells in order to perform CHESS and to identify mutants with the increased functional expression in E. coli cells. This approach can benefit the studies, in which the use of E. coli cells is preferable [49].
Recently, the YDDS (yeast direct detergent screening) method for selecting receptors has been reported, which allowed to identify GPCRs resistant to the short-chain detergents upon their expression in yeast cells [57].
Using machine learning to predict point mutations: CompoMug algorithm. The use of methods employing artificial intelligence systems has led to significant breakthroughs in many scientific areas and allowed to solve the problems that otherwise could not have been efficiently solved by the classical approaches. The CompoMug algorithm consists of four modules based on the analysis of available data, sequence analysis, structure analysis, and machine learning, respectively, which combine several approaches to compile a list of candidate point mutations that could improve the stability of GPCRs. The data- and sequence-based modules work only with the information on the target receptor sequence, while the structure- and machine learning-based modules work with the structural information. This algorithm generates a priority list of point mutations (with the efficiency of each mutation estimated using internal algorithm criteria in each module), that can be tested experimentally to create receptors that can be purified as homogenous stable protein samples suitable for structural studies. The CompoMug algorithm uses various types of information related to the GPCR stability, e.g., the data on the stabilizing mutations transferred between GPCRs, sequence-based information on the protein evolution (e.g., conservation of absolute and relative positions of amino acids), and structural data (e.g., distances between the contacts and residue energies). All information is encoded numerically to compile the GPCR stability descriptors. Machine learning algorithms were applied to the descriptors calculated for the known GPCR-stabilizing point mutations in order to obtain a model for comprehensive prediction. The method has been iteratively improved with the accumulation of data on the stabilizing effect of point mutations for new GPCRs with regard to the experimental feedback data. The method was implemented using modern machine learning algorithms and computational biology. The software distribution tool and its description are available at: CompoMug 2.0 https://gitlab.com/pp_lab/CompoMug. CompoMug is a computational platform for GPCR thermostabilization and can be further refined with the accumulation of experimental data [58].
Deletion of unordered regions from the receptor amino acid sequence. Flexible regions of proteins often hinder crystallization and reduce the quality of the obtained data. The most flexible regions in GPCRs are the N- and C-terminal sequences; therefore, they are often deleted with only 10-20 amino acid residues being left. A long N-terminal fragment of the receptor can contain glycosylation sites, which influence protein processing in the cytoplasmic membrane, thereby affecting its release [59]. The C-terminal part can also affect protein expression and its monodispersity [60]. Unfortunately, it is impossible to predict the effect of deletion of the N- and C-terminal regions on the level of receptor expression. At the same time, one should take into consideration the functional importance of glycosylation sites and sites providing receptor interaction with ligands, G proteins and other partners [61, 62]. In practice, it is preferable to delete the unordered regions in multiple rounds (1 to 5 residues per each) and to test the effect of these deletions experimentally.
In class A GPCRs, the third intracellular loop (ICL3) is very flexible [63]. To improve crystallization, it is often replaced by a fusion protein, e.g., the thermostable region of apocytochrome (BRIL), flavodoxin, T4 lysozyme, rubredoxin, glycogen synthase, etc. [64] (see the section “Use of Fusion Partners”).
Insertion of additional elements into the receptor amino acid sequence. Insertions into the receptor amino acid sequence can serve different purposes. Expression of GPCRs and their translocation to the cell surface are often increased by adding the HA-tag (MKTIIALSYIFCLVFA) [65, 66], a fragment of hemagglutinin (major surface protein of the influenza virus). When located at the protein N-terminus, the HA-tag provides more efficient translocation of protein to the endoplasmic reticulum and its further transport to the cell membrane surface [65]. The HA-tag can be used in both SF9 insect cells [67] and human HEK293 cells [66, 68]. Another fragment of hemagglutinin sequence (YPYDVPDYA) was used as an epitope for the immunohistochemical studies of GPCRs [69-71].
To facilitate receptor purification by affinity chromatography, receptor molecules are often modified by special tags, such as the FLAG-tag (DYKDDDDK/A) and/or His-tag (5-10 histidine residues) at the N-terminus [72]. Both His-tag and FLAG-tag can be inserted into the C-terminal part of the receptor as well, however, the affinity resin for receptor purification should be taken into account. These tags can also be used to analyze the level of protein expression by immunoblotting and flow cytometry. Since the N-terminus of a GPCR is always located outside the cell, the labeling of intact cells requires FLAG-tag to be added to the protein N-terminus [73, 74].
Maltose-binding protein (MBP) is also used for the modification of the GPCR sequence, but less frequently [75]. This rather large protein (42.5 kDa) can be used not only as a tag for affinity chromatography, but as a good fusion partner to increase the expression of GPCRs in bacterial cells [29, 47, 76, 77].
Simultaneous introduction of MBP and thioredoxin A (TrxA) in the NTR1 construct increased the amount of protein expressed in the E. coli membrane and allowed to obtain the crystals of the receptor [47]. Addition of glutathione S-transferase (GST) to the N-terminus of the CXCL8 chemokine receptor (CXCR1) made it possible to express this protein in amounts sufficient for crystallization, although within the inclusion bodies [78]. Various fusion partners and expression systems are discussed in detail in the sections “Use of Fusion Partners” and “Expression Systems for GPCR Production”.
GPCR isolation can be facilitated by the introduction of the Strep-tag, a small peptide of eight amino acid residues (WSHPQFEK), which was found by chance as a peptide selectively binding to streptavidin [79, 80].
Affinity chromatography techniques are discussed in the section “Purification and Stabilization of GPCRs”.
Since polypeptide tags are unordered sequences, their presence in the receptor construct can hinder analysis of the protein spatial structure. The tags could be removed using highly specific proteases. The commonly used enzymes are TEV (tobacco etch virus) protease and PreScission protease (a variant of HRV 3C protease). The recognition site for TEV protease is the ENLYFQ/S sequence that is cleaved between the glutamine and serine residues. Instead of serine, there can be glycine, alanine, methionine, cysteine, and histidine residues. The recognition site for the PreScission protease is LEVLFQ/GP, which is hydrolyzed strictly between the glutamine and glycine residues, which makes this protease more specific than TEV protease.
In some cases, the receptor sequence and the introduced fusion proteins, tags, protease recognition sites are separated by linkers several amino acid residues long to facilitate the access to the epitopes (for antibodies) and protease recognition sites, as well as to modify the flexibility of the resulting recombinant protein [81].
Use of fusion partners. GPCRs are dynamic proteins that change their conformation depending on the stage of the signaling process [82]. Also, as integral membrane proteins, GPCRs have a low content of hydrophilic regions that are necessary for the formation of crystals. These features are the major obstacles in obtaining diffraction-quality crystals of GPCRs [64].
Chimeric proteins consisting of GPCR and a partner protein are created to increase the stability of the receptor and the probability of contact formation in the protein crystal. The partner protein has to satisfy certain criteria, such as the existence of its high-resolution structure, small size (~5-20 kDa), simple folding, absence of posttranslational modifications, and molecule hydrophilicity [83]. Another requirement is that if the fusion partner is inserted between the receptor TM domains 5 and 6, the distance between its N- and C-termini protein should be less than 15 Å [64].
As a rule, the partner protein is incorporated into the ICL3 [13, 84], because it is conformationally heterogenous and connects the most mobile TM domains 5 and 6. Less often, the partner protein is fused to the receptor N-terminus [85]. It is also possible to insert the partner protein into other parts of the receptor molecule, e.g., intracelular loop 2 (ICL2) [86] or C-terminus [87]. In some cases, several partner proteins have been used simultaneously [87, 88].
None of the known fusion partners provides a universal solution for the problem of GPCR crystallization. Typically, several partner proteins are tested with the same receptor and several insertion sites are tried for the same partner protein. In most cases, the partner protein is added together with the introduction of stabilizing point mutations in the receptor. The proteins used most frequently for the fusion with GPCRs (Fig. 1) contain various elements of the secondary structure. They might consist of loops (rubredoxin), β-sheets (xylanase), α-helices [C-terminal fragment of T4 lysozyme (CtermT4L) and its modifications (mT4L), thermostabilized apocytochrome b562 (BRIL)], or a combination of these elements [flavodoxin, catalytic domain of glycogen synthase from Pyrococcus abyssi (PGS)] [64, 89].

Common fusion partners of GPCRs. Images were taken from GPCR structures from PDB database with each of these fusion partners: rubredoxin – 4MBS; TrxA – 6IBL; mT4L – 4U16; T4L – 2RH1; BRIL – 5NM4; flavodoxin – 5TGZ; PGS – 5U09. The pie chart represents the frequency of their use as fusion proteins.
The first GPCR structures with fusion proteins were obtained with T4L, an easily crystallized well-folded protein. Currently, the most often used protein is BRIL. Unlike T4L, BRIL provides more rigid fixation of the TM helices 5 and 6 due to the smooth transition into the α-helices of BRIL. The best resolution for the GPCR structure obtained by X-ray crystallography is 1.7 Å (PDB ID: 5NM4), which was achieved by using GPCR fused with BRIL and containing thermostabilizing point mutations.
TrxA and MBP are used mainly to enhance GPCR expression in E. coli cells. TrxA was also used to increase the protein expression level in a cell-free translation system [90]. In most cases, these partner proteins are cleaved off during purification [47, 87, 91]. However, the fusion with the partner proteins can facilitate the cryo-EM studies of GPCRs. For example, TrxA was added to the N-terminus of A2AR to increase the molecular mass of the receptor without affecting its pharmacological properties [92, 93].
The use of alternative partner proteins has increased the number of successful crystallizations and published structures. For example, addition of the DARPin D12 protein to the seventh TM helix of GPCR [94]. This partner protein was used for crystallization of NTSR1 and α1B-adrenergic receptor [95, 96] produced by expression in E. coli cells. DARPin D12 is a compact protein containing the ankyrin repeat motif forming the helix–turn–helix conformation and promoting protein–protein interactions [97]. It has been shown that the force of protein–protein interactions of DARPin is sufficient to form protein crystals under various conditions [98].
EXPRESSION SYSTEMS FOR GPCR PRODUCTION
The photoreceptor protein rhodopsin is the only GPCR expressed with a high density in cell membranes in a multicellular organism [99] and, therefore, it was isolated directly from the discs of the external segment of bovine (Bos taurus) retinal rod cells [100] in amounts sufficient for crystallization. As a result, the first 3D structure of GPCR resolved by X-ray structure analysis was published in 2000 [101]. Later, the structure of rhodopsin from the retina of Japanese common squid (Todarodes pacificus) was solved [102]. However, the members of the GPCR superfamily are typically expressed in cell membranes in extremely small amounts, which makes their purification from tissues impractical and technically difficult. In addition, for most receptors, production of high-quality crystals requires insertion of modifications in the receptor amino acid sequence, which is impossible for the receptors isolated from natural sources. Therefore, obtaining sufficient amounts of protein for its characterization and further structural studies is one of the key problems in the structural biology of GPCRs.
Multiple expression strategies are used to increase the yield of the purified receptor up to milligrams, the most common being heterologous expressed in various systems. As most other proteins, GPCRs have been expressed in bacterial, yeast, insect, and mammalian cells, as well as in cell-free expression systems.
Bacterial expression systems. E. coli is an extensively studied and cost-efficient bacterial expression system that has shown itself effective in the production of recombinant proteins, including those used in the structural studies. However, the use of E. coli cells for the production of GPCRs has been significantly limited. The reasons for this include significant differences in the lipid composition of prokaryotic and eukaryotic membranes (since lipids are important regulators of the receptor function), the absence of well-developed machinery for protein folding and processing, and the absence of most of posttranslational modifications typical for mammalian membrane proteins, which results to the impaired folding and function of expressed membrane proteins [103]. Typical GPCR modifications are glycosylation, palmitoylation, phosphorylation, etc., that are important for the correct protein folding and intracellular transport of the receptors [16]. In addition, protein overexperssion in bacterial cells often leads to the formation of insoluble protein aggregates (inclusion bodies) [104]. The latter per se are not necessarily a disadvantage, because proteins in inclusion bodies are relatively pure and unavailable from digestion with cellular proteases. However, protein recovery from inclusion bodies requires a great deal of efforts to select conditions for the in vitro protein refolding and is often unsuccessful. In addition, the reducing nature of the E. coli cytoplasm is unfavorable for the formation of disulfide bonds. There have been numerous attempts to overcome the limitations of expression of eukaryotic membrane proteins in E. coli cells, e.g., by using partner proteins directing GPCR expression to the cytoplasmic membrane.
Despite the disadvantages of bacterial expression systems, the attempts of bacterial expression of GPCRs have been made more than once, resulting, for example, in obtaining the NTR1 structure in 2014 [47]. The N- and C-termini of the receptor were fused with MBP and TrxA, respectively, to provide the high expression level of the recombinant protein. In addition, numerous mutations were introduced into the protein sequence in order to crystallize it, which affected the functional properties of NTR1 (at the same time, the properties of the receptor expressed in insect cells were not impaired [44]).
Expression in yeast cells combines the advantages of bacterial expression systems (low cost of reagents, simplicity of genetic manipulations, rapid biomass growth) and the possibility of some posttranslational modifications [105]. Pichia pastoris and Saccharomyces cerevisiae strains have been well characterized genetically. Protein expression in Pichia is typically preferred [106], as it has provided much higher (10-100 times) biomass yield compared to the same volume of S. cerevisiae culture [107]. Pichia strains have been succussfully used for the production of recombinant membrane proteins, including GPCRs [108-111].
The shortcomings of the yeast expression system include the difficulties with cell lysis and disintegration due to the rigidness of yeast cell wall, as well as the fact that protein glycosylation profile in yeast is different from that in mammalian cells [107].
Expression in insect cells is the most common approach in the structural studies of GPCRs, because expressed proteins undergo most of necessary posttranslational modifications occurring in mammalian cells. However, N-glycosylation in insect cells involves attachment of simple unbranched glycoproteins, in contrast to mammalian cells, where complex glycoproteins with branched oligosaccharide chains are formed [112-114].
The temperature conditions for the cultivation of insect and mammalian cells are different, which may influence the cell membrane composition. The membranes of insect cells are characterized by a lower cholesterol content, higher phosphatidylinositol content, and the absence of phosphatidylserine [115]. Such differences can result in lower yields of the expressed receptor. Nevertheless, the cell culture medium can be enriched with the necessary lipids, which enhanced the expression of some receptors, e.g., turkey β1AR [116] and human dopamine D3 receptor [117].
Despite the disadvantages described above, expression in insect cells yields functional proteins that can be used in structural studies.
The highest number of proteins with the solved structures have been obtained in Spodoptera frugiperda cells (cell lines Sf9 and Sf21); a lesser number of proteins have been produced in Trichoplusia ni cells (Hi5 cell line). It is recommended first to test the level of protein expression in small volumes of cell culture, because it can be significantly different in the tested cell lines. Thus, expression of turkey β1AR in Hi5 cells was much higher than in other cell lines [116].
Expression in insect cells is typically performed using the baculovirus system. The expression cassette with the target gene is incorporated through the site-specific transposition into a baculovirus shuttle vector (bacmid) [118] produced in E. coli cells. Subsequent transfection of insect cells with the recombinant bacmid results in the production of baculoviral progeny, which is then used to infect the cells for the expression of protein of interest.
The shortcomings of this method include relatively expensive cultivation medium, cell lysis as a result of infection by the recombinant baculovirus (which can lead to proteolytic degradation of the target protein), and the differences in the glycosylation profile of the produced proteins with the proteins synthesized in mammalian cells. The advantages of this method are its simplicity, high protein yields in suspension culture, the use of serum-free medium for cultivation, and correct folding of membrane proteins (after optimization) [119]. The overall process, from bacmid production to protein expression, takes about 3 weeks on average.
Expression in mammalian cells provides the most native environment for the production of human receptors. Due to correct protein folding and existence of mechanisms providing posttranslational modifications of human proteins, expression in mammalian cells is widely used in molecular biology. HEK293 (human embryo kidney) and CHO (Chinese hamster ovary) cells are the best studied and most popular in GPCR research. The limitations for protein production in mammalian cells include the use of expensive culturing media and antibiotics, as well as the time-consuming selection of cell lines providing stable expression and high yield of the target protein [120]. In this context, mammalian expression systems are typically used in the studies of receptor role in signal transduction, when the high levels of protein expression are not required [121, 122] or in the cases when production of functinally active receptor in other expression systems (e.g., insect cells) has failed [123, 124].
Cell-free expression systems. Cell-free protein synthesis in vitro is based on the use of cell extracts for the protein transcription and translation [125] or translation only [126, 127]. Such systems include high-molecular components (ribosomes, DNA template or mRNA [126], enzymes) and various substrates, such as amino acids, nucleoside triphosphates (NTPs), and energy sources [128]. Membrane-simulating components (e.g., micelle-forming detergents, liposomes, nanodiscs) are often added to ensure expression of membrane proteins in a soluble form [129, 130]. Expression in the absence of membrane-simulating components is also possible [129], but might result in the production of insoluble aggragates. Although it often provides higher protein yields [131], the recombinant protein has to be solubilized, which may have a negative effect on its properties [130].
One of the advantages of the cell-free system is that it can be used for the synthesis of proteins that are toxic for cells [90, 132, 133]. It also allows tight control of the reaction components [127], as well as medium adaptation using various additives [130]. Moreover, cell-free expression systems make it possible to include uncommon amino acids into the produced protein to impart new properties to the latter [132]. For example, labeled amino acids can be introduced to the protein sequence for further protein characterization by NMR spectroscopy [131, 132]. Also, affinity purification can be performed immediately after the expression [133, 134], in opposite to cell-based systems that require cell disintegration and membrane solubilization for the isolation of membrane proteins [135]. Because the proteins can be expressed in small volumes, it makes easy to evaluate the results of optimization of expression conditions by high-throughput screening with a plate reader [131, 136].
The composition of the cell extract can vary depending on the centrifugal force used for the fractionation of cell components [134]. Cell extract from E. coli cells is most commonly used for the synthesis of recombinant proteins, because it is cost-efficient, easy to use, and provides high protein yields (mg/ml) [134, 135]. However, when choosing the source of cell extract, one should take into consideration the required protein yield, as well as the ability of the system to provide correct protein folding and necessary posttranslational modifications [137]. For example, the extracts of eukaryotic cells, such as insect cells [138, 139], rabbit reticulocytes [140], and Chinese hamster ovary cells [141], ensure protein glycosylation. Also, E. coli extracts can be optimized by adding chaperones, stabilizing compounds, and redox agents that contribute to the correct folding of eukaryotic proteins [142-145]. E. coli strains capable of some types of glycosylation have been obtained as well [146-148]. Other important factors for consideration when choosing an expression system, are how easy the system is to work with and its cost.
Numerous attempts have been made to express GPCRs in the cell-free systems. The produced receptors demonstrated similar protein–ligand binding affinities compared to the receptors expressed in the cell-based systems [149-154]. Moreover, it was possible to obtain conformation-specific antibodies against class A and C GPCRs expressed in wheat germ extract [155]. However, a considerable portion of the synthesized receptors might be nonfunctional [154], because even the near-optimal conditions can affect the formation of the functional protein [129].
Apparently, this is the reason why there are only a few X-ray structures solved for membrane proteins synthesized in cell-free expression systems [135, 156, 157].
Despite the advantages of protein synthesis in cell extracts, cell-free expression system has not become very popular in GPCR research, as they require a lot of effort for the optimization of expression of functional GPCRs.
PURIFICATION AND STABILIZATION OF GPCRs
The structural studies of GPCRs are hindered by the low stability, amphiphilic nature, and conformational mobility (a quality essential for the signal transduction) of the receptors. High-affinity ligands and antibody fragments can be added to the receptors to stabilize them and to reduce their conformational variability.
Methods for GPCR purification. GPCR purification is a complex multistage process often combining several purification techniques, such as gel filtration, ion exchange chromatography, affinity chromatography, and immunoaffinity chromatography.
Affinity chromatography is a common method for purification of recombinant proteins. This technique is based on the specific interaction between a target protein and a ligand bound to the stationary phase. Below, we describe the ligands used for GPCR purification.
Metal chelate affinity chromatography uses the interaction between an immobilized metal ion and free electron donor groups of amino acid side chains. Typically, a resin with immobilized Ni2+ or Co2+ is used as the stationary phase, while the receptor is modified with the His-tag at the N- or C-terminus. The imidazole rings of the His-tag bind to the chelated ions of divalent metals, thus allowing to separate the target protein from the cell lysate components, even in the presence of detergents used for GPCR solubilization. Resins with immobilized cobalt ions are believed to have a lower protein binding ability and a higher specificity than resins with immobilized nickel ions [158]. Because of a higher purity of the resulting protein sample, Co2+-containing resins are often preferred for the GPCR purification.
The first stages of GPCR purification are often performed using other chromatography techniques. For example, proteins can be purified using the binding between Strep-tag (WSHPQFEL) incorporated into the receptor amino acid sequence and immobilized avidin (Strep-Tacin) [80, 159, 160]. It is also possible to use amylose resins for purification of MBP-tagged proteins [75, 161, 162].
Immunoaffinity chromatography is based on the antibody–protein interaction. Very often, the second [163, 164] or even the first [165, 166] step in the GPCR purification is chromatography with anti-FLAG antibodies [72]. At present, calcium-dependent antibodies M1 [73], calcium-independent antibodies M2 [167], M5 [168], and L5 [169], and other anti-FLAG antibodies are commercailly available; however, only agarose beads with immobilized M1 and M2 are produced. To use immobilized M1 antibodies, the FLAG-tag should be at the protein N-terminus, while M2 antibodies bind both to the N-terminal and C-terminal FLAG-tag; however, the chromatography on M1 antibodies is preferable [170].
Rhodopsin has been studied using a special resin with 1D4 antibodies specifically binding to its C-terminal sequence (TETSQVAPA) [171, 172], which was used for purification of other recombinant GPCRs [173]. Another tag that can be employed in the immunoaffinity chromatography of GPCRs is the C-reactive protein, which binds to specific anti-C-reactive protein antibodies immobilized on agarose [12].
