
Abstract
A much debated question is whether aging is the cumulative consequence of degenerative factors insufficiently opposed by natural selection, or, on the contrary, an ordered process, genetically determined and regulated, modeled by natural selection, and for which the definition of phenoptotic phenomenon would be entirely appropriate. In this review, theoretical arguments and empirical data about the two hypotheses are exposed, with more evidence in support of the thesis of aging as a form of phenoptosis. However, as the thesis of aging as an adaptive and programmed phenomenon necessarily requires the existence of specific mechanisms that determine to age, such as the subtelomere–telomere theory proposed for this purpose, the evidence supporting the mechanisms described by this theory is reported. In particular, it is highlighted that the recent interpretation of the role of TERRA sequences in the context of subtelomere–telomere theory is a fundamental point in supporting the hypothesized mechanisms. Furthermore, some characteristics of the mechanisms proposed by the theory, such as epigenetic modifications in aging, gradual cell senescence, cell senescence, limits in cell duplications, and fixed size of the telomeric heterochromatin hood, are exposed in their compatibility with both the thesis of aging as phenoptotic phenomenon and the opposite thesis. In short, aging as a form of phenoptosis appears a scientifically sound hypothesis while the opposite thesis should clarify the meaning of various phenomena that appear to invalidate it.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
It has been 25 years since the neologism and concept of phenoptosis as “programmed death of an organism” was defined and proposed [1, 2]. Subsequently: (i) the aging manifested by individuals of our and many other species, precisely described as “increasing mortality with increasing chronological age in populations in the wild” [3], has been indicated as a form of “slow phenoptosis” [4]; (ii) the definition of phenoptosis was extended: “Phenoptosis is the death of an individual caused by its own actions or by actions of close relatives (siblicide; in particular, the parent-caused death of an offspring or filial infanticide) and not caused primarily by accidents or diseases or external factors, which is determined, regulated or influenced by genes favored by natural selection” [5]; (iii) it was underlined how this type of phenomenon is very widespread in nature [5, 6].
The concept of phenoptosis allows to embrace with a single term a series of very heterogeneous phenomena in which, in countless species, an individual dies or exposes itself to the risk of dying in well-defined phases of his life cycle or on particular occasions [5, 7].
Phenoptotic phenomena imply two general common features:
– Natural selection, at a strictly individual level, cannot explain genes that favor characteristics for which the individual dies or reduces his chances of survival. Consequently, any type of phenoptosis must be determined by natural selection acting at a supra-individual level;
– Phenoptotic phenomena must be caused by particular mechanisms determined and regulated by specific genes whose existence is allowed by the afore-mentioned supra-individual selective mechanisms.
The concept of phenoptosis appears in contrast with Darwin’s ideas if limited to the expression “survival of the fittest,” which is in the title of the fifth edition of his book [8]. Without further clarification, it excludes the possibility that natural selection could favor anything that kills or damages the individual. However, it is important to remember that Darwin did not exclude the possibility that natural selection could favor characters harmful to the individual: “A tribe including many members who... were always ready to aid one another, and to sacrifice themselves for the common good, would be victorious over most other tribes; and this would be natural selection” [9], p. 500. Therefore, it would be unfair to believe that the concept of phenoptosis is incompatible with Darwin’s ideas or is an overcoming of them. It is more accurate to conceive the category of phenoptotic phenomena as something underestimated in the past, not described as a set of phenomena that could be enclosed under a single term, and perhaps unduly conceived as a series of rare and curious exceptions to the queen’s rule of the “survival of the fittest.”
However, as shown in the discussion of the present work, the concept of phenoptosis must overcome deep-rooted preconceived ideas that are proved to be groundless by both theoretical arguments and empirical evidence.
The idea of aging as a programmed phenomenon is sometimes described as untenable, unsupported by evidence and with strong arguments against it. For instance, “[e]volutionary considerations suggest aging is caused not by active gene programming but by evolved limitations in somatic maintenance, resulting in a build-up of damage... there is scant evidence for the existence of such a program, and there are powerful arguments why it should not exist” [10].
On the contrary, the topic of this article is whether aging can be correctly defined as a form of phenoptosis, that is to say, whether it is well-grounded to conceive of aging as an adaptive phenomenon favored by natural selection, determined and regulated by specific mechanisms.
In particular, Part I of the “Discussion” section analyzes: (i) whether it is correct to accept some arguments commonly proposed against the idea of aging as an adaptive and programmed phenomenon; (ii) the plausibility of some arguments and facts in support of the opposite idea of aging as a non-adaptive and non-programmed phenomenon.
Afterwards, Part II debates the existence or non-existence of specific mechanisms that determine aging, which are indispensable in the case of aging as a phenoptotic phenomenon and unjustifiable in the opposite case.
DISCUSSION
Part I. Arguments and evidence about the two opposite interpretations of aging.
A. Popular arguments against the hypothesis of aging as an adaptive phenomenon. There are three arguments often proposed as preliminary objections against the possibility of aging as an adaptive phenomenon, which is an essential condition to propose aging as a type of phenoptosis. However, these arguments are contradicted by the evidence and do not appear sustainable:
A.1. Aging as a universal and inevitable phenomenon. A popular idea is that aging is a universal and so inevitable phenomenon for any species (e.g.: “Unlike any disease, age changes… occur in every multicellular animal that reaches a fixed size at reproductive maturity... occur in virtually all animate and inanimate matter” [11]). According to this conception, it is certainly possible to age at a more or less rapid pace but non-aging species do not exist. Consequently, since aging is a universal and completely natural phenomenon, it does not require particular justifications.