Ligand chromatography. Strictly speaking, the previously described techniques also represent ligand chromatography, as they involve biospecific interactions between proteins and immobilized ligands. Here, we will describe the type of affinity chromatography that does not require introduction of special tags in the receptor amino acid sequence. In the case of ligand-specific chromatography, a ligand selectively interacting with the studied receptor is cross-linked to an inert carrier, thereby making it possible to separate functional ligand-binding receptors from the nonfunctional ones [72]. The best known technique is alprenolol affinity chromatography. Alprenolol is a nonselective beta-blocker and antagonist of serotonine receptors 5HT1A and 5-HT1B [174]. A resin with immobilized alprenolol was used to purification of β1AR [12, 175-178]. To get rid of alprenolol, different chromatographic methods could be combined [179, 180]. Ligand chromatography was used for purification of untagged M2 muscarinic receptor [181].
The final stage of purification of recombinant GPCRs and their complexes can be affinity chromatography on immobilized lectins [12]. Concavalin A (ConA) is a protein that binds α-D-mannose and α-D-glucose with a high specificity; therefore, it binds only glycosylated membrane proteins [59].
Ion-exchange chromatography is used for GPCR purification less often than affinity chromatography. It is based on the electrostatic interactions between the charged side groups of amino acids and oppositely charged immobilized groups. The strength of binding is proportional to the total protein charge. Ion exchange chromatography has been used to purify GPCRs [47, 182], as well as G proteins, nanobodies, antibodies [180, 183], and other proteins used in the studies of GPCRs [184].
Gel filtration, or size-exclusion chromatography (SEC), is a method for the separation of proteins according to their size and shape. For globular proteins, the size of the molecule directly depends on its molecular mass, which allows to separate proteins by their mass [185]. The resin for gel filtration is synthesized by cross-linking dextran molecules into beads of a particular size that act as molecular sieves. The pores of the beads retain smaller molecules, while larger molecules do not enter the pores and pass throught the resin much faster [186]. The tecnique is described in more detail in the section “Characterization of Protein Preparations and Quality Control”.
Although preparative gel filtration is rarely used as the final purification step before receptor crystallization [20], it is commonly used to purify large GPCR complexes with G proteins, antibodies, etc. [161, 184, 187-189].
Membrane modelling systems in GPCR studies; their pros and cons. Each GPCR in the cell membrane creates a unique physicochemical environment necessary for its functioning. Correct GPCR folding, interaction with ligands, and signal transduction depend on the membrane thickness, curvature, lipid composition, electrostatic potential, as well as membrane pressure on the receptor. All these factors should be taken into account when choosing a membrane modelling system (MMS) [190]. Below, we discuss the most popular MMSs used in the studies of GPCR structure and function.
Detergent micelles. Detergents are amphiphilic compounds consisting of the hydrophilic head and hydrophobic tail. When a detergent is added to the GPCR-containing membranes, detergent molecules substitute for the membrane lipids and form a micelle around the protein (Fig. 2a). Delipidation is the main cause of GPCR destabilization and inactivation [191]. Detergent molecules are more mobile than lipids, which means that the micelle geometry is unstable; the protein can determine the thickness and shape of the micelle hydrophobic part, as well as adopt some nonphysiological conformations [192]. Therefore, studying GPCRs in micelles often requires introduction of stabilizing mutations into the protein. Detergents can also bind to ligands, G proteins, and other molecules, hindering the interactions between these molecules and GPCR.

Main membrane modelling systems.
Based on the charge, detergents are divided into nonionic, ionic, and zwitterionic. Nonionic detergents [e.g., DDM (n-dodecyl-β-D-maltoside), CHS (cholesteryl hemisuccinate), and LMNG (lauryl maltose neopentyl glycol)] are very efficient for solubilization [193]. Ionic detergents, e.g., SDS (sodium dodecyl sulfate), have a more severe impact on the protein molecule and can be used for both protein solubilization [194] and denaturation [195]. Zwitterionic detergents are used less commonly; however, they allow to obtain GPCRs with the melting temperature close to that in a DDM+CHS mixture [196].
Branched detergents better stabilize GPCRs due to their structure and more efficient packaging. The branched detergent LMNG forms a hydrogen bond between its two heads, thus restricting the mobility of the receptor. In the case of unbranched DDM, two detergent molecules cannot form stable hydrogen bonds between them; the receptor in such micelle is more flexible and, therefore, binds the ligands more efficiently [197].
Detergents are characterized by the critical micelle concentration (CMC) which is defined as the detergent concentration above which micelles form. The CMC of a detergent depends on the temperature and properties of the solvent [198]. The CMC for different detergents can vary within a rather broad range: in water, the CMC for DDM is 0.17 mM and CMC for LMNG is 0.01 mM (i.e., more than 15 times less than for DDM). The CMC should be taken into account when choosing a detergent, inter alia, for the structural studies. Usually, for successful solubilization, the detergent is added to the protein in excess, which results in the presence of free detergent molecules or even empty micelles in the solution [199]. Solubilized receptors can have the same size as empty micelles and therefore might be mistaken for them, which significantly hinders the analysis by cryo-EM [200]. Free detergent also decreases the signal/noise ratio. During crystallization in lipid cubic phase (LCP; see below), free detergent molecules can prevent the phase formation [201]. Therefore, it is necessary to get rid of free detergent, which it is difficult to achieve for detergents with low CMC values using traditional methods (e.g., dialysis or gel filtration). New methods are currently being developed for the removal of free detergent from the GPCR solutions [200]. A detergent with a very high CMC can be added to the protein sample to form a layer at the water–air interface and to prevent protein adsorption on it [202, 203]. However, receptors are more efficiently incorporated into micelles formed from the detergents with lower CMCs (due to longer hydrophobic tails) [204]. It makes such detergents more attractive for the micelle formation. For example, the structures of the modified β2-adrenergic receptor (β2AR) in a complex with the G protein and beta-arrestin [205], A2AR complex with the G protein [93], and rhodopsin complex with the G protein [206] were obrtained using the LMNG micelles. However, there is no universal solution; the detergent should be selected for each receptor and in each experiment.
Incorporation of GPCRs into detergent micelles is currently the most popular method of their solubilization due to simplicity of micelle formation and diversity and relatively low cost of detergents. Other MMSs, unless otherwise specified, require preliminary protein solubilization into detergent micelles and, therefore, have the same shortcomings, e.g., the necessity for additional protein stabilization by mutations.
Amphiphilic polymers. Amphiphilic polymers (amphipols, Apols; Fig. 2b) are long molecules with alternating hydrophilic and hydrophobic regions. The molecular mass of such an amphiphilic polymer can be up to 34 kDa [207]. Amphiphilic polymers show high affinity to the protein hydrophobic part and wrap around it; at the same time, the concentration of free amphiphilic polymers in solution is very low. Amphiphilic polymers have a much milder impact on protein than detergents and are ineffective in solubilization of membrane proteins [208]. The possibility of GPCR stabilization by a biotinylated amphiphilic polymer has been shown for the rhodopsin-like growth hormone secretagogue receptor (GHSR) [209].
SMALP polymers. SMA (styrene maleic acid) is a polymer of styrene (hydrophobic) and maleic (hydrophilic) acids (Fig. 2d). It can solubilize receptors directly from the membrane, allowing to bypass the detergent stage. Styrene acid penetrates into the membrane and fragments it, while maleic acid stays outside the formed SMALP (styrene maleic acid lipid particle) [210]. The distinguishing feature of this MMS is that GPCR is isolated together with a surrounding membrane region. On the one hand, it allows to study GPCRs in the presence of native lipids and to analyze their effect on the receptor function. On the other hand, it makes impossible to control the lipid environment of the receptor. SMALPs were used to isolate β2AR from HEK293T cells [211] and to study the interaction between A2AR and its ligands [212].
Lipid–protein nanodiscs. Lipid–protein nanodisc (Fig. 2c) consists of two molecules of the modified apolipoprotein ApoA-1 wrapped around the lipid bilayer in the antiparallel orientation. By varying the length of ApoA-1, nanodiscs of different sizes (8-16 nm in diameter) can be obtained. Nanodiscs have a higher stability than micelles, and their structure is more similar to the membrane one [213], so they can be used to study the interactions between GPCRs and G proteins [214]. The nanodisc technology continues to evolve and new techniques of nanodisc assembly are being developed, e.g., covalent circularization using the treatment with sortase [215] or the use of DNA-origami barrels to obtain nanodiscs up to 90 nm in diameter [216].
SapNP nanodiscs. Saposins A, B, C, and D are small proteins involved in the sphingolipid metabolism in the body [217]. They consist of four alpha-helices and form disc-shaped particles in the presence of lipids [218]. Similar to SMALP, saposins can solubilize proteins directly from the membrane. They have different lipid-binding specificity, with saposin A binding to the largest number of lipids. Therefore, it is more often used for assembling SapNP (saposin nanoparticle) discs. The proteins in SapA discs are more thermostable than in detergent micelles [219].
Bicelles. Bicelle consists of a flat phospholipid bilayer rimmed with a detergent (Fig. 2e). The most popular bicelle components are DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) as the phospholipid and DHPC (1,2-dihexanoyl-sn-glycero-3-phosphocholine) as the detergent; the ratio between these components determines the size of a bicelle [220]. Bicelles can be used to study the interactions of GPCRs with G proteins and ligands, as it has been done for the human neuropeptide Y receptor type 2 (Y2R) [221]. Bicelles are also used in protein crystallization [222, 223].
Liposomes. When lipids are dried and then rehydrated, they self-arrange into liposomes, which are small bilayer vesicles (Fig. 2f). Multilamellar liposomes can reach up to 50 µm in diameter. Multilamellar liposomes are usually undesirable; therefore, they are additionally treated to obtain unilamellar liposomes. Liposomes are produced by extrusion, sonication, and homogenization [224, 225] and can be used to study multicomponent systems and to reconstruct entire GPCR-triggered signaling pathways. Among all the above-described MMSs, liposomes are the most similar to the cell membrane in their physical properties [226, 227].
Lipid cubic phase (LCP) is a lipid bilayer, which under certain conditions, self-arranges into an infinite periodic surface that divides the space into two non-overlapping areas (Fig. 2g). LCP serves as a matrix consisting of a lipid bilayer; it provides diffusion of protein molecules within itself and facilitates crystallization. This MMS has become very popular as it allowed to crystallize some GPCRs and retinal-containing membrane proteins. Successful crystallization in LCP depends on several factors. Firstly, the lipid bilayer facilitates ordered positioning of hydrophilic and hydrophobic parts of adjacent protein molecules, allowing them to form a crystal consisting of multiple membrane protein layers. Such crystals usually diffract well, because the contacts between the protein molecules are formed by both hydrophilic and hydrophobic regions, which contributes to better ordering. Secondly, the lipid bilayer simulates the natural environment of membrane protein, thereby increasing its stability. Thus, crystallization in LCP or its less ordered analogs (e.g., lipidic-sponge phase) makes it possible to eliminate the major difficulties associated with membrane proteins. In addition, LCP serves as a filter that cuts off large molecular aggregates, thereby facilitating protein crystallization [228]. Depending on the selected conditions (mainly, the concentrations of components in the solution and temperature), some of lipids can assemble into the structures with different space group symmetry (the most comon are the cubic phases Im3m and Pn3m and the lamellar phase) [229]. The most popular lipid for the LCP formation is monoolein; however, using other lipids allows to alter the properties of the LCP (see above) and to vary the sizes of the elementary cell from 30 to 240 Å [230] in order to select the phase parameters for a particular protein to facilitate its correct incorporation into the lipid bilayer [231]. GPCR crystals grown in the LCP may be too small to obtain a high-resolution structure even while using the microfocus stations at the synchrotron radiation sources, so these crystals can be studied by serial femtosecond crystallography with XFEL (X-ray free electron laser) or micro-electron diffraction [232, 233].
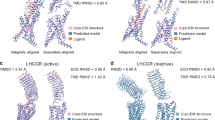
Using ligands for receptor stabilization. In the absence of ligands, GPCRs can exist in numerous conformational states [82]. Ligand binding results in the conformational changes that may stabilize the receptor in the ligand-bound state, which is determined by the type of ligand used (antagonist, inverse agonist, or agonist). The ligands providing the best receptor stabilization are selected before crystallization, e.g., by the thermal shift analysis with a fluorescent dye interacting with protein cysteines [234]. Among 450 GPCR structures solved by X-ray crystallography, 429 were crystallized with exogenous ligands; 301 out of 337 cryo-EM structures were also resolved in the presence of ligands (according to GPCRdb [235], August 2022).
So far, only several receptors have been successfully crystallized in the absence of a ligand or without stabilization by the antibody fragments. The first ligand-free structure was solved in 2008 for the bovine opsin [236]. The other structures appeared much later, e.g., of the zebrafish (Danio rerio) lipid receptor LPA6 in 2017. The structure of this receptor was shown to have a lateral vertical cleft between TM4 and TM5, that contained a hydrophobic molecule, probably, endogenous lipid, detergent, or monoolein, which was present in abundance during crystallization. Supposedly, this cleft is a part of the ligand-binding pocket and is intended for the acyl chain of lysophosphatidic acid, a natural ligand of the LPA6 receptor [237]. The structure of the class F human Frizzled 4 receptor (FZD4) with deleted extracellular cysteine-rich domain (CRD) and stabilized by 4 mutations appeared in 2018 [238]. Human orphan receptor GPR52 stabilized by point mutations was crystallized in the absence of ligands. It was found that GPR52 has a uniquely folded extracellular loop 2 (ECL2) that operates as a built-in agonist [239]. Nevertheless, for most GPCRs, ligands proved to be necessary for the receptor stabilization and crystallization, which emphasizes their importance and the necessity of further search for ligands for difficult-to-crystallize receptors.
Using antibody fragments for stabilization of GPCR conformational states. Strategies of using antibody fragments instead of or in addition to partner proteins proved to be successful for some receptors. Antibody fragments can form contacts in the crystal, stabilize proteins in a specific state (thus making protein samples homogeneous), serve as allosteric modulators, and increase receptor thermostability [240]. Antibody fragments aimed at stabilization of receptors in a certain conformational state in complexes with ligands are obtained by immunization of laboratory animals, followed by the generation and selection of antibody-producing hybridomas and purification of monoclonal antibodies [11, 240]. Purified antibodies are fragmented into the antigen-binding Fab domain and crystallizable Fc fragment using papain [241].
For the first time, the monoclonal antibody Fab fragment was used for the crystallization of β2AR (PDB ID: 2R4R) in a complex with its partial agonist in order to decrease the conformational mobility of flexible protein domains and to increase the polar surface for the formation of contacts in crystal, what resulted in a structure with a resolution of 3.4 Å [183]. The structures of the 5-HT2B receptor, sphingosine-1-phosphate receptor 3 (S1PR3), and angiotensin II type 2 receptor (AT2R) in the active conformation were also obtained using Fab fragments recognizing the extracellular domains of these receptors [240, 242-244]. Receptors that were crystallized in the inactive state with the help of Fab fragments include the class B glucagon receptor with T4L incorporated in the ICL2 [86], A2AR receptor [109], and others [11, 38, 240, 245, 246].
In addition to Fab fragments targeting the receptor epitopes, the antibody fragments against the fusion partner BRIL were used to crystallize the ligand-bound glucagon-like peptide-1 receptor. Because anti-BRIL Fab fragments can be employed to facilitate crystallization of different GPCRs fused with BRIL without having direct effect on the receptor, they can be used to study both active and inactive receptor states [11].
In later studies, protein crystallization was performed in the presence of nanobodies (Nb), which are recombinant antigen-binding domains of unique camelid (Tylopoda) antibodies lacking the light chains [247]. The term nanobodies was chosen because of their small size (25% of typical Fab fragment) [248]. Originaly, nanobodies have been used for stabilization of the active conformations of receptors in complexes with agonists, because crystallization of GPCRs in the active state is a difficult task due to their conformational flexibility and instability. For example, the first structure of β2AR in the active state was solved using the Nb80 nanobody that served as an alternative to the Gs protein [248]. Later, Nb80 has been subjected to molecular evolution with the creation of Nb6B9, which was used to obtain several more receptor structures, including those with low-affinity agonists [179, 249]. Some Nbs have been found to stabilize GPCRs in the inactive conformation [250, 251].
Production of antibodies against the fixed states of individual receptors is a labor-, time-, and money-consuming task [252]. One of the approaches used to facilitate this process was successful transfer of the Nb6 epitope from the kappa opioid receptor κOP to other GPCRs [253]. Originally, Nb6 was obtained against the ICL3 of the κOP receptor [254]. Replacing the ICL3 of the target receptor with the ICL3 of κOP ensured Nb6 binding with the target GPCR, which makes not necessary the production of new specific antibodies against the studied receptor.
Using salts and chemical agents for purification and stabilization of receptors. The buffers used in GPCR studies often contain chemical agents to maintain the protein in a stable and monomeric state. The most common of them are glycerol and sodium chloride, which are present at almost all stages of receptor purification. Depending on the structural research method (crystallization or cryo-EM), it might be necessary to treat the receptor with other salts and small molecules, such as magnesium chloride, potassium chloride, ATP, and iodoacetamide.
Sodium chloride determines the ionic strength of the solution and may help to maintain the receptor in the monomeric state by abolishing polar interactions between the protein molecules [255]. The concentration of sodium chloride in buffers for different receptors can vary from trace amounts (below 6 mM) [37] to 800 mM [256, 257].
Many receptors are stable within a broad range of NaCl concentration [242, 257-259], which allows NaCl to be used as a tool in GPCR purification. For example, cells with the overexpressed receptor are washed in the buffer with a zero concentration of NaCl and low concentrations of KCl and MgCl2. KCl and MgCl2 provide the ionic strength necessary to maintain the receptor stability, while the absence of NaCl outside the cells causes an osmotic influx of water into the cells, which later facilitates their disintegration. Consequently, the membranes of already disintegrated cells are washed with a buffer with high NaCl concentration (1000 mM) to remove soluble intracellular and peripheral membrane proteins (high salt concentrations cause their aggregation) [260], while GPCRs incorporated in the membrane remain in the native conformation. In addition to NaCl, urea can be used to remove peripheral membrane proteins [44, 261].
Glycerol is another additive stabilizing GPCRs. Glycerol is amphiphilic and shields the hydrophobic regions on the surface of protein molecule from the aqueous solution, thereby maintaining correct receptor conformation [262]. It is especially important in the case when GPCR is extracted from the membrane by solubilization and is embedded in detergent micelles. As mentioned above, detergent molecules are more mobile than native lipids in the membrane; in addition, there is a continuous exchange of detergent molecules between micelles and surrounding buffer. Therefore, the hydrophobic amino acids hidden by lipids in the native membrane might become exposed, at least transiently, to the solution. In order to maintain the receptor stability in micelles, glycerol is added to a concentration of 10-30% during receptor solubilization [259, 263]. In many GPCR purification protocols, glycerol concentration in the washing buffer is 10% [257-259]. Glycerol is also a cryoprotectant [264] and is added to the buffers for the long-term storage of cell membranes or to purified receptors before freezing [259].
Iodoacetamide is used for the purification of GPCR preparations intended for crystallization, which requires the protein to be in an extremely monomeric state. Iodoacetamide is an alkylating agent that irreversibly modifies the -SH group of cysteine, preventing further formation of disulfide bonds. If the receptor contains cysteine residues exposed to the solution, two receptor molecules can form a disulfide bond through these cysteines after solubilization, thus disturbing the monomeric state of the protein. Iodoacetamide prevents the formation of such covalently linked dimers [265], for which the membranes containining the receptor are incubated with iodacetamide at a conentration of ~2 mg/ml immediately before solubilization [258, 259].
ATP is also used for GPCR purification to increase the monomeric state of the receptor. All GPCRs have a rather complex structure and often require molecular chaperones for correct folding [266]. Individual chaperones, e.g., Hsp70, regulate the process of GPCR signaling, ensuring dissociation of G proteins from the receptors [267]. Even after membrane washing and solubilization, some receptors can still be bound to chaperones, many of which are ATP-dependent. When interacting with ATP, chaperones change their conformation and can be detached from GPCR. Therefore, ATP is added to the washing buffer for the solubilized receptor [258, 259].
CHARACTERIZATION OF PROTEIN SAMPLES AND QUALITY CONTROL
Purified protein samples used for the structural and functional studies can be characterized by analytical gel filtration, thermal shift assay, and nano differential scanning fluorimetry.
Analytical gel filtration, or size-exclusion chromatography (SEC), is a chromatographic technique providing separation of macromolecules in solution according to the ratio of the molecule hydrodynamic radius to the average pore size of chromatography resin [268] packed into a chromatography column. When proteins in a buffer pass through the column, smaller molecules enter the particle pores, so their retention time increases, while large proteins are eluted without entering the pores. As a result, proteins are fractionated according to their size [269]. Eluted proteins are usually monitored with a UV absorption detector.
In fluorescence-detection SEC (FSEC), the target proteins are covalently linked to the green fluorescent protein (GFP) or some other, and the SEC profile is recorded using the fluorophore emission. Since FSEC uses unique GFP signal, the experiment requires neither protein purification, nor large-scale protein production. The measurements can be performed after solubilization of intact cells or unpurified membranes, which significantly simplifies precrystallization screening required for selecting the optimal conditions of protein production [270].
SEC is one of the most useful tools for monitoring the monodispersity and stability of target proteins. A monodisperse and correctly folded protein is usually eluted as a symmetric Gaussian peak, while a polydisperse, unstable, or unfolded protein is eluted as several asymmetric peaks (Fig. 3). SEC elution profiles can also provide information on the impurities present in the sample; therefore, SEC is widely used for the analysis of the homogeneity, stability, and purity of proteins and their complexes (i.e., basic indicators for the suitability of protein samples for the structural studies) [271].

Example of the SEC profile of a protein sample (absorbance at 280 nm plotted vs. retention time). Monomeric protein is eluted as a symmetric Gaussian peak 1. The fractions with higher molecular masses, presumably protein dimers (peak 2) and protein aggregates (peak 3) are eluted before the monomeric protein.
The homogeneity and oligomeric state of proteins can be determined by comparing the retention times of the protein–detergent complex and molecular mass standrads. However, it should be taken into account that the molecular mass standards for SEC are soluble proteins or small molecules, whereas membrane proteins form complexes with detergent molecules, which can potentially increase their hydrodynamnic radius and lead to the overestimation of the protein molecular mass [272]. This problem can be solved by using light scattering techniques, e.g., MALS.
Multi-angle light scattering (MALS) is a well-proven method for studying protein interactions, which can be used both for single proteins and protein complexes. In a typical MALS experiment, scattering of a laser beam by the protein solution is measured at several angles in the plane perpendicular to the incident light. The total scattering intensity depends on the protein molar mass and concentration, whereas the angular dependence is related to the root mean square (rms) radius of the molecule. Analysis of the angular variation of scattered intensity allows to determine the root mean square radius, molecular mass, and concentration of macromolecules in the sample [273].
MALS is especially efficient in combination with SEC. As protein complexes are eluted from a SEC column, they can be immediately analyzed with a MALS detector. This makes it possible to fractionate different proteins or protein complexes present in the sample with simultaneous measurement of their molecular masses [274].
Thermal shift assay. Expression and purification of recombinant proteins can be considerably improved by the addition of stabilizing buffers or ligands that reduce the tendency of expressed proteins to form aggregates during purification and storage in vitro. Moreover, for recombinant proteins, high protein stability correlates with its ability for crystallization [275]. Stabilizing buffers and additives are identified by their capacity to increase the protein melting temperature during its thermal denaturation.
One of the methods for assessing protein thermostability is thermal shift assay (TSA), also referred to as differential scanning fluorimetry. Protein preparation for TSA involves protein incubation with specific fluorescent dyes (e.g., SYPRO Orange) that increase their quantum yield upon binding with the protein hydrophobic regions that become available for the interaction during protein denaturation. Thermal denaturation induced by a gradual temperature elevation can be monitored as the increase in the dye fluorescence [276]. Protein stability is evaluated based on the melting temperature (Tm), which can be increased by changing the buffer or introducing more additives to the buffer solution. This increase in the melting temperature is referred to as the thermal shift indicating an increase in the protein stability. The thermal shift can be also used to identify the ligands that stabilize receptors by binding to them [277]. Thus, the dye N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM) was first used in the TSA of human apelin receptor (APJ) [234]. Therefore, TSA can be used to analysis the stability of GPCR variants with different partner proteins, point mutations, and lengths of the N- and C-termini, as well as to search for new ligands by screening compound libraries.
Nano differential scanning fluorimetry (nanoDSF). In contrast to TSA, nanoDSF does not involve protein labeling with a fluorescent dye. NanoDSF tracks the changes in the intrinsic fluorescence of tryptophan residues in the protein molecule caused by alterations in the protein 3D structure induced by temperature changes [278]. Intrinsic tryptophan fluorescence responds to the changes in the protein microenvironment due to the solvatochromic properties (i.e., environment-dependent changes in the fluorescence parameters) of the indole ring. The fluorescence maximum of tryptophan in a nonpolar environment is 330 nm (excitation wavelength, 280 nm). In the polar environment, the fluorescence intensity usually decreases due to the static and dynamic quenching by the solvent molecules, while the emission peak shifts toward the red region of the spectrum (approximately to 350 nm) [279]. This usually occurs when tryptophan residues normally hidden in the protein hydrophobic core are exposed to the aqueous environment during denaturation. The temperature required to unfold 50% of the protein (Tm) can be determined from the extent of the red shift (sometimes, blue shift) of the tryptophan fluorescence (Fig. 4).

Example of nanoDSF data. Solid lines, the ratio of sample fluorescence at 350 nm to fluorescence at 330 nm; dashed lines, the first derivative of the ratio; blue curves, the data for protein melting without the ligand; red curves, the data for protein melting with the ligand; vertical dash-dotted lines, the melting temperature of the samples. Protein stabilization due to the ligand binding results in the increase of the melting temperature.
Since nanoDSF can be used to monitor protein thermostability from changes in its melting temperature, it is employed to search for the optimal conditions of protein storage and crystallization that ensure the highest protein stability [280]. NanoDSF also allows to detect the protein–ligand binding and can help in obtaining valuable information on the ligand-binding site and the mechanism of protein action [281].
FORMATION OF RECEPTOR COMPLEXES WITH MAIN INTERACTION PARTNERS
Formation of GPCR complexes for crystallographic studies. In order to understand the molecular mechanisms of GPCR function, it is essential to know the structures of receptor complexes not only with ligands, but also with protein molecules interacting with the receptor and involved in the signal transduction inside the cell, such as G protein-coupled receptor kinases (GRKs) [265], G proteins, arrestins, etc.
According to GPCRdb, 450 GPCR structures have been obtained by X-Ray crystallography. Among them, 53 are receptor complexes with antibodies, antibody fragments, and nanobodies; 18 and 4 are complexes with G proteins and visual arrestins, respectively.
Antibodies and antibody fragments are often used in the structural studies of GPCRs (see “Using Antibody Fragments for Stabilization of GPCR Conformational States” section). For example, nanobodies have been used as chaperones in crystallization of receptor; however, each receptor requires development of a specific unique nanobody [282]. Protocol for the nanobody–receptor complex formation and crystallization is the following: a receptor is mixed with the excess of a nanobody; the reaction is carried out at 4°C for 1-8 h; the complex is purified and concentrated; aliquots (7-8 µl) of the complex are frozen in liquid nitrogen and defrosted immediately before mixing with lipids and following crystallization [179, 283-285]. If the receptor is thermostable, the procedure of complex formation can be performed at room temperature and more rapidly [249]. The complex can be concentrated up to 40-50 mg protein/ml [286] or crystallized without preliminary freezing [180]. The protocol for receptor–antibody fragment complexes formation is principally the same: the antibodies are mixed with the receptor, which may be purified, immobilized on the affine resin during purification, or even can still be in the membrane before solubilization. The mixture is kept on ice for 1-8 h and then the receptor–antibody fragment complex is purified by gel filtration, concentrated, and used for crystallization [11, 240, 243, 246, 287].
Heterotrimeric G proteins are a family of important proteins involved in transmitting signals through GPCRs. G protein is composed of three subunits: Gα, Gβ, and Gγ [288]. During receptor activation, G protein dissociates into the Gα and Gβγ subunits. Based on the homology and associated downstream signaling pathways, G proteins are classified into four families according to their Gα subunits: Gs (Gs and Golf), Gi/o (Gi1, Gi2, Gi3, Go, Gt1, Gt2, Gt3, and Gz), Gq/11 (Gq, G11, G14, and G15), and G12/13 (G12 and G13) [289]. Gs subunits activate adenylate cyclase, while Gi/o subunits inhibit it; Gq/11 subunits activate phospholipase C-β, and G12/13 subunits activate small GTPases [290]. There are also 5 different types of Gβ and 12 types of Gγ subunits which trigger the associated signaling pathways and provide a great variety of heterotrimeric G proteins. Gα subunits are GTPases that use the energy of guanosine triphosphate (GTP) hydrolysis into guanosine diphosphate (GDP) and trigger intracellular signaling cascades [291-293].
The first crystal structure of the GPCR complex with the G protein was published in 2008. Rhodopsin solubilized from bovine retina was mixed with the chemically synthesized C-terminal fragment of the Gα subunit at a molar ratio of 1 : 4, incubated on ice, and illuminated by green light (500 ± 20 nm) for complex formation, followed by crystallization using the hanging drop technique [236].
The next solved crystal structure of the β2AR complex with the Gs heterotrimer and nanobody 35 (Nb35) has become the crowning achievement of ten years of studies of GPCRs. The structure was obtained for the receptor at the moment of its activation and signal transduction via the G protein. The G protein subunits (recombinant bovine Gα and Gγ and rat Gβ) were expressed separately, combined into a complex, and then mixed with the ligand–receptor complex with a slight excess of Gs. The mixture was incubated for 3 h at room temperature with the addition of apyrase for the hydrolysis of GDP released from Gs during the complex formation to prevent dissociation of the receptor–Gs complex. The complex was separated from the unbound G proteins using chromatography on anti-FLAG resin and from the unbound receptors using SEC. One hour before crystallization, the ligand–T4L–β2AR–Gs complex was mixed with a slight molar excess of Nb35 and kept at room temperature. Nb35 was used for stabilization of the Gs complex, which improved its affinity to the receptor. Crystallization of the ligand–T4L–β2AR–Gs complex was performed by the LCP crystallization technique [85].
To facilitate crystallization of the GPCR–Gs complex, mini-Gs (construct 414) was designed, which is a shortened variant of Gαs with eight-point mutations [294] capable of the protein stablization in the absence of Gβγ [92, 295]. Mini-Gs increased the receptor affinity to the agonist (similar to the heterotrimeric Gs) and demonstrates the same sensitivity to Na+ (allosteric modulator). The complex of A2AR with the NECA agonist and mini-Gs proved to be more thermostable than the receptor–NECA complex. The complex was formed by mixing the purified A2AR with a 1.2 molar excess of mini-Gs in the presence of apyrase. The mixture was incubated overnight on ice, and the complex was purified by SEC before crystallization [294].
Mini-G versions have been designed for other Gα subunits [92]. The protocol for the formation of GPCR complexes with mini-G proteins is rather universal [171].
All above-described crystallization procedures have been developed for the nucleotide-free complexes, so additional experiments were required to obtain the receptor–Gs–GDP complexes. The group of Brian Kobilka [296] inserted the amino acid sequence of the GDP-binding region of Gαs (hereinafter, GsCT) into the β2AR instead of the ICL3. The GsCT sequence was inserted via optimized linkers between TM5 and T4L, T4L and GsCT, GsCT and TM6. A disulfide bridge was introduced between GsCT and TM5, which stabilized the interaction between the receptor and the G protein. This method yielded high-quality crystals that allowed to understand the interaction between the receptor and nucleotide-free Gαs. Gi/oCT peptides created by analogy with GsCT have been used to study structural changes associated with signal transduction from the visual receptors to G proteins [297-304].
The latest crystal structure of the GPCR–G protein complex was published in 2021 [305]. The structure of the complex consisting of the D1 dopamine receptor, its ligand, Gs heterotrimeric protein, and Nb35 was compared to that of the analogous β2AR complex and studied for the interaction between the receptor and G protein. The reaction of complex formation in this work was carried out on the anti-FLAG resin. The complex components were added to the resin and the mixture was incubated for 3 h at 4°C with gentle shaking. Next, the receptors and the assembled complexes were eluted from the resin, followed by the final complex purification by gel filtration.
To ensure the assembly of the GPCR complex with G proteins, it is necessary to add apyrase in order to remove nucleotides that can cause the dissociation of the complex [85]. Copper phenanthroline can be used to catalyze the formation of disulfide bridges between the receptor and the G protein [296]. The disulfide-mediated protein aggregation during the complex formation can be prevented by tris(2-carboxyethyl)phosphine (TCEP), but not with iodoacetamide, because the latter blocks cysteines in subunits α and β of Gs and causes complex dissociation [85].
Other important components of the GPCR-mediated signal transduction are arrestins. These proteins are involved in receptor desensitization and removal from the cell membrane, as well as in the triggering of the G protein-independent cellular pathways. The arrestin family includes the subfamilies of visual arrestins (arrestin 1 and arrestin 4) and non-visual β-arrestins (β-arrestin 1 and β-arrestin 2, also referred to as arrestin 2 and arrestin 3, respectively). Also, the family of α-arrestins structurally related to β-arrestins has been identified. Type 1 and 2 β-arrestins, which are typically used in structural studies, are expressed in most mammalian tissues and cell types [306].
At present, four structures of the visual rhodopsin–arrestin 1 complexes have been solved. Three of them were obtained by the co-expression of both proteins in a single polypeptide chain composed of the full-sized human rhodopsin with T4 lysozyme at the N-terminus and type 1 arrestin connected by the flexible AAAGSAGSAGSAGSA linker to protein C-terminus. All-trans-retinal was added to the complex during the LCP preparation [15, 307]. Another rhodopsin–arrestin 1 complex was assembled from the rhodopsin isolated from bovine retina and chemically synthesized ArrFL-1 peptide (arrestin 1 loop region). The solubilized receptor was mixed with the peptide at a molar ratio of 1 : 12; the mixture was incubated on ice for 5 min and illuminated by light at 530 nm. The complex was crystallized by the hanging drop technique [308].
Studying the complex structure by cryo-EM. The first receptor structure obtained in 2017 by cryo-EM was the structure of calcitonin receptor [309]. The same year, Jacque Dubochet, Joachim Frank, and Richard Henderson were awarded the Nobel Prize in Chemistry “for developing cryo-electron microscopy for the high-resolution structure determination of biomolecules in solution”. After the decades of improving this technique, its application to the structural biology of GPCRs has become a tremendous success. According to GPCRdb, 337 GPCR structures have been solved by cryo-EM in just 5 years.
The procedure used in the first work was as follows: the receptor modified with the N-terminal FLAG-tag and C-terminal His-tag, Gαs and Gβ1γ2 were coexpressed in the baculovirus system in Hi5 insect cells [310]. For this, the cell culture was simultaneously infected with the viral suspensions at the 1 : 2 : 2 ratio, respectively. After the expression for 48 h, the cells were centrifuged and resuspended, and complex formation was triggered by adding the ligand (salmon calcitonin), Nb35, and apyrase. Nb35 bound to the Gαs–Gβ complex, thereby stabilizing the G protein. The reaction of complex formation was performed for 1 h at room temperature, followed by solubilization of the receptors and multistage purification of the complexes.
Due to the limitations of cryo-EM technique, which requires a certain minimum mass and shape of a studied object, this method was first suitable for solving the structures of only class C GPCR complexes with small ligands due to the presence of large extramembrane domains in these proteins [311-315]. For other GPCR classes, the structure of the receptor–ligand complex in the absence of other molecules could be obtained only in the case of dimerization (as for rhodopsin [172]) or by using large protein ligands, such as chorionic gonadotropin [316].
Coexpression allows to obtain protein complexes in both bacterial [317-319] and eukaryotic [320-323] expression systems.The most common approach in the GPCR structural studies is cell transfection with several vectors [320-323]. Coexpression of proteins in a single reading frame is used less frequently [313].
The formation of protein complexes from purified components [12, 184, 324], before protein purification [325], or before receptor solubilization [326, 327] produced similar results. It is possible that the authors of the latter works optimized the protocol of complex purification for their purposes.
Cryo-EM has made it possible to obtain the structure of µ-opioid receptor (µOR) in a complex with Gi1 protein [328]. µOR transmits signals via the subfamily of Gi/o proteins that inhibit adenylate cyclase [329]. The activation of opioid receptor results in pain suppression [330]; therefore, understanding the structure of the agonist–µOR–Gi1 complex was especially important. The Gi1 heterotrimer in the absence of the bound nucleotide was stabilized using a single-chain variable fragment (scFv) that interacted with the Gαi and Gβ subunits, thus stabilizing the G protein but producing no effect on the interactions between the G protein and the receptor. This work was the first one to solve the structure of the GPCR complex with the G protein other than from the Gs subfamily. The experimental protocol for the ligand–GPCR–Gi complex formation was not much different from the procedures for obtaining receptor complexes with Gs proteins: coexpressed G protein subunits were purified, concentrated, and added to the purified concentrated receptor–ligand complex. The reaction of complex formation was carried out for 1 h at room temperature and then for another 1 h in the presence of apyrase. The complex was purified and treated with proteases, then scFv16 was added. The resulting complex was purified by gel filtration, concentrated, and used for the structural studies.
The Gα12/13 subfamily contains only two members: Gα12 and Gα13 [331]. About 30 receptors bind G proteins of this subfamily during activation [332]. G12 and G13 activate the guanine nucleotide exchange factor (GEF) regulating monomeric small GTPases of the Rho family [333]. The mechanisms of the receptor interaction with G12 and G13 had remained unclear for a long time, until the structures of the complexes of type 2 sphingosine-1-phosphate receptor (S1PR2) [334] and adhesion receptor ADGRG1 (GPR56) [335] were obtained in 2022. The G13 heterotrimer in [334] was unstable; therefore, it was decided to create a chimeric Gα13 subunit, in which the αN helix of the wild-type Gα13 was replaced with the αN helix of Gαi [336], and the G protein was stabilized using scFv16. The purified and concentrated components were added in a particular order ‒ first, the receptor and the G protein and then scFv16. In [335], a mini-Gα13 variant was designed by analogy with the previously created thermostable mini-Gα12 [336].
The studies of GPCR complexes with the Gαq protein are particularly difficult case. When Gq/11 proteins bind to GPCR, they activate β-phospholipase, which in turn triggers a long cascade of reactions [337-339]. The structures of the receptor–Gq/11 complex were obtained only after introduction of stabilizing deletions and mutations, as well as modifications ensuring Gαq binding to scFv16. Mini-Gq constructs were created, in which the N-terminal sequence of Gαq/11 was replaced with the N-terminal sequences of other Gα subunits [92, 340, 341]. Gαq/11 subunits in all obtained complexes were chimeric [342-344].
β-Arrestins can desensitize GPCRs [345] or perform signaling functions when bound to GPCRs [346-348]. In contrast to visual arrestins used for the crystallization of protein complexes, β-arrestins are much more flexible and therefore, hinder the X-ray analysis of the complex structure. Cryo-EM was used to establish the structure of the arrestin 2 (Arr2) complex with NTSR1 [349]. The construct consisting of the receptor, β-arrestin, and light chain of Fab30 (BRIL-NTSR1–Arr2–Fab30L) connected via linkers was coexpressed with the heavy chain of Fab30H and GRK5 kinase in Sf9 insect cells. The receptor was solubilized, and the receptor–β-arrestin–heavy and light Fab30 chain complex was purified, concentrated, and used for further studies. GRK5 was necessary for the successful complex formation (the role of phosphorylation in the attachment of β-arrestins to the receptors will be discussed below). The receptor–β-arrestin complex can also be formed from purified and concentrated components, using sortase to attach the phosphorylated peptide to the C-terminus of the receptor [350] or to treat the GRK receptor [351].
Before the attachment of β-arrestins, the C-terminus and ICLs of the receptors undergo selective phosphorylation by kinases of the GRK family that includes seven members [352]. This process determines the direction and dynamics of biochemical signaling and the fate of the receptor; therefore, the structure of the GPCR–GRK complex is of particular interest. At present, such structure was obtained for the complex of visual rhodopsin with GRK1 using cross-linking and kinase inhibitors for the complex stabilization [184].
GPCRs bind G proteins during activation and dissociate from them after signal transduction. Next, GSK phosphorylates the intracellular domains of the receptor, and the receptor is desensitized by β-arrestins. However, it has been noted that sometimes the receptor continues signaling [353-355], so it was hypothesized and then proven that some GPCRs interact simultaneously with β-arrestins and G proteins [356]. The structure of such mega complex was obtained in 2019 [357]. For this, β2AR with the substituted C-terminal sequence of the type 2 vasopressin receptor (β2V2R), β-arrestin 1 (β-arr1), and GRK2 were coexpressed. Before centrifugation, the cells were heated to 37°C with the addition of a ligand for the complex formation. Before solubilization, an excess of Fab30 was added to the membranes and the mixture was incubated for 30 min at room temperature. The receptor was solubilized and Gs heterotrimer, Nb35, and Nb32K were added in excess to the purified β2V2R–β-arr1–GRK2–Fab30 complex. The complex was formed at room temperature within an hour. This publication was supplemented with a short article with comments on the complex structure [205].
CONCLUSION
Here, we reviewed modern approaches to obtaining homogenous stabilized GPCRs. First and foremost, genetic modifications are introduced into the wild-type receptors to delete unordered regions of the protein amino acid sequence, reduce their conformational mobility, eliminate heterogenous posttranslational modifications, and add fusion proteins. The purpose of these modifications is to increase the protein stability, facilitate the formation of contacts in the crystal, or create an additional volume and certain protein shape for the cryo-EM studies. The resulting constructs are expressed in the systems providing correct protein folding. According to GPCRdb, the baculovirus system using insect cells has been proven to be the most successful for GPCR expression. The following stages include receptor purification and stabilization in suspensions with membrane-modeling media and characterization of its homogeneity and stability. We also discussed the issues related to production of GPCR complexes with their main interaction partners, such as G proteins, β-arrestins, and GRKs.
Abbreviations
- BRIL:
-
thermostabilized apocytochrome b562
- cryo-EM:
-
cryoelectron microscopy
- GDP:
-
guanosine diphosphate
- GPCR:
-
G protein-coupled receptor
- GPCRdb:
-
database containing information on modern studies of GPCR (https://gpcrdb.org/)
- GRK:
-
G protein-coupled receptor kinase
- LCP:
-
lipid cubic phase
- MBP:
-
maltose-binding protein
- Nb:
-
nanobody
References
Sriram, K., and Insel, P. A. (2018) G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol. Pharmacol., 93, 251-258, https://doi.org/10.1124/mol.117.111062.
Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schiöth, H. B., and Gloriam, D. E. (2017) Trends in GPCR drug discovery: new agents, targets and indications, Nat. Rev. Drug Discov., 16, 829-842, https://doi.org/10.1038/nrd.2017.178.
Van Montfort, R. L. M., and Workman, P. (2017) Structure-based drug design: aiming for a perfect fit, Essays Biochem., 61, 431-437, https://doi.org/10.1042/EBC20170052.
Kermani, A. A. (2021) A guide to membrane protein X-ray crystallography, FEBS J., 288, 5788-5804, https://doi.org/10.1111/febs.15676.
Callaway, E. (2020) Revolutionary cryo-EM is taking over structural biology, Nature, 578, 201, https://doi.org/10.1038/d41586-020-00341-9.
Martynowycz, M. W., and Gonen, T. (2021) Studying membrane protein structures by MicroED, Methods Mol. Biol., 2302, 137-151, https://doi.org/10.1007/978-1-0716-1394-8_8.
Danmaliki, G. I., and Hwang, P. M. (2020) Solution NMR spectroscopy of membrane proteins, Biochim. Biophys. Acta Biomembr., 1862, 183356, https://doi.org/10.1016/j.bbamem.2020.183356.
Zhang, K., Zhang, J., Gao, Z. G., Zhang, D., Zhu, L., Han, G. W., Moss, S. M., Paoletta, S., Kiselev, E., Lu, W., et al. (2014) Structure of the human P2Y12 receptor in complex with an antithrombotic drug, Nature, 508, 115-118, https://doi.org/10.1038/nature13083.
Yasuda, S., Kajiwara, Y., Toyoda, Y., Morimoto, K., Suno, R., Iwata, S., Kobayashi, T., Murata, T., and Kinoshita, M. (2017) Hot-spot residues to be mutated common in G protein-coupled receptors of class A: identification of thermostabilizing mutations followed by determination of three-dimensional structures for two example receptors, J. Phys. Chem. B, 121, 6341-6350, https://doi.org/10.1021/acs.jpcb.7b02997.
Katritch, V., Fenalti, G., Abola, E. E., Roth, B. L., Cherezov, V., and Stevens, R. C. (2014) Allosteric sodium in class A GPCR signaling, Trends Biochem. Sci., 39, 233-244, https://doi.org/10.1016/j.tibs.2014.03.002.
Wu, F., Yang, L., Hang, K., Laursen, M., Wu, L., Han, G. W., Ren, Q., Roed, N. K., Lin, G., Hanson, M. A., et al. (2020) Full-length human GLP-1 receptor structure without orthosteric ligands, Nat. Commun., 11, 1272, https://doi.org/10.1038/s41467-020-14934-5.
Tsutsumi, N., Qu, Q., Mavri, M., Baggesen, M. S., Maeda, S., Waghray, D., Berg, C., Kobilka, B. K., Rosenkilde, M. M., Skiniotis, G., et al. (2021) Structural basis for the constitutive activity and immunomodulatory properties of the Epstein-Barr virus-encoded G protein-coupled receptor BILF1, Immunity, 54, 1405-1416, https://doi.org/10.1016/j.immuni.2021.06.001.
Cherezov, V., Rosenbaum, D. M., Hanson, M. A., Rasmussen, S. G. F., Foon, S. T., Kobilka, T. S., Choi, H. J., Kuhn, P., Weis, W. I., Kobilka, B. K., et al. (2007) High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor, Science, 318, 1258-1265, https://doi.org/10.1126/science.1150577.
Zhang, C., Srinivasan, Y., Arlow, D. H., Fung, J. J., Palmer, D., Zheng, Y., Green, H. F., Pandey, A., Dror, R. O., Shaw, D. E., et al. (2012) High-resolution crystal structure of human protease-activated receptor 1, Nature, 492, 387-392, https://doi.org/10.1038/nature11701.
Kang, Y., Zhou, X. E., Gao, X., He, Y., Liu, W., Ishchenko, A., Barty, A., White, T. A., Yefanov, O., Han, G. W., et al. (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser, Nature, 523, 561-567, https://doi.org/10.1038/nature14656.
Patwardhan, A., Cheng, N., and Trejo, J. (2021) Post-translational modifications of G protein-coupled receptors control cellular signaling dynamics in space and time, Pharmacol. Rev., 73, 120-151, https://doi.org/10.1124/pharmrev.120.000082.
Matsumura, M., Signor, G., and Matthews, B. W. (1989) Substantial increase of protein stability by multiple disulphide bonds, Nature, 342, 291-293, https://doi.org/10.1038/342291a0.
Darby, N., and Creighton, T. E. (1995) Disulfide bonds in protein folding and stability, Methods Mol. Biol., 40, 219-252, https://doi.org/10.1385/0-89603-301-5:219.
Zavodszky, M., Chen, C.-W., Huang, J.-K., Zolkiewski, M., Wen, L., and Krishnamoorthi, R. (2001) Disulfide bond effects on protein stability: designed variants of Cucurbita maxima trypsin inhibitor-V, Protein Sci., 10, 149-160, https://doi.org/10.1110/ps.26801.
Standfuss, J., Xie, G., Edwards, P. C., Burghammer, M., Oprian, D. D., and Schertler, G. F. X. (2007) Crystal structure of a thermally stable rhodopsin mutant, J. Mol. Biol., 372, 1179-1188, https://doi.org/10.1016/j.jmb.2007.03.007.
Xie, G., Gross, A. K., and Oprian, D. D. (2003) An opsin mutant with increased thermal stability, Biochemistry, 42, 1995-2001, https://doi.org/10.1021/bi020611z.
Standfuss, J., Zaitseva, E., Mahalingam, M., and Vogel, R. (2008) Structural impact of the E113Q counterion mutation on the activation and deactivation pathways of the G protein-coupled receptor rhodopsin, J. Mol. Biol., 380, 145-157, https://doi.org/10.1016/j.jmb.2008.04.055.
Salam, N. K., Adzhigirey, M., Sherman, W., Pearlman, D. A., and Thirumalai, D. (2014) Structure-based approach to the prediction of disulfide bonds in proteins, Prot. Engin. Design Select., 27, 365-374, https://doi.org/10.1093/protein/gzu017.
Chrencik, J. E., Roth, C. B., Terakado, M., Kurata, H., Omi, R., Kihara, Y., Warshaviak, D., Nakade, S., Asmar-Rovira, G., Mileni, M., et al. (2015) Crystal structure of antagonist bound human lysophosphatidic acid receptor 1, Cell, 161, 1633-1643, https://doi.org/10.1016/j.cell.2015.06.002.
Song, G., Yang, D., Wang, Y., De Graaf, C., Zhou, Q., Jiang, S., Liu, K., Cai, X., Dai, A., Lin, G., et al. (2017) Human GLP-1 receptor transmembrane domain structure in complex with allosteric modulators, Nature, 546, 312-315, https://doi.org/10.1038/nature22378.
Morrison, K. L., and Weiss, G. A. (2001) Combinatorial alanine-scanning, Curr. Opin. Chem. Biol., 5, 302-307, https://doi.org/10.1016/S1367-5931(00)00206-4.
Cunningham, B. C., and Wells, J. A. (1989) High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis, Science, 244, 1081-1085, https://doi.org/10.1126/science.2471267.
Blaber, M., Zhang, X. J., and Matthews, B. W. (1993) Structural basis of amino acid α helix propensity, Science, 260, 1637-1640, https://doi.org/10.1126/science.8503008.
Serrano-Vega, M. J., Magnani, F., Shibata, Y., and Tate, C. G. (2008) Conformational thermostabilization of the β1-adrenergic receptor in a detergent-resistant form, Proc. Natl. Acad. Sci. USA, 105, 877-882, https://doi.org/10.1073/pnas.0711253105.
Magnani, F., Shibata, Y., Serrano-Vega, M. J., and Tate, C. G. (2008) Co-evolving stability and conformational homogeneity of the human adenosine A2a receptor, Proc. Natl. Acad. Sci. USA, 105, 10744-10749, https://doi.org/10.1073/pnas.0804396105.
Lomasneys, J. W., Leeb-lundberg, L. M. F., Cotecchiag, S., Regan, J. W., Debernardisli, J. F., Caron, M. G., and Lefkowitz, R. J. (1986) Mammalian a1-adrenergic receptor, J. Biol. Chem., 261, 7710-7716, https://doi.org/10.1016/S0021-9258(19)57458-0.
Huang, H., and Tao, Y. X. (2014) Functions of the dry motif and intracellular loop 2 of human melanocortin 3 receptor, J. Mol. Endocrinol., 53, 319-330, https://doi.org/10.1530/JME-14-0184.
Shibata, Y., Gvozdenovic-Jeremic, J., Love, J., Kloss, B., White, J. F., Grisshammer, R., and Tate, C. G. (2013) Optimising the combination of thermostabilising mutations in the neurotensin receptor for structure determination, Biochim. Biophys. Acta Biomembr., 1828, 1293-1301, https://doi.org/10.1016/j.bbamem.2013.01.008.
Robertson, N., Jazayeri, A., Errey, J., Baig, A., Hurrell, E., Zhukov, A., Langmead, C. J., Weir, M., and Marshall, F. H. (2011) The properties of thermostabilised G protein-coupled receptors (StaRs) and their use in drug discovery, Neuropharmacology, 60, 36-44, https://doi.org/10.1016/j.neuropharm.2010.07.001.
Magnani, F., Serrano-Vega, M. J., Shibata, Y., Abdul-Hussein, S., Lebon, G., Miller-Gallacher, J., Singhal, A., Strege, A., Thomas, J. A., and Tate, C. G. (2016) A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies, Nat. Protocols, 11, 1554-1571, https://doi.org/10.1038/nprot.2016.088.
Warne, T., Serrano-Vega, M. J., Baker, J. G., Moukhametzianov, R., Edwards, P. C., Henderson, R., Leslie, A. G. W., Tate, C. G., and Schertler, G. F. X. (2008) Structure of a β1-adrenergic G-protein-coupled receptor, Nature, 454, 486-491, https://doi.org/10.1038/nature07101.
Sato, T., Baker, J., Warne, T., Brown, G. A., Leslie, A. G. W., Congreve, M., and Tate, C. G. (2015) Pharmacological analysis and structure determination of 7-methylcyanopindolol-bound β1-adrenergic receptor, Mol. Pharmacol., 88, 1024-1034, https://doi.org/10.1124/mol.115.101030.
Cheng, R. K. Y., Fiez-Vandal, C., Schlenker, O., Edman, K., Aggeler, B., Brown, D. G., Brown, G. A., Cooke, R. M., Dumelin, C. E., Doré, A. S., et al. (2017) Structural insight into allosteric modulation of protease-activated receptor 2, Nature, 545, 112-115, https://doi.org/10.1038/nature22309.
Rappas, M., Ali, A. A. E., Bennett, K. A., Brown, J. D., Bucknell, S. J., Congreve, M., Cooke, R. M., Cseke, G., De Graaf, C., Doré, A. S., et al. (2020) Comparison of Orexin 1 and Orexin 2 ligand binding modes using X-ray crystallography and computational analysis, J. Med. Chem., 63, 1528-1543, https://doi.org/10.1021/acs.jmedchem.9b01787.
Brown, A. J. H., Bradley, S. J., Marshall, F. H., Brown, G. A., Bennett, K. A., Brown, J., Cansfield, J. E., Cross, D. M., de Graaf, C., Hudson, B. D., et al. (2021) From structure to clinic: design of a muscarinic M1 receptor agonist with potential to treatment of Alzheimer’s disease, Cell, 184, 5886-5901.e22, https://doi.org/10.1016/j.cell.2021.11.001.
Warne, T., Moukhametzianov, R., Baker, J. G., Nehmé, R., Edwards, P. C., Leslie, A. G. W., Schertler, G. F. X., and Tate, C. G. (2011) The structural basis for agonist and partial agonist action on a β1-adrenergic receptor, Nature, 469, 241-245, https://doi.org/10.1038/nature09746.
Doré, A. S., Robertson, N., Errey, J. C., Ng, I., Hollenstein, K., Tehan, B., Hurrell, E., Bennett, K., Congreve, M., Magnani, F., et al. (2011) Structure of the adenosine A 2A receptor in complex with ZM241385 and the xanthines XAC and caffeine, Structure, 19, 1283-1293, https://doi.org/10.1016/j.str.2011.06.014.
Moukhametzianov, R., Warne, T., Edwards, P. C., Serrano-Vega, M. J., Leslie, A. G. W., Tate, C. G., and Schertler, G. F. X. (2011) Two distinct conformations of helix 6 observed in antagonist-bound structures of a β1-adrenergic receptor, Proc. Natl. Acad. Sci. USA, 108, 8228-8232, https://doi.org/10.1073/pnas.1100185108.
White, J. F., Noinaj, N., Shibata, Y., Love, J., Kloss, B., Xu, F., Gvozdenovic-Jeremic, J., Shah, P., Shiloach, J., Tate, C. G., et al. (2012) Structure of the agonist-bound neurotensin receptor, Nature, 490, 508-513, https://doi.org/10.1038/nature11558.
Warne, T., Edwards, P. C., Leslie, A. G. W., and Tate, C. G. (2012) Crystal structures of a stabilized β1-adrenoceptor bound to the biased agonists bucindolol and carvedilol, Structure, 20, 841-849, https://doi.org/10.1016/j.str.2012.03.014.
Christopher, J. A., Brown, J., Doré, A. S., Errey, J. C., Koglin, M., Marshall, F. H., Myszka, D. G., Rich, R. L., Tate, C. G., Tehan, B., et al. (2013) Biophysical fragment screening of the β1-adrenergic receptor: Identification of high affinity arylpiperazine leads using structure-based drug design, J. Med. Chem., 56, 3446-3455, https://doi.org/10.1021/jm400140q.
Egloff, P., Hillenbrand, M., Klenk, C., Batyuk, A., Heine, P., Balada, S., Schlinkmann, K. M., Scott, D. J., Schütz, M., and Plückthun, A. (2014) Structure of signaling-competent neurotensin receptor 1 obtained by directed evolution in Escherichia coli, Proc. Natl. Acad. Sci. USA, 111, E655-E662, https://doi.org/10.1073/pnas.1317903111.
Miller-Gallacher, J. L., Nehmé, R., Warne, T., Edwards, P. C., Schertler, G. F. X., Leslie, A. G. W., and Tate, C. G. (2014) The 2.1 Å resolution structure of cyanopindolol-bound β1-adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand-free receptor, PLoS One, 9, e92727, https://doi.org/10.1371/journal.pone.0092727.
Waltenspühl, Y., Jeliazkov, J. R., Kummer, L., and Plückthun, A. (2021) Directed evolution for high functional production and stability of a challenging G protein-coupled receptor, Sci. Rep., 11, 8630, https://doi.org/10.1038/s41598-021-87793-9.
Sarkar, C. A., Dodevski, I., Kenig, M., Dudli, S., Mohr, A., Hermans, E., and Plückthun, A. (2008) Directed evolution of a G protein-coupled receptor for expression, stability, and binding selectivity, Proc. Natl. Acad. Sci. USA, 105, 14808-14813, https://doi.org/10.1073/pnas.0803103105.
Scott, D. J., and Plückthun, A. (2013) Direct molecular evolution of detergent-stable G protein-coupled receptors using polymer encapsulated cells, J. Mol. Biol., 425, 662-677, https://doi.org/10.1016/j.jmb.2012.11.015.
Schlinkmann, K. M., Hillenbrand, M., Rittner, A., Künz, M., Strohner, R., and Plückthun, A. (2012) Maximizing detergent stability and functional expression of a GPCR by exhaustive recombination and evolution, J. Mol. Biol., 422, 414-428, https://doi.org/10.1016/j.jmb.2012.05.039.
Schütz, M., Schöppe, J., Sedlák, E., Hillenbrand, M., Nagy-Davidescu, G., Ehrenmann, J., Klenk, C., Egloff, P., Kummer, L., and Plückthun, A. (2016) Directed evolution of G protein-coupled receptors in yeast for higher functional production in eukaryotic expression hosts, Sci. Rep., 6, 21508, https://doi.org/10.1038/srep21508.
Waltenspühl, Y., Schöppe, J., Ehrenmann, J., Kummer, L., and Plückthun, A. (2020) Crystal structure of the human oxytocin receptor, Sci. Adv., 6, eabb5419, https://doi.org/10.1126/sciadv.abb5419.
Ehrenmann, J., Schöppe, J., Klenk, C., Rappas, M., Kummer, L., Doré, A. S., and Plückthun, A. (2018) High-resolution crystal structure of parathyroid hormone 1 receptor in complex with a peptide agonist, Nat. Struct. Mol. Biol., 25, 1086-1092, https://doi.org/10.1038/s41594-018-0151-4.
Claff, T., Yu, J., Blais, V., Patel, N., Martin, C., Wu, L., Han, G. W., Holleran, B. J., Van der Poorten, O., White, K. L., et al. (2019) Elucidating the active δ-opioid receptor crystal structure with peptide and small-molecule agonists, Sci. Adv., 5, eaax9115, https://doi.org/10.1126/sciadv.aax9115.
Meltzer, M., Zvagelsky, T., Hadad, U., Papo, N., and Engel, S. (2022) Yeast-based directed-evolution for high-throughput structural stabilization of G protein-coupled receptors (GPCRs), Sci. Rep., 12, 8657, https://doi.org/10.1038/s41598-022-12731-2.
Popov, P., Peng, Y., Shen, L., Stevens, R. C., Cherezov, V., Liu, Z. J., and Katritch, V. (2018) Computational design of thermostabilizing point mutations for G protein-coupled receptors, eLife, 7, e34729, https://doi.org/10.7554/eLife.34729.
Arey, B. J. (2012) Glycosylation. The Role of Glycosylation in Receptor Signaling, IntechOpen, London, pp. 273-286, https://doi.org/10.5772/50262.
Pándy-Szekeres, G., Munk, C., Tsonkov, T. M., Mordalski, S., Harpsøe, K., Hauser, A. S., Bojarski, A. J., and Gloriam, D. E. (2018) GPCRdb in 2018: Adding GPCR structure models and ligands, Nucleic Acids Res., 46, D440-D446, https://doi.org/10.1093/nar/gkx1109.
Hamby, S. E., and Hirst, J. D. (2008) Prediction of glycosylation sites using random forests, BMC Bioinform., 9, 500, https://doi.org/10.1186/1471-2105-9-500.
Xie, H. L., Fu, L., and Nie, X. Du. (2013) Using ensemble SVM to identify human GPCRs N-linked glycosylation sites based on the general form of Chou’s PseAAC, Prot. Engin. Design Select., 26, 735-742, https://doi.org/10.1093/protein/gzt042.
West, G. M., Chien, E. Y. T., Katritch, V., Gatchalian, J., Chalmers, M. J., Stevens, R. C., and Griffin, P. R. (2011) Ligand-dependent perturbation of the conformational ensemble for the GPCR β 2 adrenergic receptor revealed by HDX, Structure, 19, 1424-1432, https://doi.org/10.1016/j.str.2011.08.001.
Chun, E., Thompson, A. A., Liu, W., Roth, C. B., Griffith, M. T., Katritch, V., Kunken, J., Xu, F., Cherezov, V., Hanson, M. A., et al. (2012) Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors, Structure, 20, 967-976, https://doi.org/10.1016/j.str.2012.04.010.
Guan, X. M., Kobilka, T. S., and Kobilka, B. K. (1992) Enhancement of membrane insertion and function in a type IIIb membrane protein following introduction of a cleavable signal peptide, J. Biol. Chem., 267, 21995-21998, https://doi.org/10.1016/S0021-9258(18)41623-7.
Quitterer, U., Pohl, A., Langer, A., Koller, S., and AbdAlla, S. (2011) A cleavable signal peptide enhances cell surface delivery and heterodimerization of Cerulean-tagged angiotensin II AT1 and bradykinin B2 receptor, Biochem. Biophys. Res. Commun., 409, 544-549, https://doi.org/10.1016/j.bbrc.2011.05.041.
Zhang, S., Hansen, D. T., Martin-Garcia, J. M., Zook, J. D., Pan, S., Craciunescu, F. M., Burnett, J. C., and Fromme, P. (2022) Purification, characterization, and preliminary serial crystallography diffraction advances structure determination of full-length human particulate guanylyl cyclase A receptor, Sci. Rep., 12, 11824, https://doi.org/10.1038/s41598-022-15798-z.
Koehl, A., Hu, H., Feng, D., Sun, B., Zhang, Y., Robertson, M. J., Chu, M., Kobilka, T. S., Laeremans, T., Steyaert, J., et al. (2019) Structural insights into the activation of metabotropic glutamate receptors., Nature, 566, 79-84, https://doi.org/10.1038/s41586-019-0881-4.
Watkins, L. R., and Orlandi, C. (2021) In vitro profiling of orphan G protein coupled receptor (GPCR) constitutive activity, Br. J. Pharmacol., 178, 2963-2975, https://doi.org/10.1111/bph.15468.
Sherrill, J. D., and Miller, W. E. (2006) G protein-coupled receptor (GPCR) kinase 2 regulates agonist-independent Gq/11 signaling from the mouse cytomegalovirus GPCR M33, J. Biol. Chem., 281, 39796-39805, https://doi.org/10.1074/jbc.M610026200.
Böhme, I., and Beck-Sickinger, A. G. (2009) Illuminating the life of GPCRs, Cell Commun. Signal., 7, 16, https://doi.org/10.1186/1478-811X-7-16.
Kobilka, B. K. (1995) Amino and carboxyl terminal modifications to facilitate the production and purification of a G protein-coupled receptor, Anal. Biochem., 231, 269-271, https://doi.org/10.1006/abio.1995.1533.
Prickett, K. S., Amberg, D. C., and Hopp, T. P. (1989) A calcium-dependent antibody for identification and purification of recombinant proteins, BioTechniques, 7, 580-589.
Beerepoot, P., Lam, V. M., and Salahpour, A. (2013) Measurement of G protein-coupled receptor surface expression, J. Receptors Signal Transduct., 33, 162-165, https://doi.org/10.3109/10799893.2013.781625.
Shao, Z., Shen, Q., Yao, B., Mao, C., Chen, L.-N., Zhang, H., Shen, D.-D., Zhang, C., Li, W., Du, X., et al. (2022) Identification and mechanism of G protein-biased ligands for chemokine receptor CCR1, Nat. Chem. Biol., 18, 264-271, https://doi.org/10.1038/s41589-021-00918-z.
Bertin, B., Freissmuth, M., Breyer, R. M., Schutz, W., Strosberg, A. D., and Marullo, S. (1992) Functional expression of the human serotonin 5-HT1A receptor in Escherichia coli. Ligand binding properties and interaction with recombinant G protein α-subunits, J. Biol. Chem., 267, 8200-8206, https://doi.org/10.1016/S0021-9258(18)42427-1.
Grisshammer, R., Duckworth, R., and Henderson, R. (1993) Expression of a rat neurotensin receptor in Escherichia coli, Biochem. J., 295, 571-576, https://doi.org/10.1042/bj2950571.
Park, S. H., Das, B. B., Casagrande, F., Tian, Y., Nothnagel, H. J., Chu, M., Kiefer, H., Maier, K., De Angelis, A. A., Marassi, F. M., et al. (2012) Structure of the chemokine receptor CXCR1 in phospholipid bilayers, Nature, 491, 779-783, https://doi.org/10.1038/nature11580.
Schmidt, T. G. M., and Skerra, A. (1993) The random peptide library-assisted engineering of a c-terminal affinity peptide, useful for the detection and purification of a functional Ig Fv fragment, Protein Eng. Design Select., 6, 109-122, https://doi.org/10.1093/protein/6.1.109.
Zhu, Y., Lin, X., Zong, X., Han, S., Wang, M., Su, Y., Ma, L., Chu, X., Yi, C., Zhao, Q., et al. (2022) Structural basis of FPR2 in recognition of Aβ42 and neuroprotection by humanin, Nat. Commun., 13, 1775, https://doi.org/10.1038/s41467-022-29361-x.
Klein, J. S., Jiang, S., Galimidi, R. P., Keeffe, J. R., Bjorkman, P. J., and Regan, L. (2014) Design and characterization of structured protein linkers with differing flexibilities, Protein Engin. Design Select., 27, 325-330, https://doi.org/10.1093/protein/gzu043.
Lohse, M. J., Maiellaro, I., and Calebiro, D. (2014) Kinetics and mechanism of G protein-coupled receptor activation, Curr. Opinion Cell Biol., 27, 87-93, https://doi.org/10.1016/j.ceb.2013.11.009.
Xiang, J., Chun, E., Liu, C., Jing, L., Al-Sahouri, Z., Zhu, L., and Liu, W. (2016) Successful Strategies to Determine High-Resolution Structures of GPCRs, Trends Pharmacol. Sci., 37, 1055-1069, https://doi.org/10.1016/j.tips.2016.09.009.
Rosenbaum, D. M., Cherezov, V., Hanson, M. A., Rasmussen, S. G. F. F., Thian, F. S., Kobilka, T. S., Choi, H.-J. J., Yao, X.-J. J., Weis, W. I., Stevens, R. C., et al. (2007) GPCR engineering yields high-resolution structural insights into β 2-adrenergic receptor function, Science, 318, 1266-1273, https://doi.org/10.1126/science.1150609.
Rasmussen, S. G. F., Devree, B. T., Zou, Y., Kruse, A. C., Chung, K. Y., Kobilka, T. S., Thian, F. S., Chae, P. S., Pardon, E., Calinski, D., et al. (2011) Crystal structure of the β 2 adrenergic receptor-Gs protein complex, Nature, 477, 549-557, https://doi.org/10.1038/nature10361.
Zhang, H., Qiao, A., Yang, D., Yang, L., Dai, A., De Graaf, C., Reedtz-Runge, S., Dharmarajan, V., Zhang, H., Han, G. W., et al. (2017) Structure of the full-length glucagon class B G-protein-coupled receptor, Nature, 546, 259-264, https://doi.org/10.1038/nature22363.
Yeliseev, A., Zoubak, L., and Gawrisch, K. (2007) Use of dual affinity tags for expression and purification of functional peripheral cannabinoid receptor, Protein Express. Purif., 53, 153-163, https://doi.org/10.1016/j.pep.2006.12.003.
Johansson, L. C., Stauch, B., McCorvy, J. D., Han, G. W., Patel, N., Huang, X.-P., Batyuk, A., Gati, C., Slocum, S. T., Li, C., et al. (2019) XFEL structures of the human MT2 melatonin receptor reveal the basis of subtype selectivity, Nature, 569, 289-292, https://doi.org/10.1038/s41586-019-1144-0.
Yin, J., Mobarec, J. C., Kolb, P., and Rosenbaum, D. M. (2015) Crystal structure of the human OX2 orexin receptor bound to the insomnia drug suvorexant, Nature, 519, 247-250, https://doi.org/10.1038/nature14035.
Ishihara, G., Goto, M., Saeki, M., Ito, K., Hori, T., Kigawa, T., Shirouzu, M., and Yokoyama, S. (2005) Expression of G protein coupled receptors in a cell-free translational system using detergents and thioredoxin-fusion vectors, Protein Express. Purif., 41, 27-37, https://doi.org/10.1016/j.pep.2005.01.013.
Krepkiy, D., Wong, K., Gawrisch, K., and Yeliseev, A. (2006) Bacterial expression of functional, biotinylated peripheral cannabinoid receptor CB2, Protein Express. Purif., 49, 60-70, https://doi.org/10.1016/j.pep.2006.03.002.
Nehmé, R., Carpenter, B., Singhal, A., Strege, A., Edwards, P. C., White, C. F., Du, H., Grisshammer, R., and Tate, C. G. (2017) Mini-G proteins: Novel tools for studying GPCRs in their active conformation, PLoS One, 12, e0175642, https://doi.org/10.1371/journal.pone.0175642.
García-Nafría, J., Lee, Y., Bai, X., Carpenter, B., and Tate, C. G. (2018) Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein, eLife, 7, e35946, https://doi.org/10.7554/eLife.35946.
Mittl, P. R., Ernst, P., and Plückthun, A. (2020) Chaperone-assisted structure elucidation with DARPins, Curr. Opin. Struct. Biol., 60, 93-100, https://doi.org/10.1016/j.sbi.2019.12.009.
Deluigi, M., Klipp, A., Klenk, C., Merklinger, L., Eberle, S. A., Morstein, L., Heine, P., Mitt, P. R. E., Ernst, P., Kamenecka, T. M., et al. (2021) Complexes of the neurotensin receptor 1 with small-molecule ligands reveal structural determinants of full, partial, and inverse agonism, Sci. Adv., 7, 5504-5531, https://doi.org/10.1126/sciadv.abe5504.
Deluigi, M., Morstein, L., Schuster, M., Klenk, C., Merklinger, L., Cridge, R. R., de Zhang, L. A., Klipp, A., Vacca, S., Vaid, T. M., et al. (2022) Crystal structure of the α1B-adrenergic receptor reveals molecular determinants of selective ligand recognition, Nat. Commun., 13, 382, https://doi.org/10.1038/s41467-021-27911-3.
Li, J., Mahajan, A., and Tsai, M. D. (2006) Ankyrin repeat: A unique motif mediating protein-protein interactions, Biochemistry, 45, 15168-15178, https://doi.org/10.1021/bi062188q.
Wu, Y., Batyuk, A., Honegger, A., Brandl, F., Mittl, P. R. E., and Plückthun, A. (2017) Rigidly connected multispecific artificial binders with adjustable geometries, Sci. Rep., 7, 11217, https://doi.org/10.1038/s41598-017-11472-x.
Okada, T., Takeda, K., and Kouyama, T. (1998) Highly selective separation of rhodopsin from bovine rod outer segment membranes using combination of divalent cation and alkyl(thio)glucoside, Photochem. Photobiol., 67, 495-499, https://doi.org/10.1111/j.1751-1097.1998.tb09445.x.
Okada, T., Le Trong, I., Fox, B. A., Behnke, C. A., Stenkamp, R. E., and Palczewski, K. (2000) X-ray diffraction analysis of three-dimensional crystals of Bovine Rhodopsin obtained from mixed micelles, J. Struct. Biol., 130, 73-80, https://doi.org/10.1006/jsbi.1999.4209.
Palczewski, K., Kumasaka, T., Hori, T., Behnke, C. A., Motoshima, H., Fox, B. A., Trong, I. Le, Teller, D. C., Okada, T., Stenkamp, R. E., et al. (2000) Crystal structure of rhodopsin: a G protein-coupled receptor, Science, 289, 739-745, https://doi.org/10.1126/science.289.5480.739.
Murakami, M., and Kouyama, T. (2008) Crystal structure of squid rhodopsin, Nature, 453, 363-367, https://doi.org/10.1038/nature06925.
Sahdev, S., Khattar, S. K., and Saini, K. S. (2008) Production of active eukaryotic proteins through bacterial expression systems: A review of the existing biotechnology strategies, Mol. Cell. Biochem., 307, 249-264, https://doi.org/10.1007/s11010-007-9603-6.
Qu, X., Wang, D., and Wu, B. (2020) GPCRs. Chapter 1 – Progress in GPCR Structure Determination, Elsevier, pp. 3-22, https://doi.org/10.1016/B978-0-12-816228-6.00001-5.
Ledesma, L., Sandoval, E., Cruz-Martínez, U., Escalante, A. M., Mejía, S., Moreno-Álvarez, P., Ávila, E., García, E., Coello, G., and Torres-Quiroz, F. (2018) YAAM: Yeast Amino Acid Modifications Database, Database, 2018, bax099, https://doi.org/10.1093/database/bax099.
Byrne, B. (2015) Pichia pastoris as an expression host for membrane protein structural biology, Curr. Opin. Struct. Biol., 32, 9-17, https://doi.org/10.1016/j.sbi.2015.01.005.
He, Y., Wang, K., and Yan, N. (2014) The recombinant expression systems for structure determination of eukaryotic membrane proteins, Protein Cell, 5, 658-672, https://doi.org/10.1007/s13238-014-0086-4.
Shimamura, T., Shiroishi, M., Weyand, S., Tsujimoto, H., Winter, G., Katritch, V., Abagyan, R., Cherezov, V., Liu, W., Han, G. W., et al. (2011) Structure of the human histamine H 1 receptor complex with doxepin, Nature, 475, 65-72, https://doi.org/10.1038/nature10236.
Hino, T., Arakawa, T., Iwanari, H., Yurugi-Kobayashi, T., Ikeda-Suno, C., Nakada-Nakura, Y., Kusano-Arai, O., Weyand, S., Shimamura, T., Nomura, N., et al. (2012) G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody, Nature, 482, 237-240, https://doi.org/10.1038/nature10750.
Eddy, M. T., Lee, M. Y., Gao, Z. G., White, K. L., Didenko, T., Horst, R., Audet, M., Stanczak, P., McClary, K. M., Han, G. W., et al. (2018) Allosteric coupling of drug binding and intracellular signaling in the A2A adenosine receptor, Cell, 172, 68-80.e12, https://doi.org/10.1016/j.cell.2017.12.004.
Hori, T., Okuno, T., Hirata, K., Yamashita, K., Kawano, Y., Yamamoto, M., Hato, M., Nakamura, M., Shimizu, T., Yokomizo, T., et al. (2018) Na+-mimicking ligands stabilize the inactive state of leukotriene B 4 receptor BLT1, Nat. Chem. Biol., 14, 262-269, https://doi.org/10.1038/nchembio.2547.
Harrison, R. L., and Jarvis, D. L. (2006) Protein N-glycosylation in the Baculovirus-insect cell expression system and engineering of insect cells to produce “mammalianized” recombinant glycoproteins, Adv. Virus Res., 68, 159-191, https://doi.org/10.1016/S0065-3527(06)68005-6.
Thomas, J., and Tate, C. G. (2014) Quality control in eukaryotic membrane protein overproduction, J. Mol. Biol., 426, 4139-4154, https://doi.org/10.1016/j.jmb.2014.10.012.
Ghaderi, D., Zhang, M., Hurtado-Ziola, N., and Varki, A. (2012) Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation, Biotechnol. Genet. Engin. Rev., 28, 147-176, https://doi.org/10.5661/bger-28-147.
Milić, D., and Veprintsev, D. B. (2015) Large-scale production and protein engineering of G protein-coupled receptors for structural studies, Front. Pharmacol., 6, 66, https://doi.org/10.3389/fphar.2015.00066.
Warne, T., Chirnside, J., and Schertler, G. F. X. (2003) Expression and purification of truncated, non-glycosylated turkey beta-adrenergic receptors for crystallization, Biochim. Biophys. Acta Biomembr., 1610, 133-140, https://doi.org/10.1016/S0005-2736(02)00716-2.
Chien, E. Y. T., Liu, W., Zhao, Q., Katritch, V., Han, G. W., Hanson, M. A., Shi, L., Newman, A. H., Javitch, J. A., Cherezov, V., et al. (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist, Science, 330, 1091-1095, https://doi.org/10.1126/science.1197410.
Luckow, V. A., Lee, S. C., Barry, G. F., and Olins, P. O. (1993) Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli, J. Virol., 67, 4566-4579, https://doi.org/10.1128/jvi.67.8.4566-4579.1993.
Contreras-Gómez, A., Sánchez-Mirón, A., García-Camacho, F., Molina-Grima, E., and Chisti, Y. (2014) Protein production using the baculovirus-insect cell expression system, Biotechnol. Progress, 30, 1-18, https://doi.org/10.1002/btpr.1842.
Chaudhary, S., Pak, J. E., Gruswitz, F., Sharma, V., and Stroud, R. M. (2012) Overexpressing human membrane proteins in stably transfected and clonal human embryonic kidney 293S cells, Nat. Protocols, 7, 453-466, https://doi.org/10.1038/nprot.2011.453.
Fricker, S. P., Anastassov, V., Cox, J., Darkes, M. C., Grujic, O., Idzan, S. R., Labrecque, J., Lau, G., Mosi, R. M., Nelson, K. L., et al. (2006) Characterization of the molecular pharmacology of AMD3100: A specific antagonist of the G-protein coupled chemokine receptor, CXCR4, Biochem. Pharmacol., 72, 588-596, https://doi.org/10.1016/j.bcp.2006.05.010.
Cheng, Z., Garvin, D., Paguio, A., Stecha, P., Wood, K., and Fan, F. (2012) Luciferase reporter assay system for deciphering GPCR pathways, Curr. Chem. Genomics, 4, 84-91, https://doi.org/10.2174/1875397301004010084.
Tate, C. G. (2001) Overexpression of mammalian integral membrane proteins for structural studies, FEBS Lett., 504, 94-98, https://doi.org/10.1016/S0014-5793(01)02711-9.
Andréll, J., and Tate, C. G. (2013) Overexpression of membrane proteins in mammalian cells for structural studies, Mol. Membr. Biol., 30, 52-63, https://doi.org/10.3109/09687688.2012.703703.
Mikami, S., Kobayashi, T., Masutani, M., Yokoyama, S., and Imataka, H. (2008) A human cell-derived in vitro coupled transcription/translation system optimized for production of recombinant proteins, Protein Express. Purif., 62, 190-198, https://doi.org/10.1016/j.pep.2008.09.002.
Goren, M. A., Nozawa, A., Makino, S., Wrobel, R. L., and Fox, B. G. (2009) Chapter 37 cell-free translation of integral membrane proteins into unilamelar liposomes, Methods Enzymol., 463, 647-673, https://doi.org/10.1016/S0076-6879(09)63037-8.
Kögler, L. M., Stichel, J., and Beck-Sickinger, A. G. (2019) Structural investigations of cell-free expressed G protein-coupled receptors, Biol. Chem., 401, 97-116, https://doi.org/10.1515/hsz-2019-0292.
Schwarz, D., Junge, F., Durst, F., Frölich, N., Schneider, B., Reckel, S., Sobhanifar, S., Dötsch, V., and Bernhard, F. (2007) Preparative scale expression of membrane proteins in Escherichia coli-based continuous exchange cell-free systems, Nat. Protoc., 2, 2945-2957, https://doi.org/10.1038/nprot.2007.426.
Proverbio, D., Henrich, E., Orbán, E., Dötsch, V., and Bernhard, F. (2014) Membrane Protein Quality Control in Cell-Free Expression Systems: Tools, Strategies and Case Studies, in Membrane Proteins Production for Structural Analysis, pp. 45-70, https://doi.org/10.1007/978-1-4939-0662-8_2.
Zemella, A., Thoring, L., Hoffmeister, C., and Kubick, S. (2015) Cell-free protein synthesis: pros and cons of prokaryotic and eukaryotic systems, ChemBioChem, 16, 2420-2431, https://doi.org/10.1002/cbic.201500340.
Klammt, C., Schwarz, D., Löhr, F., Schneider, B., Dötsch, V., and Bernhard, F. (2006) Cell-free expression as an emerging technique for the large scale production of integral membrane protein, FEBS J., 273, 4141-4153, https://doi.org/10.1111/j.1742-4658.2006.05432.x.
Quast, R. B., Mrusek, D., Hoffmeister, C., Sonnabend, A., and Kubick, S. (2015) Cotranslational incorporation of non-standard amino acids using cell-free protein synthesis, FEBS Lett., 589, 1703-1712, https://doi.org/10.1016/j.febslet.2015.04.041.
Klammt, C., Schwarz, D., Eifler, N., Engel, A., Piehler, J., Haase, W., Hahn, S., Dötsch, V., and Bernhard, F. (2007) Cell-free production of G protein-coupled receptors for functional and structural studies, J. Struct. Biol., 158, 482-493, https://doi.org/10.1016/j.jsb.2007.01.006.
Henrich, E., Dötsch, V., and Bernhard, F. (2015) Screening for lipid requirements of membrane proteins by combining cell-free expression with nanodiscs, Methods Enzymol., 556, 351-369, https://doi.org/10.1016/bs.mie.2014.12.016.
Boland, C., Li, D., Shah, S. T. A., Haberstock, S., Dötsch, V., Bernhard, F., and Caffrey, M. (2014) Cell-free expression and in meso crystallisation of an integral membrane kinase for structure determination, Cell. Mol. Life Sci., 71, 4895-4910, https://doi.org/10.1007/s00018-014-1655-7.
Umbach, S., Dötsch, V., and Bernhard, F. (2022) Cell-free expression of GPCRs into nanomembranes for functional and structural studies, Methods Mol. Biol., 2507, 405-424, https://doi.org/10.1007/978-1-0716-2368-8_22.
Carlson, E. D., Gan, R., Hodgman, C. E., and Jewett, M. C. (2012) Cell-free protein synthesis: applications come of age, Biotechnol. Adv., 30, 1185-1194, https://doi.org/10.1016/j.biotechadv.2011.09.016.
Tarui, H., Imanishi, S., and Hara, T. (2000) A novel cell-free translation/glycosylation system prepared from insect cells, J. Biosci. Bioengin., 90, 508-514, https://doi.org/10.1016/S1389-1723(01)80031-1.
Suzuki, T., Ito, M., Ezure, T., Shikata, M., Ando, E., Utsumi, T., Tsunasawa, S., and Nishimura, O. (2006) N-terminal protein modifications in an insect cell-free protein synthesis system and their identification by mass spectrometry, Proteomics, 6, 4486-4495, https://doi.org/10.1002/pmic.200600126.
Hancock, J. F. (1995) Reticulocyte lysate assay for in vitro translation and posttranslational modification of Ras proteins, Methods Enzymol., 255, 60-65, https://doi.org/10.1016/S0076-6879(95)55009-7.
Brödel, A. K., Sonnabend, A., and Kubick, S. (2014) Cell-free protein expression based on extracts from CHO cells, Biotechnol. Bioengin., 111, 25-36, https://doi.org/10.1002/bit.25013.
Foshag, D., Henrich, E., Hiller, E., Schäfer, M., Kerger, C., Burger-Kentischer, A., Diaz-Moreno, I., García-Mauriño, S. M., Dötsch, V., Rupp, S., et al. (2018) The E. coli S30 lysate proteome: a prototype for cell-free protein production, New Biotechnol., 40, 245-260, https://doi.org/10.1016/j.nbt.2017.09.005.
Yin, G., and Swartz, J. R. (2004) Enhancing multiple disulfide bonded protein folding in a cell-free system, Biotechnol. Bioengin., 86, 188-195, https://doi.org/10.1002/bit.10827.
Kim, D. M., and Swartz, J. R. (2004) Efficient production of a bioactive, multiple disulfide-bonded protein using modified extracts of Escherichia coli, Biotechnol.Bioengin., 85, 122-129, https://doi.org/10.1002/bit.10865.
Goerke, A. R., and Swartz, J. R. (2008) Development of cell-free protein synthesis platforms for disulfide bonded proteins, Biotechnol. Bioengin., 99, 351-367, https://doi.org/10.1002/bit.21567.
Jaroentomeechai, T., Stark, J. C., Natarajan, A., Glasscock, C. J., Yates, L. E., Hsu, K. J., Mrksich, M., Jewett, M. C., and Delisa, M. P. (2018) Single-pot glycoprotein biosynthesis using a cell-free transcription-translation system enriched with glycosylation machinery, Nat. Commun., 9, 2686, https://doi.org/10.1038/s41467-018-05110-x.
Schoborg, J. A., Hershewe, J. M., Stark, J. C., Kightlinger, W., Kath, J. E., Jaroentomeechai, T., Natarajan, A., DeLisa, M. P., and Jewett, M. C. (2018) A cell-free platform for rapid synthesis and testing of active oligosaccharyltransferases, Biotechnol. Bioengin., 115, 739-750, https://doi.org/10.1002/bit.26502.
Kightlinger, W., Lin, L., Rosztoczy, M., Li, W., Delisa, M. P., Mrksich, M., and Jewett, M. C. (2018) Design of glycosylation sites by rapid synthesis and analysis of glycosyltransferases article, Nat. Chem. Biol., 14, 627-635, https://doi.org/10.1038/s41589-018-0051-2.
Suzuki, Y., Ogasawara, T., Tanaka, Y., Takeda, H., Sawasaki, T., Mogi, M., Liu, S., and Maeyama, K. (2018) Functional G-protein-coupled receptor (GPCR) synthesis: The pharmacological analysis of human histamine H1 receptor (HRH1) synthesized by a wheat germ cell-free protein synthesis system combined with asolectin glycerosomes, Front. Pharmacol., 9, 38, https://doi.org/10.3389/fphar.2018.00038.
Yang, J. P., Cirico, T., Katzen, F., Peterson, T. C., and Kudlicki, W. (2011) Cell-free synthesis of a functional G protein-coupled receptor complexed with nanometer scale bilayer discs, BMC Biotechnol., 11, 57, https://doi.org/10.1186/1472-6750-11-57.
Sonnabend, A., Spahn, V., Stech, M., Zemella, A., Stein, C., and Kubick, S. (2017) Production of G protein-coupled receptors in an insect-based cell-free system, Biotechnol. Bioengin., 114, 2328-2338, https://doi.org/10.1002/bit.26346.
Junge, F., Luh, L. M., Proverbio, D., Schäfer, B., Abele, R., Beyermann, M., Dötsch, V., and Bernhard, F. (2010) Modulation of G-protein coupled receptor sample quality by modified cell-free expression protocols: A case study of the human endothelin A receptor, J. Struct. Biol., 172, 94-106, https://doi.org/10.1016/j.jsb.2010.05.004.
Zemella, A., Grossmann, S., Sachse, R., Sonnabend, A., Schaefer, M., and Kubick, S. (2017) Qualifying a eukaryotic cell-free system for fluorescence based GPCR analyses, Sci. Rep., 7, 3740, https://doi.org/10.1038/s41598-017-03955-8.
Rues, R. B., Dong, F., Dötsch, V., and Bernhard, F. (2018) Systematic optimization of cell-free synthesized human endothelin B receptor folding, Methods, 147, 73-83, https://doi.org/10.1016/j.ymeth.2018.01.012.
Takeda, H., Ogasawara, T., Ozawa, T., Muraguchi, A., Jih, P. J., Morishita, R., Uchigashima, M., Watanabe, M., Fujimoto, T., Iwasaki, T., et al. (2015) Production of monoclonal antibodies against GPCR using cell-free synthesized GPCR antigen and biotinylated liposome-based interaction assay, Sci. Rep., 5, 11333, https://doi.org/10.1038/srep11333.
Wada, T., Shimono, K., Kikukawa, T., Hato, M., Shinya, N., Kim, S. Y., Kimura-Someya, T., Shirouzu, M., Tamogami, J., Miyauchi, S., et al. (2011) Crystal structure of the eukaryotic light-driven proton-pumping rhodopsin, Acetabularia rhodopsin II, from marine alga, J. Mol. Biol., 411, 986-998, https://doi.org/10.1016/j.jmb.2011.06.028.
Chen, Y. J., Pornillos, O., Lieu, S., Ma, C., Chen, A. P., and Chang, G. (2007) X-ray structure of EmrE supports dual topology model, Proc. Natl. Acad. Sci. USA, 104, 18999-19004, https://doi.org/10.1073/pnas.0709387104.
Bornhorst, J. A., and Falke, J. J. (2000) Purification of proteins using polyhistidine affinity tags, Methods Enzymol., 326, 245-254, https://doi.org/10.1016/S0076-6879(00)26058-8.
Qu, X., Qiu, N., Wang, M., Zhang, B., Du, J., Zhong, Z., Xu, W., Chu, X., Ma, L., Yi, C., et al. (2022) Structural basis of tethered agonism of the adhesion GPCRs ADGRD1 and ADGRF1, Nature, 604, 779-785, https://doi.org/10.1038/s41586-022-04580-w.
Bous, J., Orcel, H., Floquet, N., Leyrat, C., Lai-Kee-Him, J., Gaibelet, G., Ancelin, A., Saint-Paul, J., Trapani, S., Louet, M., et al. (2021) Cryo-electron microscopy structure of the antidiuretic hormone arginine-vasopressin V2 receptor signaling complex, Sci. Adv., 7, eabg5628, https://doi.org/10.1126/sciadv.abg5628.
Cong, Z., Chen, L. N., Ma, H., Zhou, Q., Zou, X., Ye, C., Dai, A., Liu, Q., Huang, W., Sun, X., et al. (2021) Molecular insights into ago-allosteric modulation of the human glucagon-like peptide-1 receptor, Nat. Commun., 12, 3763, https://doi.org/10.1038/s41467-021-24058-z.
Ma, S., Shen, Q., Zhao, L. H., Mao, C., Zhou, X. E., Shen, D. D., de Waal, P. W., Bi, P., Li, C., Jiang, Y., et al. (2020) Molecular basis for hormone recognition and activation of corticotropin-releasing factor receptors, Mol. Cell, 77, 669-680.e4, https://doi.org/10.1016/j.molcel.2020.01.013.
Wang, H., Hetzer, F., Huang, W., Qu, Q., Meyerowitz, J., Kaindl, J., Hübner, H., Skiniotis, G., Kobilka, B. K., and Gmeiner, P. (2022) Structure-based evolution of G protein-biased µ-opioid receptor agonists, Angewandte Chemie, 61, e202200269, https://doi.org/10.1002/anie.202200269.
Cao, J., Belousoff, M. J., Liang, Y.-L., Johnson, R. M., Josephs, T. M., Fletcher, M. M., Christopoulos, A., Hay, D. L., Danev, R., Wootten, D., et al. (2022) A structural basis for amylin receptor phenotype, Science, 375, eabm9609, https://doi.org/10.1126/science.abm9609.
Bokoch, M. P., Zou, Y., Rasmussen, S. G. F., Liu, C. W., Nygaard, R., Rosenbaum, D. M., Fung, J. J., Choi, H. J., Thian, F. S., Kobilka, T. S., et al. (2010) Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor, Nature, 463, 108-112, https://doi.org/10.1038/nature08650.
Rosenbaum, D. M., Zhang, C., Lyons, J. A., Holl, R., Aragao, D., Arlow, D. H., Rasmussen, S. G. F., Choi, H. J., Devree, B. T., Sunahara, R. K., et al. (2011) Structure and function of an irreversible agonist-β2 adrenoceptor complex, Nature, 469, 236-242, https://doi.org/10.1038/nature09665.
Brizzard, B. L., Chubet, R. G., and Vizard, D. L. (1994) Immunoaffinity purification of FLAG® epitope-tagged bacterial alkaline phosphatase using a novel monoclonal antibody and peptide elution, BioTechniques, 16, 730-735.
Slootstra, J. W. (1997) Identification of new tag sequences with differential and selective recognition properties for the anti-FLAG monoclonal antibodies Ml, M2 and M5, Mol. Divers., 2, 156-164, https://doi.org/10.1007/BF01682203.
Park, S. H., Cheong, C., Idoyaga, J., Kim, J. Y., Choi, J. H., Do, Y., Lee, H., Jo, J. H., Oh, Y. S., Im, W., et al. (2008) Generation and application of new rat monoclonal antibodies against synthetic FLAG and OLLAS tags for improved immunodetection, J. Immunol. Methods, 331, 27-38, https://doi.org/10.1016/j.jim.2007.10.012.
Gerace, E., and Moazed, D. (2015) Affinity Pull-Down of Proteins Using Anti-FLAG M2 Agarose Beads, Methods Enzymol., 559, 99-110, https://doi.org/10.1016/bs.mie.2014.11.010.
Tsai, C. J., Pamula, F., Nehmé, R., Mühle, J., Weinert, T., Flock, T., Nogly, P., Edwards, P. C., Carpenter, B., Gruhl, T., et al. (2018) Crystal structure of rhodopsin in complex with a mini-Go sheds light on the principles of G protein selectivity, Sci. Adv., 4, 7052, https://doi.org/10.1126/sciadv.aat7052.
Zhao, D. Y., Pöge, M., Morizumi, T., Gulati, S., Van Eps, N., Zhang, J., Miszta, P., Filipek, S., Mahamid, J., Plitzko, J. M., et al. (2019) Cryo-EM structure of the native rhodopsin dimer in nanodiscs, J. Biol. Chem., 294, 14215-14230, https://doi.org/10.1074/jbc.RA119.010089.
Wong, J. P., Reboul, E., Molday, R. S., and Kast, J. (2009) A carboxy-terminal affinity tag for the purification and mass spectrometric characterization of integral membrane proteins, J. Proteome Res., 8, 2388-2396, https://doi.org/10.1021/pr801008c.
Langlois, M., Brémont, B., Rousselle, D., and Gaudy, F. (1993) Structural analysis by the comparative molecular field analysis method of the affinity of β-adrenoreceptor blocking agents for 5-HT1A and 5-HT1B receptors, Eur. J. Pharmacol. Mol. Pharmacol., 244, 77-87, https://doi.org/10.1016/0922-4106(93)90061-D.
Sun, R., Mak, S., Haschemi, J., Horn, P., Boege, F., and Luppa, P. B. (2019) Nanodiscs incorporating native β1 adrenergic receptor as a novel approach for the detection of pathological autoantibodies in patients with dilated cardiomyopathy, J. Appl. Lab. Med., 4, 391-403, https://doi.org/10.1373/jalm.2018.028225.
Parker, E. M., Kameyama, K., Higashijima, T., and Ross, E. M. (1991) Reconstitutively active G protein-coupled receptors purified from baculovirus-infected insect cells, J. Biol. Chem., 266, 519-527, https://doi.org/10.1016/S0021-9258(18)52467-4.
Stadel, J. M. (1985) Photoaffinity labeling of β-adrenergic receptors, Pharmacol. Ther., 31, 57-77, https://doi.org/10.1016/0163-7258(85)90037-3.
Caron, M. G., Srinivasan, Y., Pitha, J., Kociolek, K., and Lefkowitz, R. J. (1979) Affinity chromatography of the β adrenergic receptor, J. Biol. Chem., 254, 2923-2927, https://doi.org/10.1016/S0021-9258(17)30162-X.
Ring, A. M., Manglik, A., Kruse, A. C., Enos, M. D., Weis, W. I., Garcia, K. C., and Kobilka, B. K. (2013) Adrenaline-activated structure of β 2-adrenoceptor stabilized by an engineered nanobody, Nature, 502, 575-579, https://doi.org/10.1038/nature12572.
Masureel, M., Zou, Y., Picard, L. P., van der Westhuizen, E., Mahoney, J. P., Rodrigues, J. P. G. L. M., Mildorf, T. J., Dror, R. O., Shaw, D. E., Bouvier, M., et al. (2018) Structural insights into binding specificity, efficacy and bias of a β2AR partial agonist, Nat. Chem. Biol., 14, 1059-1066, https://doi.org/10.1038/s41589-018-0145-x.
Haga, K., Kruse, A. C., Asada, H., Yurugi-Kobayashi, T., Shiroishi, M., Zhang, C., Weis, W. I., Okada, T., Kobilka, B. K., Haga, T., et al. (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist, Nature, 482, 547-551, https://doi.org/10.1038/nature10753.
Edwards, P. C., Li, J., Burghammer, M., Hugh McDowell, J., Villa, C., Hargrave, P. A., and Schertler, G. F. X. (2004) Crystals of native and modified bovine rhodopsins and their heavy atom derivatives, J. Mol. Biol., 343, 1439-1450, https://doi.org/10.1016/j.jmb.2004.08.089.
Rasmussen, S. G. F., Choi, H. J., Rosenbaum, D. M., Kobilka, T. S., Thian, F. S., Edwards, P. C., Burghammer, M., Ratnala, V. R. P., Sanishvili, R., Fischetti, R. F., et al. (2007) Crystal structure of the human β2 adrenergic G-protein-coupled receptor, Nature, 450, 383-387, https://doi.org/10.1038/nature06325.
Chen, Q., Plasencia, M., Li, Z., Mukherjee, S., Patra, D., Chen, C. L., Klose, T., Yao, X. Q., Kossiakoff, A. A., Chang, L., et al. (2021) Structures of rhodopsin in complex with G-protein-coupled receptor kinase 1, Nature, 595, 600-605, https://doi.org/10.1038/s41586-021-03721-x.
Whitaker, J. R. (1963) Determination of molecular weights of proteins by gel filtration of sephadex, Anal. Chem., 35, 1950-1953, https://doi.org/10.1021/ac60205a048.
Andrews, P. (1964) Estimation of the molecular weights of proteins by Sephadex gel-filtration, Biochem. J., 91, 222-233, https://doi.org/10.1042/bj0910222.
Zhou, F., Zhang, H., Cong, Z., Zhao, L.-H., Zhou, Q., Mao, C., Cheng, X., Shen, D.-D., Cai, X., Ma, C., et al. (2020) Structural basis for activation of the growth hormone-releasing hormone receptor, Nat. Commun., 11, 5205, https://doi.org/10.1038/s41467-020-18945-0.
Yuan, Y., Jia, G., Wu, C., Wang, W., Cheng, L., Li, Q., Li, Z., Luo, K., Yang, S., Yan, W., et al. (2021) Structures of signaling complexes of lipid receptors S1PR1 and S1PR5 reveal mechanisms of activation and drug recognition, Cell Res., 31, 1263-1274, https://doi.org/10.1038/s41422-021-00566-x.
Isaikina, P., Tsai, C.-J., Dietz, N., Pamula, F., Grahl, A., Goldie, K. N., Guixà-González, R., Branco, C., Paolini-Bertrand, M., Calo, N., et al. (2021) Structural basis of the activation of the CC chemokine receptor 5 by a chemokine agonist, Sci. Adv., 7, eabg8685, https://doi.org/10.1126/sciadv.abg8685.
Jones, A. J. Y., Gabriel, F., Tandale, A., and Nietlispach, D. (2020) Structure and dynamics of GPCRs in lipid membranes: physical principles and experimental approaches, Molecules, 25, 4729, https://doi.org/10.3390/molecules25204729.
Popot, J. L. (2010) Amphipols, nanodiscs, and fluorinated surfactants: Three nonconventional approaches to studying membrane proteins in aqueous solutions, Annu. Rev. Biochem., 79, 737-775, https://doi.org/10.1146/annurev.biochem.052208.114057.
Zhou, H. X., and Cross, T. A. (2013) Influences of membrane mimetic environments on membrane protein structures, Annu. Rev. Biophys., 42, 361-392, https://doi.org/10.1146/annurev-biophys-083012-130326.
Vanaken, T., Foxall-Vanaken, S., Castleman, S., and Ferguson-Miller, S. (1986) Alkyl glycoside detergents: synthesis and applications to the study of membrane proteins, Methods Enzymol., 125, 27-35, https://doi.org/10.1016/S0076-6879(86)25005-3.
Tulumello, D. V., and Deber, C. M. (2009) SDS micelles as a membrane-mimetic environment for transmembrane segments, Biochemistry, 48, 12096-12103, https://doi.org/10.1021/bi9013819.
Bhuyan, A. K. (2010) On the mechanism of SDS-induced protein denaturation, Biopolymers, 93, 186-199, https://doi.org/10.1002/bip.21318.
Heim, B., Handrick, R., Hartmann, M. D., and Kiefer, H. (2021) Refolding and characterization of two G protein-coupled receptors purified from E. coli inclusion bodies, PLoS One, 16, e0247689, https://doi.org/10.1371/journal.pone.0247689.
Lee, S., Ghosh, S., Jana, S., Robertson, N., Tate, C. G., and Vaidehi, N. (2020) How do branched detergents stabilize GPCRs in micelles? Biochemistry, 59, 2125-2134, https://doi.org/10.1021/acs.biochem.0c00183.
Makowska, J., Wyrzykowski, D., Pilarski, B., and Chmurzyński, L. (2015) Thermodynamics of sodium dodecyl sulphate (SDS) micellization in the presence of some biologically relevant pH buffers, J. Therm. Anal. Calorimetry, 121, 257-261, https://doi.org/10.1007/s10973-015-4644-7.
Seddon, A. M., Curnow, P., and Booth, P. J. (2004) Membrane proteins, lipids and detergents: not just a soap opera, Biochim. Biophys. Acta Biomembr., 1666, 105-117, https://doi.org/10.1016/j.bbamem.2004.04.011.
Singh, S. K., and Sigworth, F. J. (2015) Cryo-EM: spinning the micelles away, Structure, 23, 1561, https://doi.org/10.1016/j.str.2015.08.001.
Birch, J., Axford, D., Foadi, J., Meyer, A., Eckhardt, A., Thielmann, Y., and Moraes, I. (2018) The fine art of integral membrane protein crystallisation, Methods, 147, 150-162, https://doi.org/10.1016/j.ymeth.2018.05.014.
Kampjut, D., Steiner, J., and Sazanov, L. A. (2021) Cryo-EM grid optimization for membrane proteins, iScience, 24, 102139, https://doi.org/10.1016/j.isci.2021.102139.
Maeda, S., Yamamoto, H., Kinch, L. N., Garza, C. M., Takahashi, S., Otomo, C., Grishin, N. V., Forli, S., Mizushima, N., and Otomo, T. (2020) Structure, lipid scrambling activity and role in autophagosome formation of ATG9A, Nat. Struct. Mol. Biol., 27, 1194-1201, https://doi.org/10.1038/s41594-020-00520-2.
Ratkeviciute, G., Cooper, B. F., and Knowles, T. J. (2021) Methods for the solubilisation of membrane proteins: the micelle-aneous world of membrane protein solubilisation, Biochem. Soc. Trans., 49, 1763-1777, https://doi.org/10.1042/BST20210181.
Nguyen, A. H., and Lefkowitz, R. J. (2021) Signaling at the endosome: cryo-EM structure of a GPCR-G protein-beta-arrestin megacomplex, FEBS J., 288, 2562-2569, https://doi.org/10.1111/febs.15773.
Tsai, C.-J., Marino, J., Adaixo, R., Pamula, F., Muehle, J., Maeda, S., Flock, T., Taylor, N. M., Mohammed, I., Matile, H., et al. (2019) Cryo-EM structure of the rhodopsin-Gαi-βγ complex reveals binding of the rhodopsin C-terminal tail to the gβ subunit, eLife, 8, e46041, https://doi.org/10.7554/eLife.46041.
Tribet, C., Audebert, R., and Popot, J. L. (1996) Amphipols: polymers that keep membrane proteins soluble in aqueous solutions, Proc. Natl. Acad. Sci. USA, 93, 15047-15050, https://doi.org/10.1073/pnas.93.26.15047.
Popot, J. L., Althoff, T., Bagnard, D., Banéres, J. L., Bazzacco, P., Billon-Denis, E., Catoire, L. J., Champeil, P., Charvolin, D., Cocco, M. J., et al. (2011) Amphipols from A to Z, Annu. Rev. Biophys., 40, 379-408, https://doi.org/10.1146/annurev-biophys-042910-155219.
Bosco, M., Damian, M., Chauhan, V., Roche, M., Guillet, P., Fehrentz, J. A., Bonneté, F., Polidori, A., Banères, J. L., and Durand, G. (2020) Biotinylated non-ionic amphipols for GPCR ligands screening, Methods, 180, 69-78, https://doi.org/10.1016/j.ymeth.2020.06.001.
Wheatley, M., Charlton, J., Jamshad, M., Routledge, S. J., Bailey, S., La-Borde, P. J., Azam, M. T., Logan, R. T., Bill, R. M., Dafforn, T. R., et al. (2016) GPCR-styrene maleic acid lipid particles (GPCR-SMALPs): Their nature and potential, Biochem. Soc. Transact., 44, 619-623, https://doi.org/10.1042/BST20150284.
Tedesco, D., Maj, M., Malarczyk, P., Cingolani, A., Zaffagnini, M., Wnorowski, A., Czapiński, J., Benelli, T., Mazzoni, R., Bartolini, M., et al. (2021) Application of the SMALP technology to the isolation of GPCRs from low-yielding cell lines, Biochim. Biophys. Acta Biomembr., 1863, 183641, https://doi.org/10.1016/j.bbamem.2021.183641.
Routledge, S. J., Jamshad, M., Little, H. A., Lin, Y.-P., Simms, J., Thakker, A., Spickett, C. M., Bill, R. M., Dafforn, T. R., Poyner, D. R., et al. (2020) Ligand-induced conformational changes in a SMALP-encapsulated GPCR, Biochim. Biophys. Acta Biomembr., 1862, 183235, https://doi.org/10.1016/j.bbamem.2020.183235.
Denisov, I. G., and Sligar, S. G. (2016) Nanodiscs for structural and functional studies of membrane proteins, Nat. Struct. Mol. Biol., 23, 481-486, https://doi.org/10.1038/nsmb.3195.
Cai, Y., Liu, Y., Culhane, K. J., DeVree, B. T., Yang, Y., Sunahara, R. K., and Yan, E. C. Y. (2017) Purification of family B G protein-coupled receptors using nanodiscs: application to human glucagon-like peptide-1 receptor, PLoS One, 12, e0179568, https://doi.org/10.1371/journal.pone.0179568.
Nasr, M. L., Baptista, D., Strauss, M., Sun, Z. Y. J., Grigoriu, S., Huser, S., Plückthun, A., Hagn, F., Walz, T., Hogle, J. M., et al. (2016) Covalently circularized nanodiscs for studying membrane proteins and viral entry, Nat. Methods, 14, 49-52, https://doi.org/10.1038/nmeth.4079.
Zhao, Z., Zhang, M., Hogle, J. M., Shih, W. M., Wagner, G., and Nasr, M. L. (2018) DNA-Corralled nanodiscs for the structural and functional characterization of membrane proteins and viral entry, J. Am. Chem. Soc., 140, 10639-10643, https://doi.org/10.1021/jacs.8b04638.
Kishimoto, Y., Hiraiwa, M., and O’Brien, J. S. (1992) Saposins: Structure, function, distribution, and molecular genetics, J. Lipid Res., 33, 1255-1267, https://doi.org/10.1016/S0022-2275(20)40540-1.
Popovic, K., Holyoake, J., Pomès, R., and Privé, G. G. (2012) Structure of saposin A lipoprotein discs, Proc. Natl. Acad. Sci. USA, 109, 2908-2912, https://doi.org/10.1073/pnas.1115743109.
Flayhan, A., Mertens, H. D. T., Ural-Blimke, Y., Martinez Molledo, M., Svergun, D. I., and Löw, C. (2018) Saposin lipid nanoparticles: a highly versatile and modular tool for membrane protein research, Structure, 26, 345-355.e5, https://doi.org/10.1016/j.str.2018.01.007.
Dürr, U. H. N., Gildenberg, M., and Ramamoorthy, A. (2012) The magic of bicelles lights up membrane protein structure, Chem. Rev., 112, 6054-6074, https://doi.org/10.1021/cr300061w.
Schmidt, P., Bender, B. J., Kaiser, A., Gulati, K., Scheidt, H. A., Hamm, H. E., Meiler, J., Beck-Sickinger, A. G., and Huster, D. (2018) Improved in vitro folding of the Y2 G protein-coupled receptor into bicelles, Front. Mol. Biosci., 4, 100, https://doi.org/10.3389/fmolb.2017.00100.
Murugova, T. N., Ivankov, O. I., Ryzhykau, Y. L., Soloviov, D. V., Kovalev, K. V., Skachkova, D. V., Round, A., Baeken, C., Ishchenko, A. V., Volkov, O. A., et al. (2022) Mechanisms of membrane protein crystallization in “bicelles”, Sci. Rep., 12, 11109, https://doi.org/10.1038/s41598-022-13945-0.
Ujwal, R., and Bowie, J. U. (2011) Crystallizing membrane proteins using lipidic bicelles, Methods, 55, 337-341, https://doi.org/10.1016/j.ymeth.2011.09.020.
Goddard, A. D., Dijkman, P. M., Adamson, R. J., Dos Reis, R. I., and Watts, A. (2015) Reconstitution of membrane proteins: a GPCR as an example, Methods Enzymol., 556, 405-424, https://doi.org/10.1016/bs.mie.2015.01.004.
Ong, S. G. M., Chitneni, M., Lee, K. S., Ming, L. C., and Yuen, K. H. (2016) Evaluation of extrusion technique for nanosizing liposomes, Pharmaceutics, 8, 36, https://doi.org/10.3390/pharmaceutics8040036.
Akbarzadeh, A., Rezaei-Sadabady, R., Davaran, S., Joo, S. W., Zarghami, N., Hanifehpour, Y., Samiei, M., Kouhi, M., and Nejati-Koshki, K. (2013) Liposome: classification, preparation, and applications, Nanoscale Res. Lett., 8, 102, https://doi.org/10.1186/1556-276X-8-102.
Nakhaei, P., Margiana, R., Bokov, D. O., Abdelbasset, W. K., Jadidi Kouhbanani, M. A., Varma, R. S., Marofi, F., Jarahian, M., and Beheshtkhoo, N. (2021) Liposomes: structure, biomedical applications, and stability parameters with emphasis on cholesterol, Front. Bioengin. Biotechnol., 9, 748, https://doi.org/10.3389/fbioe.2021.705886.
Cherezov, V. (2011) Lipidic cubic phase technologies for membrane protein structural studies, Curr. Opin. Struct. Biol., 21, 559-566, https://doi.org/10.1016/j.sbi.2011.06.007.
Caffrey, M. (2015) A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes, Acta Crystallogr. Sect. F Struct. Biol. Commun., 71, 3-18, https://doi.org/10.1107/S2053230X14026843.
Cherezov, V., Clogston, J., Misquitta, Y., Abdel-Gawad, W., and Caffrey, M. (2002) Membrane protein crystallization in meso: lipid type-tailoring of the cubic phase, Biophys. J., 83, 3393-3407, https://doi.org/10.1016/S0006-3495(02)75339-3.
Cherezov, V., Abola, E., and Stevens, R. C. (2010) Recent progress in the structure determination of GPCRs, a membrane protein family with high potential as pharmaceutical targets, Methods Mol. Biol., 654, 141-168, https://doi.org/10.1007/978-1-60761-762-4_8.
Ishchenko, A., Gati, C., and Cherezov, V. (2018) Structural biology of G protein-coupled receptors: new opportunities from XFELs and cryoEM, Curr. Opin. Struct. Biol., 51, 44-52, https://doi.org/10.1016/j.sbi.2018.03.009.
Martynowycz, M. W., Shiriaeva, A., Ge, X., Hattne, J., Nannenga, B. L., Cherezov, V., and Gonen, T. (2021) MicroED structure of the human adenosine receptor determined from a single nanocrystal in LCP, Proc. Natl. Acad. Sci. USA, 118, e2106041118, https://doi.org/10.1073/pnas.2106041118.
Alexandrov, A. I., Mileni, M., Chien, E. Y. T., Hanson, M. A., and Stevens, R. C. (2008) Microscale fluorescent thermal stability assay for membrane proteins, Structure, 16, 351-359, https://doi.org/10.1016/j.str.2008.02.004.
Kooistra, A. J., Mordalski, S., Pándy-Szekeres, G., Esguerra, M., Mamyrbekov, A., Munk, C., Keserű, G. M., and Gloriam, D. E. (2021) GPCRdb in 2021: Integrating GPCR sequence, structure and function, Nucleic Acids Res., 49, D335-D343, https://doi.org/10.1093/nar/gkaa1080.
Park, J. H., Scheerer, P., Hofmann, K. P., Choe, H. W., and Ernst, O. P. (2008) Crystal structure of the ligand-free G-protein-coupled receptor opsin, Nature, 454, 183-187, https://doi.org/10.1038/nature07063.
Taniguchi, R., Inoue, A., Sayama, M., Uwamizu, A., Yamashita, K., Hirata, K., Yoshida, M., Tanaka, Y., Kato, H. E., Nakada-Nakura, Y., et al. (2017) Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA 6, Nature, 548, 356-360, https://doi.org/10.1038/nature23448.
Yang, S., Wu, Y., Xu, T. H., de Waal, P. W., He, Y., Pu, M., Chen, Y., DeBruine, Z. J., Zhang, B., Zaidi, S. A., et al. (2018) Crystal structure of the Frizzled 4 receptor in a ligand-free state, Nature, 560, 666-670, https://doi.org/10.1038/s41586-018-0447-x.
Lin, X., Li, M., Wang, N., Wu, Y., Luo, Z., Guo, S., Han, G. W., Li, S., Yue, Y., Wei, X., et al. (2020) Structural basis of ligand recognition and self-activation of orphan GPR52, Nature, 579, 152-157, https://doi.org/10.1038/s41586-020-2019-0.
Asada, H., Horita, S., Hirata, K., Shiroishi, M., Shiimura, Y., Iwanari, H., Hamakubo, T., Shimamura, T., Nomura, N., Kusano-Arai, O., et al. (2018) Crystal structure of the human angiotensin II type 2 receptor bound to an angiotensin II analog, Nat. Struct. Mol. Biol., 25, 570-576, https://doi.org/10.1038/s41594-018-0079-8.
Coulter, A., and Harris, R. (1983) Simplified preparation of rabbit fab fragments, J. Immunol. Methods, 59, 199-203, https://doi.org/10.1016/0022-1759(83)90031-5.
Ishchenko, A., Wacker, D., Kapoor, M., Zhang, A., Han, G. W., Basu, S., Patel, N., Messerschmidt, M., Weierstall, U., Liu, W., et al. (2017) Structural insights into the extracellular recognition of the human serotonin 2B receptor by an antibody, Proc. Natl. Acad. Sci. USA, 114, 8223-8228, https://doi.org/10.1073/pnas.1700891114.
Maeda, S., Shiimura, Y., Asada, H., Hirata, K., Luo, F., Nango, E., Tanaka, N., Toyomoto, M., Inoue, A., Aoki, J., et al. (2021) Endogenous agonist-bound S1PR3 structure reveals determinants of G protein-subtype bias, Sci. Adv., 7, eabf5325, https://doi.org/10.1126/sciadv.abf5325.
Asada, H., Inoue, A., Ngako Kadji, F. M., Hirata, K., Shiimura, Y., Im, D., Shimamura, T., Nomura, N., Iwanari, H., Hamakubo, T., et al. (2020) The crystal structure of angiotensin II type 2 receptor with endogenous peptide hormone, Structure, 28, 418-425.e4, https://doi.org/10.1016/j.str.2019.12.003.
Shiimura, Y., Horita, S., Hamamoto, A., Asada, H., Hirata, K., Tanaka, M., Mori, K., Uemura, T., Kobayashi, T., Iwata, S., et al. (2020) Structure of an antagonist-bound ghrelin receptor reveals possible ghrelin recognition mode, Nat. Commun., 11, 4160, https://doi.org/10.1038/s41467-020-17554-1.
Im, D., Inoue, A., Fujiwara, T., Nakane, T., Yamanaka, Y., Uemura, T., Mori, C., Shiimura, Y., Kimura, K. T., Asada, H., et al. (2020) Structure of the dopamine D2 receptor in complex with the antipsychotic drug spiperone, Nat. Commun., 11, 6442, https://doi.org/10.1038/s41467-020-20221-0.
Hamers-Casterman, C., Atarhouch, T., Muyldermans, S., Robinson, G., Hammers, C., Songa, E. B., Bendahman, N., and Hammers, R. (1993) Naturally occurring antibodies devoid of light chains, Nature, 363, 446-448, https://doi.org/10.1038/363446a0.
Rasmussen, S. G. F., Choi, H. J., Fung, J. J., Pardon, E., Casarosa, P., Chae, P. S., Devree, B. T., Rosenbaum, D. M., Thian, F. S., Kobilka, T. S., et al. (2011) Structure of a nanobody-stabilized active state of the β2 adrenoceptor, Nature, 469, 175-181, https://doi.org/10.1038/nature09648.
Weichert, D., Kruse, A. C., Manglik, A., Hiller, C., Zhang, C., Hübner, H., Kobilka, B. K., and Gmeiner, P. (2014) Covalent agonists for studying G protein-coupled receptor activation, Proc. Natl. Acad. Sci. USA, 111, 10744-10748, https://doi.org/10.1073/pnas.1410415111.
Staus, D. P., Strachan, R. T., Manglik, A., Pani, B., Kahsai, A. W., Kim, T. H., Wingler, L. M., Ahn, S., Chatterjee, A., Masoudi, A., et al. (2016) Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation, Nature, 535, 448-452, https://doi.org/10.1038/nature18636.
Che, T., English, J., Krumm, B. E., Kim, K., Pardon, E., Olsen, R. H. J., Wang, S., Zhang, S., Diberto, J. F., Sciaky, N., et al. (2020) Nanobody-enabled monitoring of kappa opioid receptor states, Nat. Commun., 11, 1145, https://doi.org/10.1038/s41467-020-14889-7.
Ghosh, E., Kumari, P., Jaiman, D., and Shukla, A. K. (2015) Methodological advances: the unsung heroes of the GPCR structural revolution, Nat. Rev. Mol. Cell Biol., 16, 69-81, https://doi.org/10.1038/nrm3933.
Robertson, M. J., Papasergi-Scott, M. M., He, F., Seven, A. B., Meyerowitz, J. G., Panova, O., Peroto, M. C., Che, T., and Skiniotis, G. (2022) Structure determination of inactive-state GPCRs with a universal nanobody, Nat. Struct. Mol. Biol., 29, 1188-1195, https://doi.org/10.1038/s41594-022-00859-8.
Che, T., Majumdar, S., Zaidi, S. A., Ondachi, P., McCorvy, J. D., Wang, S., Mosier, P. D., Uprety, R., Vardy, E., Krumm, B. E., et al. (2018) Structure of the nanobody-stabilized active state of the kappa opioid receptor, Cell, 172, 55-67.e15, https://doi.org/10.1016/j.cell.2017.12.011.
Collins, K. D. (2004) Ions from the Hofmeister series and osmolytes: Effects on proteins in solution and in the crystallization process, Methods, 34, 300-311, https://doi.org/10.1016/j.ymeth.2004.03.021.
Gusach, A., Luginina, A., Marin, E., Brouillette, R. L., Besserer-Offroy, É., Longpré, J. M., Ishchenko, A., Popov, P., Patel, N., Fujimoto, T., et al. (2019) Structural basis of ligand selectivity and disease mutations in cysteinyl leukotriene receptors, Nat. Commun., 10, 5573, https://doi.org/10.1038/s41467-019-13348-2.
Michaelian, N., Sadybekov, A., Besserer-Offroy, É., Han, G. W., Krishnamurthy, H., Zamlynny, B. A., Fradera, X., Siliphaivanh, P., Presland, J., Spencer, K. B., et al. (2021) Structural insights on ligand recognition at the human leukotriene B4 receptor 1, Nat. Commun., 12, 2971, https://doi.org/10.1038/s41467-021-23149-1.
Luginina, A., Gusach, A., Marin, E., Mishin, A., Brouillette, R., Popov, P., Shiriaeva, A., Besserer-Offroy, É., Longpré, J.-M., Lyapina, E., et al. (2019) Structure-based mechanism of cysteinyl leukotriene receptor inhibition by antiasthmatic drugs, Sci. Adv., 5, eaax2518, https://doi.org/10.1126/sciadv.aax2518.
Batyuk, A., Galli, L., Ishchenko, A., Han, G. W., Gati, C., Popov, P. A., Lee, M. Y., Stauch, B., White, T. A., Barty, A., et al. (2016) Native phasing of x-ray free-electron laser data for a G protein-coupled receptor, Sci. Adv., 2, e160029, https://doi.org/10.1126/sciadv.1600292.
Holik, H., Heß, H., Müller, W., and Lüdtke, O. (2013) Unit Operations, In Handbook of Paper and Board: Second Edition (Holik, H., ed.), 1, 351-472, https://doi.org/10.1002/9783527652495.ch7.
Krumm, B. E., Lee, S., Bhattacharya, S., Botos, I., White, C. F., Du, H., Vaidehi, N., and Grisshammer, R. (2016) Structure and dynamics of a constitutively active neurotensin receptor, Sci. Rep., 6, 38564, https://doi.org/10.1038/srep38564.
Vagenende, V., Yap, M. G. S., and Trout, B. L. (2009) Mechanisms of protein stabilization and prevention of protein aggregation by glycerol, Biochemistry, 48, 11084-11096, https://doi.org/10.1021/bi900649t.
Krumm, B. E., White, J. F., Shah, P., and Grisshammer, R. (2015) Structural prerequisites for G-protein activation by the neurotensin receptor, Nat. Commun., 6, 7895, https://doi.org/10.1038/ncomms8895.
Jang, K., Kim, H. G., Hlaing, S. H. S., Kang, M., Choe, H.-W., and Kim, Y. J. (2022) A short review on cryoprotectants for 3D protein structure analysis, Crystals, 12, 138, https://doi.org/10.3390/cryst12020138.
Suttapitugsakul, S., Xiao, H., Smeekens, J., and Wu, R. (2017) Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS-based proteomics, Mol. BioSystems, 13, 2574-2582, https://doi.org/10.1039/C7MB00393E.
Tao, Y. X., and Conn, P. M. (2014) Chaperoning G protein-coupled receptors: From cell biology to therapeutics, Endocr. Rev., 35, 602-647, https://doi.org/10.1210/er.2013-1121.
Lim, W. K., Kanelakis, K. C., and Neubig, R. R. (2013) Regulation of G protein signaling by the 70kDa heat shock protein, Cell. Signall., 25, 389-396, https://doi.org/10.1016/j.cellsig.2012.11.002.
Barth, H. G., Jackson, C., and Boyes, B. E. (1994) Size exclusion chromatography, Anal. Chem., 66, 595-620, https://doi.org/10.1021/ac00084a022.
Duong-Ly, K. C., and Gabelli, S. B. (2014) Gel filtration chromatography (size exclusion chromatography) of proteins, Methods Enzymol., 541, 105-114, https://doi.org/10.1016/B978-0-12-420119-4.00009-4.
Kawate, T., and Gouaux, E. (2006) Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins, Structure, 14, 673-681, https://doi.org/10.1016/j.str.2006.01.013.
Gimpl, K., Klement, J., and Keller, S. (2016) Characterising protein/detergent complexes by triple-detection size-exclusion chromatography, Biol. Procedures Online, 18, 4, https://doi.org/10.1186/s12575-015-0031-9.
Newby, Z. E. R., O’Connell, J. D., Gruswitz, F., Hays, F. A., Harries, W. E. C., Harwood, I. M., Ho, J. D., Lee, J. K., Savage, D. F., Miercke, L. J. W., et al. (2009) A general protocol for the crystallization of membrane proteins for X-ray structural investigation, Nat. Protoc., 4, 619-637, https://doi.org/10.1038/nprot.2009.27.
Wyatt, P. J. (1993) Light scattering and the absolute characterization of macromolecules, Anal. Chim. Acta, 272, 1-40, https://doi.org/10.1016/0003-2670(93)80373-S.
Some, D., Amartely, H., Tsadok, A., and Lebendiker, M. (2019) Characterization of proteins by size-exclusion chromatography coupled to multi-angle light scattering (SEC-MALS), J. Visual. Exp., 2019, e59615, https://doi.org/10.3791/59615-v.
Dupeux, F., Röwer, M., Seroul, G., Blot, D., and Márquez, J. A. (2011) A thermal stability assay can help to estimate the crystallization likelihood of biological samples, Acta Crystallogr. Sect. D Biol. Crystallogr., 67, 915-919, https://doi.org/10.1107/S0907444911036225.
Huynh, K., and Partch, C. L. (2015) Analysis of protein stability and ligand interactions by thermal shift assay, Curr. Protocols Protein Sci., 79, 28.9.1-28.9.14, https://doi.org/10.1002/0471140864.ps2809s79.
Lo, M. C., Aulabaugh, A., Jin, G., Cowling, R., Bard, J., Malamas, M., and Ellestad, G. (2004) Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery, Anal. Biochem., 332, 153-159, https://doi.org/10.1016/j.ab.2004.04.031.
Real-Hohn, A., Groznica, M., Löffler, N., Blaas, D., and Kowalski, H. (2020) NanoDSF: in vitro label-free method to monitor picornavirus uncoating and test compounds affecting particle stability, Front. Microbiol., 11, 1442, https://doi.org/10.3389/fmicb.2020.01442.
Duy, C., and Fitter, J. (2006) How aggregation and conformational scrambling of unfolded states govern fluorescence emission spectra, Biophys. J., 90, 3704-3711, https://doi.org/10.1529/biophysj.105.078980.
Kotov, V., Bartels, K., Veith, K., Josts, I., Subhramanyam, U. K. T., Günther, C., Labahn, J., Marlovits, T. C., Moraes, I., Tidow, H., et al. (2019) High-throughput stability screening for detergent-solubilized membrane proteins, Sci. Rep., 9, 10379, https://doi.org/10.1038/s41598-019-46686-8.
Lea, W. A., and Simeonov, A. (2012) Differential scanning fluorometry signatures as indicators of enzyme inhibitor mode of action: case study of glutathione S-transferase, PLoS One, 7, e36219, https://doi.org/10.1371/journal.pone.0036219.
Pardon, E., Laeremans, T., Triest, S., Rasmussen, S. G. F., Wohlkönig, A., Ruf, A., Muyldermans, S., Hol, W. G. J., Kobilka, B. K., and Steyaert, J. (2014) A general protocol for the generation of Nanobodies for structural biology, Nat. Protocols, 9, 674-693, https://doi.org/10.1038/nprot.2014.039.
Wingler, L. M., McMahon, C., Staus, D. P., Lefkowitz, R. J., and Kruse, A. C. (2019) Distinctive Activation Mechanism for Angiotensin Receptor Revealed by a Synthetic Nanobody, Cell, 176, 479-490.e12, https://doi.org/10.1016/j.cell.2018.12.006.
Kruse, A. C., Ring, A. M., Manglik, A., Hu, J., Hu, K., Eitel, K., Hübner, H., Pardon, E., Valant, C., Sexton, P. M., et al. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor, Nature, 504, 101-106, https://doi.org/10.1038/nature12735.
Deshpande, I., Liang, J., Hedeen, D., Roberts, K. J., Zhang, Y., Ha, B., Latorraca, N. R., Faust, B., Dror, R. O., Beachy, P. A., et al. (2019) Smoothened stimulation by membrane sterols drives Hedgehog pathway activity, Nature, 571, 284-288, https://doi.org/10.1038/s41586-019-1355-4.
Haffke, M., Fehlmann, D., Rummel, G., Boivineau, J., Duckely, M., Gommermann, N., Cotesta, S., Sirockin, F., Freuler, F., Littlewood-Evans, A., et al. (2019) Structural basis of species-selective antagonist binding to the succinate receptor, Nature, 574, 581-585, https://doi.org/10.1038/s41586-019-1663-8.
Zhang, H.-L., Ye, H.-Q., Deng, C.-L., Liu, S.-Q., Shi, P.-Y., Qin, C.-F., Yuan, Z.-M., and Zhang, B. (2017) Generation and characterization of West Nile pseudo-infectious reporter virus for antiviral screening, Antiviral Res., 141, 38-47, https://doi.org/10.1016/j.antiviral.2017.02.006.
Gilman, A. G. (1987) G proteins: transducers of receptor-generated signals, Annu. Rev. Biochem., 56, 615-649, https://doi.org/10.1146/annurev.bi.56.070187.003151.
Hepler, J. R., Gilman, A. G. (1992) G proteins, Trends Biochem. Sci., 17, 383-387, https://doi.org/10.1016/0968-0004(92)90005-T.
Neves, S. R., Ram, P. T., and Iyengar, R. (2002) G protein pathways, Science, 296, 1636-1639, https://doi.org/10.1126/science.1071550.
Hillenbrand, M., Schori, C., Schöppe, J., and Plückthun, A. (2015) Comprehensive analysis of heterotrimeric G-protein complex diversity and their interactions with GPCRs in solution, Proc. Natl. Acad. Sci. USA, 112, E1181-E1190, https://doi.org/10.1073/pnas.1417573112.
Masuho, I., Skamangas, N. K., Muntean, B. S., and Martemyanov, K. A. (2021) Diversity of the Gβγ complexes defines spatial and temporal bias of GPCR signaling, Cell Systems, 12, 324-337.e5, https://doi.org/10.1016/j.cels.2021.02.001.
Milligan, G., and Kostenis, E. (2006) Heterotrimeric G-proteins: a short history, Br. J. Pharmacol., 147, S46-S55, https://doi.org/10.1038/sj.bjp.0706405.
Carpenter, B., and Tate, C. G. (2016) Engineering a minimal G protein to facilitate crystallisation of G protein-coupled receptors in their active conformation, Protein Engin. Design Select., 29, 583-593, https://doi.org/10.1093/protein/gzw049.
Carpenter, B., and Tate, C. (2017) Expression and purification of mini G proteins from Escherichia coli, BIO Protocol, 7, e2235, https://doi.org/10.21769/BioProtoc.2235.
Liu, X., Xu, X., Hilger, D., Aschauer, P., Tiemann, J. K. S., Du, Y., Liu, H., Hirata, K., Sun, X., Guixà-González, R., et al. (2019) Structural insights into the process of GPCR-G protein complex formation, Cell, 177, 1243-1251.e12, https://doi.org/10.1016/j.cell.2019.04.021.
Standfuss, J., Edwards, P. C., D’Antona, A., Fransen, M., Xie, G., Oprian, D. D., and Schertler, G. F. X. (2011) The structural basis of agonist-induced activation in constitutively active rhodopsin, Nature, 471, 656-660, https://doi.org/10.1038/nature09795.
Scheerer, P., Park, J. H., Hildebrand, P. W., Kim, Y. J., Krauß, N., Choe, H. W., Hofmann, K. P., and Ernst, O. P. (2008) Crystal structure of opsin in its G-protein-interacting conformation, Nature, 455, 497-502, https://doi.org/10.1038/nature07330.
Singhal, A., Guo, Y., Matkovic, M., Schertler, G., Deupi, X., Yan, E. C., and Standfuss, J. (2016) Structural role of the T94I rhodopsin mutation in congenital stationary night blindness, EMBO Rep., 17, 1431-1440, https://doi.org/10.15252/embr.201642671.
Singhal, A., Ostermaier, M. K., Vishnivetskiy, S. A., Panneels, V., Homan, K. T., Tesmer, J. J. G., Veprintsev, D., Deupi, X., Gurevich, V. V., Schertler, G. F. X., et al. (2013) Insights into congenital stationary night blindness based on the structure of G90D rhodopsin, EMBO Rep., 14, 520-526, https://doi.org/10.1038/embor.2013.44.
Mattle, D., Singhal, A., Schmid, G., Dawson, R., and Standfuss, J. (2015) Mammalian Expression, Purification, and Crystallization of Rhodopsin variants, in Rhodopsin: Methods and Protocols, pp. 39-54, https://doi.org/10.1007/978-1-4939-2330-4_3.
Park, J. H., Morizumi, T., Li, Y., Hong, J. E., Pai, E. F., Hofmann, K. P., Choe, H. W., and Ernst, O. P. (2013) Opsin, a structural model for olfactory receptors? Angewandte Chemie, 52, 11021-11024, https://doi.org/10.1002/anie.201302374.
Deupi, X., Edwards, P., Singhal, A., Nickle, B., Oprian, D., Schertler, G., and Standfuss, J. (2012) Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II, Proc. Natl. Acad. Sci. USA, 109, 119-124, https://doi.org/10.1073/pnas.1114089108.
Choe, H. W., Kim, Y. J., Park, J. H., Morizumi, T., Pai, E. F., Krau, N., Hofmann, K. P., Scheerer, P., and Ernst, O. P. (2011) Crystal structure of metarhodopsin II, Nature, 471, 651-655, https://doi.org/10.1038/nature09789.
Sun, B., Feng, D., Chu, M. L.-H., Fish, I., Lovera, S., Sands, Z. A., Kelm, S., Valade, A., Wood, M., Ceska, T., et al. (2021) Crystal structure of dopamine D1 receptor in complex with G protein and a non-catechol agonist, Nat. Commun., 12, 3305, https://doi.org/10.1038/s41467-021-23519-9.
van Gastel, J., Hendrickx, J. O., Leysen, H., Santos-Otte, P., Luttrell, L. M., Martin, B., and Maudsley, S. (2018) β-arrestin based receptor signaling paradigms: potential therapeutic targets for complex age-related disorders, Front. Pharmacol., 9, 1369, https://doi.org/10.3389/fphar.2018.01369.
Zhou, X. E., Gao, X., Barty, A., Kang, Y., He, Y., Liu, W., Ishchenko, A., White, T. A., Yefanov, O., Han, G. W., et al. (2016) X-ray laser diffraction for structure determination of the rhodopsin-arrestin complex, Sci. Data, 3, 160021, https://doi.org/10.1038/sdata.2016.21.
Szczepek, M., Beyrière, F., Hofmann, K. P., Elgeti, M., Kazmin, R., Rose, A., Bartl, F. J., von Stetten, D., Heck, M., Sommer, M. E., et al. (2014) Crystal structure of a common GPCR-binding interface for G protein and arrestin, Nat. Commun., 5, 4801, https://doi.org/10.1038/ncomms5801.
Liang, Y. L., Khoshouei, M., Radjainia, M., Zhang, Y., Glukhova, A., Tarrasch, J., Thal, D. M., Furness, S. G. B., Christopoulos, G., Coudrat, T., et al. (2017) Phase-plate cryo-EM structure of a class B GPCR-G-protein complex, Nature, 546, 118-123, https://doi.org/10.1038/nature22327.
Fu, Y., Yang, Y., Zhang, H., Farley, G., Wang, J., Quarles, K. A., Weng, Z., and Zamore, P. D. (2018) The genome of the Hi5 germ cell line from Trichoplusia ni, an agricultural pest and novel model for small RNA biology, eLife, 7, e31628, https://doi.org/10.7554/eLife.31628.
Papasergi-Scott, M. M., Robertson, M. J., Seven, A. B., Panova, O., Mathiesen, J. M., and Skiniotis, G. (2020) Structures of metabotropic GABAB receptor, Nature, 584, 310-314, https://doi.org/10.1038/s41586-020-2469-4.
Jeong, E., Kim, Y., Jeong, J., and Cho, Y. (2021) Structure of the class C orphan GPCR GPR158 in complex with RGS7-Gβ5, Nat. Commun., 12, 6805, https://doi.org/10.1038/s41467-021-27147-1.
Patil, D. N., Singh, S., Laboute, T., Strutzenberg, T. S., Qiu, X., Wu, D., Novick, S. J., Robinson, C. V., Griffin, P. R., Hunt, J. F., et al. (2022) Cryo-EM structure of human GPR158 receptor coupled to the RGS7-Gb5 signaling complex, Science, 375, 86-91, https://doi.org/10.1126/science.abl4732.
Park, J., Zuo, H., Frangaj, A., Fu, Z., Yen, L. Y., Zhang, Z., Mosyak, L., Slavkovich, V. N., Liu, J., Ray, K. M., et al. (2021) Symmetric activation and modulation of the human calcium-sensing receptor, Proc. Natl. Acad. Sci. USA, 118, e2115849118, https://doi.org/10.1073/pnas.2115849118.
Fang, W., Yang, F., Xu, C., Ling, S., Lin, L., Zhou, Y., Sun, W., Wang, X., Liu, P., Rondard, P., et al. (2022) Structural basis of the activation of metabotropic glutamate receptor 3, Cell Res., 32, 695-698, https://doi.org/10.1038/s41422-022-00623-z.
Duan, J., Xu, P., Cheng, X., Mao, C., Croll, T., He, X., Shi, J., Luan, X., Yin, W., You, E., et al. (2021) Structures of full-length glycoprotein hormone receptor signalling complexes, Nature, 598, 688-692, https://doi.org/10.1038/s41586-021-03924-2.
Bross, P., Andersen, B. A., Winter, V., Kräutle, F., Jensen, T. G., Nandy, A., Kølvraa, S., Ghisla, S., Bolund, L., and Gregersen, N. (1993) Co-overexpression of bacterial GroESL chaperonins partly overcomes non-productive folding and tetramer assembly of E. coli-expressed human medium-chain acyl-CoA dehydrogenase (MCAD) carrying the prevalent disease-causing K304E mutation, Biochim. Biophys. Acta, 1182, 264-274, https://doi.org/10.1016/0925-4439(93)90068-C.
Rivas, F. V., Tolia, N. H., Song, J. J., Aragon, J. P., Liu, J., Hannon, G. J., and Joshua-Tor, L. (2005) Purified Argonaute2 and an siRNA form recombinant human RISC, Nat. Struct. Mol. Biol., 12, 340-349, https://doi.org/10.1038/nsmb918.
Tolia, N. H., and Joshua-Tor, L. (2006) Strategies for protein coexpression in Escherichia coli, Nature Methods, 3, 55-64, https://doi.org/10.1038/nmeth0106-55.
Kost, T. A., Condreay, J. P., and Jarvis, D. L. (2005) Baculovirus as versatile vectors for protein expression in insect and mammalian cells, Nat. Biotechnol., 23, 567-575, https://doi.org/10.1038/nbt1095.
Aricescu, A. R., Lu, W., and Jones, E. Y. (2006) A time- and cost-efficient system for high-level protein production in mammalian cells, Acta Crystallogr. Sect. D Biol. Crystallogr., 62, 1243-1250, https://doi.org/10.1107/S0907444906029799.
Aricescu, A. R., Assenberg, R., Bill, R. M., Busso, D., Chang, V. T., Davis, S. J., Dubrovsky, A., Gustafsson, L., Hedfalk, K., Heinemann, U., et al. (2006) Eukaryotic expression: developments for structural proteomics, Acta Crystallogr. Sect. D Biol. Crystallogr., 62, 1114-1124, https://doi.org/10.1107/S0907444906029805.
Perrakis, A., and Romier, C. (2008) Assembly of protein complexes by coexpression in prokaryotic and eukaryotic hosts: an overview, Methods Mol. Biol., 426, 247-256, https://doi.org/10.1007/978-1-60327-058-8_15.
Seven, A. B., Barros-Álvarez, X., de Lapeyrière, M., Papasergi-Scott, M. M., Robertson, M. J., Zhang, C., Nwokonko, R. M., Gao, Y., Meyerowitz, J. G., Rocher, J. P., et al. (2021) G-protein activation by a metabotropic glutamate receptor, Nature, 595, 450-454, https://doi.org/10.1038/s41586-021-03680-3.
Draper-Joyce, C. J., Khoshouei, M., Thal, D. M., Liang, Y. L., Nguyen, A. T. N., Furness, S. G. B., Venugopal, H., Baltos, J. A., Plitzko, J. M., Danev, R., et al. (2018) Structure of the adenosine-bound human adenosine A1 receptor-Gi complex, Nature, 558, 559-565, https://doi.org/10.1038/s41586-018-0236-6.
Sun, B., Willard, F. S., Feng, D., Alsina-Fernandez, J., Chen, Q., Vieth, M., Ho, J. D., Showalter, A. D., Stutsman, C., Ding, L., et al. (2022) Structural determinants of dual incretin receptor agonism by tirzepatide, Proc. Natl. Acad. Sci. USA, 119, e2116506119, https://doi.org/10.1073/pnas.2116506119.
Zhang, H., Chen, L. N., Yang, D., Mao, C., Shen, Q., Feng, W., Shen, D. D., Dai, A., Xie, S., Zhou, Y., et al. (2021) Structural insights into ligand recognition and activation of the melanocortin-4 receptor, Cell Res., 31, 1163-1175, https://doi.org/10.1038/s41422-021-00552-3.
Koehl, A., Hu, H., Maeda, S., Zhang, Y., Qu, Q., Paggi, J. M., Latorraca, N. R., Hilger, D., Dawson, R., Matile, H., et al. (2018) Structure of the µ-opioid receptor-Gi protein complex, Nature, 558, 547-552, https://doi.org/10.1038/s41586-018-0219-7.
Connor, M., and Christie, M. J. (1999) Opioid receptor signalling mechanisms, Clin. Exp. Pharmacol. Physiol., 26, 493-499, https://doi.org/10.1046/j.1440-1681.1999.03049.x.
Raffa, R. B., Martinez, R. P., and Connelly, C. D. (1994) G-protein antisense oligodeoxyribonucleotides and µ-opioid supraspinal antinociception, Eur. J. Pharmacol., 258, R5-R7, https://doi.org/10.1016/0014-2999(94)90073-6.
Strathmann, M. P., and Simon, M. I. (1991) Gα12 and Gα13 subunits define a fourth class of G protein α subunits, Proc. Natl. Acad. Sci. USA, 88, 5582-5586, https://doi.org/10.1073/pnas.88.13.5582.
Riobo, N. A., and Manning, D. R. (2005) Receptors coupled to heterotrimeric G proteins of the G12 family, Trends Pharmacol. Sci., 26, 146-154, https://doi.org/10.1016/j.tips.2005.01.007.
Kozasa, T., Jiang, X., Hart, M. J., Sternweis, P. M., Singer, W. D., Gilman, A. G., Bollag, G., and Sternweis, P. C. (1998) p115 RhoGEF, a GTPase Activating Protein for Gα 12 and Gα 13, Science, 280, 2109-2111, https://doi.org/10.1126/science.280.5372.2109.
Chen, H., Chen, K., Huang, W., Staudt, L. M., Cyster, J. G., and Li, X. (2022) Structure of S1PR2-heterotrimeric G13 signaling complex, Sci. Adv., 8, 67, https://doi.org/10.1126/sciadv.abn0067.
Barros-Álvarez, X., Nwokonko, R. M., Vizurraga, A., Matzov, D., He, F., Papasergi-Scott, M. M., Robertson, M. J., Panova, O., Yardeni, E. H., Seven, A. B., et al. (2022) The tethered peptide activation mechanism of adhesion GPCRs, Nature, 604, 757-762, https://doi.org/10.1038/s41586-022-04575-7.
Kreutz, B., Yau, D. M., Nance, M. R., Tanabe, S., Tesmer, J. J. G., and Kozasa, T. (2006) A new approach to producing functional Gα subunits yields the activated and deactivated structures of Gα12/13 proteins, Biochemistry, 45, 167-174, https://doi.org/10.1021/bi051729t.
Singer, W. D., Brown, H. A., and Sternweis, P. C. (1997) Regulation of eukaryotic phosphatidylinositol-specific phospholipase C and phospholipase D, Annu. Rev. Biochem., 66, 475-509, https://doi.org/10.1146/annurev.biochem.66.1.475.
Taylor, S. J., Chae, H. Z., Rhee, S. G., and Exton, J. H. (1991) Activation of the β1 isozyme of phospholipase C by α subunits of the Gq class of G proteins, Nature, 350, 516-518, https://doi.org/10.1038/350516a0.
Smrcka, A. V., Hepler, J. R., Brown, K. O., and Sternweis, P. C. (1991) Regulation of polyphosphoinositide-specific phospholipase C activity by purified Gq, Science, 251, 804-807, https://doi.org/10.1126/science.1846707.
Maeda, S., Koehl, A., Matile, H., Hu, H., Hilger, D., Schertler, G. F. X., Manglik, A., Skiniotis, G., Dawson, R. J. P., and Kobilka, B. K. (2018) Development of an antibody fragment that stabilizes GPCR/G-protein complexes, Nat. Commun., 9, 3712, https://doi.org/10.1038/s41467-018-06002-w.
Wan, Q., Okashah, N., Inoue, A., Nehme, R., Carpenter, B., Tate, C. G., and Lambert, N. A. (2018) Mini G protein probes for active G protein-coupled receptors (GPCRs) in live cells, J. Biol. Chem., 293, 7466-7473, https://doi.org/10.1074/jbc.RA118.001975.
Meyerowitz, J. G., Robertson, M. J., Barros-Álvarez, X., Panova, O., Nwokonko, R. M., Gao, Y., and Skiniotis, G. (2022) The oxytocin signaling complex reveals a molecular switch for cation dependence, Nat. Struct. Mol. Biol., 29, 274-281, https://doi.org/10.1038/s41594-022-00728-4.
Xia, R., Wang, N., Xu, Z., Lu, Y., Song, J., Zhang, A., Guo, C., and He, Y. (2021) Cryo-EM structure of the human histamine H1 receptor/Gq complex, Nat. Commun., 12, 2086, https://doi.org/10.1038/s41467-021-22427-2.
Kim, K., Che, T., Panova, O., DiBerto, J. F., Lyu, J., Krumm, B. E., Wacker, D., Robertson, M. J., Seven, A. B., Nichols, D. E., et al. (2020) Structure of a Hallucinogen-Activated Gq-Coupled 5-HT2A Serotonin Receptor, Cell, 182, 1574-1588.e19, https://doi.org/10.1016/j.cell.2020.08.024.
Benovic, J. L., Kühn, H., Weyand, I., Codina, J., Caron, M. G., and Lefkowitz, R. J. (1987) Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48- kDa protein), Proc. Natl. Acad. Sci. USA, 84, 8879-8882, https://doi.org/10.1073/pnas.84.24.8879.
Luttrell, L. M., Roudabush, F. L., Choy, E. W., Miller, W. E., Field, M. E., Pierce, K. L., and Lefkowitz, R. J. (2001) Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds, Proc. Natl. Acad. Sci. USA, 98, 2449-2454, https://doi.org/10.1073/pnas.041604898.
McDonald, P. H., Chow, C.-W., Miller, W. E., Laporte, S. A., Field, M. E., Lin, F.-T., Davis, R. J., and Lefkowitz, R. J. (2000) β-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3, Science, 290, 1574-1577, https://doi.org/10.1126/science.290.5496.1574.
Luttrell, L. M., Ferguson, S. S. G., Daaka, Y., Miller, W. E., Maudsley, S., Della Rocca, G. J., Lin, F. T., Kawakatsu, H., Owada, K., Luttrell, D. K., et al. (1999) β-arrestin-dependent formation of β2 adrenergic receptor-src protein kinase complexes, Science, 283, 655-661, https://doi.org/10.1126/science.283.5402.655.
Yin, W., Li, Z., Jin, M., Yin, Y. L., de Waal, P. W., Pal, K., Yin, Y., Gao, X., He, Y., Gao, J., et al. (2019) A complex structure of arrestin-2 bound to a G protein-coupled receptor, Cell Res., 29, 971-983, https://doi.org/10.1038/s41422-019-0256-2.
Staus, D. P., Hu, H., Robertson, M. J., Kleinhenz, A. L. W., Wingler, L. M., Capel, W. D., Latorraca, N. R., Lefkowitz, R. J., and Skiniotis, G. (2020) Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc, Nature, 579, 297-302, https://doi.org/10.1038/s41586-020-1954-0.
Huang, W., Masureel, M., Qu, Q., Janetzko, J., Inoue, A., Kato, H. E., Robertson, M. J., Nguyen, K. C., Glenn, J. S., Skiniotis, G., et al. (2020) Structure of the neurotensin receptor 1 in complex with β-arrestin 1, Nature, 579, 303-308, https://doi.org/10.1038/s41586-020-1953-1.
Weiss, E. R., Ducceschi, M. H., Horner, T. J., Li, A., Craft, C. M., and Osawa, S. (2001) Species-specific differences in expression of G-protein-coupled receptor kinase (GRK) 7 and GRK1 in mammalian cone photoreceptor cells: Implications for cone cell phototransduction, J. Neurosci., 21, 9175-9184, https://doi.org/10.1523/JNEUROSCI.21-23-09175.2001.
Calebiro, D., Nikolaev, V. O., Gagliani, M. C., de Filippis, T., Dees, C., Tacchetti, C., Persani, L., and Lohse, M. J. (2009) Persistent cAMP-signals triggered by internalized G-protein-coupled receptors, PLoS Biol., 7, e1000172, https://doi.org/10.1371/journal.pbio.1000172.
Ferrandon, S., Feinstein, T. N., Castro, M., Wang, B., Bouley, R., Potts, J. T., Gardella, T. J., and Vilardaga, J. P. (2009) Sustained cyclic AMP production by parathyroid hormone receptor endocytosis, Nat. Chem. Biol., 5, 734-742, https://doi.org/10.1038/nchembio.206.
Feinstein, T. N., Yui, N., Webber, M. J., Wehbi, V. L., Stevenson, H. P., King, J. D., Hallows, K. R., Brown, D., Bouley, R., and Vilardaga, J. P. (2013) Noncanonical control of vasopressin receptor type 2 signaling by retromer and arrestin, J. Biol. Chem., 288, 27849-27860, https://doi.org/10.1074/jbc.M112.445098.
Thomsen, A. R. B., Plouffe, B., Cahill, T. J., Shukla, A. K., Tarrasch, J. T., Dosey, A. M., Kahsai, A. W., Strachan, R. T., Pani, B., Mahoney, J. P., et al. (2016) GPCR-G protein-β-Arrestin super-complex mediates sustained G protein signaling, Cell, 166, 907-919, https://doi.org/10.1016/j.cell.2016.07.004.
Nguyen, A. H., Thomsen, A. R. B., Cahill, T. J., Huang, R., Huang, L.-Y., Marcink, T., Clarke, O. B., Heissel, S., Masoudi, A., Ben-Hail, D., et al. (2019) Structure of an endosomal signaling GPCR-G protein-β-arrestin megacomplex, Nat. Struct. Mol. Biol., 26, 1123-1131, https://doi.org/10.1038/s41594-019-0330-y.
Funding
Review of Membrane model systems in GPCR studies were supported by the Ministry of Science and Higher Education of the Russian Federation (agreement 075-03-2023-106, project FSMG-2020-0003). Review of Characterization of Protein Preparations and Quality Control was supported by the Russian Science Foundation (project no. 22-74-00024, https://rscf.ru/project/22-74-00024/ [in Russian]). Other parts of the work were supported by the Russian Science Foundation (project no. 22-74-10036; https://rscf.ru/project/22-74-10036/ [in Russian]).
Author information
Authors and Affiliations
Contributions
Daria A. Dmitrieva was responsible for the “Introduction of Point Mutations”, “Alanine Scanning and StaR™ Technology”, “Directed Evolution”, “Use of Fusion Partners”, “Expression Systems for GPCR Production”, “Using Ligands for Receptor Stabilization”, “Using Antibody Fragments for Stabilization of GPCR Conformational States” sections. Tatiana V. Kotova was responsible for the “Introduction of Point Mutations”, “Alanine Scanning and StaR™ Technology” sections (with D.A.D.), “Deletion of Unordered Regions from the Receptor Amino Acid Sequence”, “Insertion of Additional Elements into the Receptor Amino Acid Sequence”, “Methods for GPCR Purification”, “Formation of Receptor Complexes with Main Interaction Partners” sections. Nadezda A. Safronova authored the “Using Salts and Chemical Agents for Purification and Stabilization of Receptors” section. Alexandra A. Sadova was responsible for the “Membrane Model Systems in GPCR Studies; Their Pros and Cons” section. Dmitrii E. Dashevskii wrote the “Characterization of Protein Preparations and Quality Control” section. Alexey V. Mishin authored the conception and the initial plan of the review, edited the whole manuscript.
Corresponding author
Ethics declarations
The authors declare no conflict of interest in financial or any other sphere.
Additional information
Translated from Uspekhi Biologicheskoi Khimii, 2023, Vol. 63, pp. 379-448.
Rights and permissions
About this article

Cite this article
Dmitrieva, D.A., Kotova, T.V., Safronova, N.A. et al. Protein Design Strategies for the Structural–Functional Studies of G Protein-Coupled Receptors. Biochemistry Moscow 88 (Suppl 1), S192–S226 (2023). https://doi.org/10.1134/S0006297923140110
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297923140110