This conception is completely disproved by the evidence. In this regard, it is sufficient to read what is described in a well-known authoritative treatise [7], and the arguments worked out in an article dedicated to phenoptosis [5] and in Chapter 2 of a recent book [6]. In short, limiting ourselves to multicellular species and a few categories of examples, a large number of species show, as their normal term of life, clearly phenoptotic phenomena other than aging as defined above [3]. Many species, both in the animal and in the vegetable kingdom, reproduce only once and then die (semelparity). Many plants and insects have annual cycles that end with the death and are clearly genetically determined. As an extreme example, many insect species do not feed in the adult stage or even have defective nutrition systems that do not allow feeding (aphagy). These cases are described in Finch’s treatise, in the chapter titled “Rapid Senescence and Sudden Death” [7].
However, the term “senescence” used for the above-said species could lead to some confusion with the case of the species where the increase in mortality is gradual, described as “Gradual Senescence with Definite Lifespan” [7], and that fall within the aforementioned definition of aging.
Another possible misunderstanding is that “Rapid Senescence and Sudden Death” necessarily involves a short life span, which is not at all true: “Various species of the thick-stemmed bamboos (Phyllostachys) have prolonged phases of vegetative growth that last for many years or decades (7, 30, 60, or 120 years) according to the species, before suddenly flowering and dying…” [7], p. 101.
However, all these cases of “Rapid Senescence” are clearly genetically determined and therefore programmed and fall within the definition of phenoptosis.
Some species do not die from phenoptosis and, on the contrary, do not show any detectable functional decline with age (“Negligible Senescence” [7]). More precisely, it is better to say that for many species there is no age-related mortality increase at ages existing in the wild. For some species, especially those where an increase in size leads to greater resistance to predation, an age-related reduction in mortality is observed (called “negative senescence” [12], which is a potentially misleading definition).
Therefore, excluding all the cases mentioned so far, the evidence shows that only a limited number of species show aging as defined above [3]. In other words, if we consider all species, aging is not a universal phenomenon but a rather rare phenomenon. The fact that our species and the animal species that are familiar to us show the phenomenon of aging is the likely origin of the misconception regarding the universality of aging.
A different concept is the inevitability of death for the individuals of any species. For example, in the case of a species with “negligible senescence,” a constant mortality rate at any age does not exclude the risk of death at any age and that all the individuals of that species will die. Furthermore, for such a species it is not possible to exclude that, in artificial conditions, at ages that do not exist in nature there is an increase in mortality in relation to age, because phenomena in any way harmful only at ages non-existent in the wild cannot be countered by natural selection [3].
A.2. Aging as a phenomenon of no selective importance. Some studies stated as clear evidence that aging has little or no importance for mortality in the wild and therefore cannot have importance for natural selection.
For example: “Ageing rarely if ever occurs in feral animals because it is unusual for them to live long enough to experience the phenomenon. The same observation can be made for prehistoric humans. Natural selection could not select for a process like ageing when few, if any, animals ever lived long enough to participate in the selection process” [11]; “… there is scant evidence that senescence contributes significantly to mortality in the wild… As a rule, wild animals simply do not live long enough to grow old. Therefore, natural selection has limited opportunity to exert a direct influence over the process of senescence” [13].
In clear contrast to this opinion, there is ample documentation of an age-related increase in mortality in populations in the wild and of the fact that this mortality increase significantly contributes to the reduction of the average life span [14, 15].
In particular, in the second study it is stated that: “The recent emergence of long-term field studies presents irrefutable evidence that senescence is commonly detected in nature. We found such evidence in 175 different animal species from 340 separate studies” [15].
Furthermore, in the analysis of the life tables of some species, it was highlighted as early as 1988 that the average life span was halved by this increase in mortality (i.e., aging) and that, moreover, excluding individuals who died before reaching maturity, the average duration of subsequent life span was reduced of two thirds [3].
Therefore, for the species whose individuals age, as age-related mortality increase exists in the wild and significantly modifies the mean duration of life, aging cannot be at all without selective importance.
A different concept is that individuals in an advanced stage of aging (for example, a centenarian for the human species) are not present in natural conditions. While this is true, it does not contradict the well-documented fact that the mortality increase in relation to age (which means an age-related decline of functions) is well detectable in natural conditions and that, consequently, at the ages in which the mortality increase exceeds a critical level, survival becomes impossible and no individuals of those ages are found.
It is necessary to avoid “the confusion of the process of senescence with the state of senility” [16], i.e., it is necessary the distinction between age-related decline in survival capacities (i.e., aging) and the ages in which the impairment of the biological decline severely impairs survival capacities. Moreover, the period in which the mortality increases and before the state of senility is certainly subject to natural selection. This was well expressed by Williams: “No one would consider a man in his thirties senile, yet, according to athletic records and life tables, senescence is rampant during this decade. Surely this part of the human life-cycle concerns natural selection” [16].
A.3. Aging as a non-adaptive phenomenon because not justifiable in terms of individual selection. The main argument against the possibility of aging as an adaptive phenomenon is that a gene that causes aging is certainly harmful to the individual and so a gene of this kind cannot be favored by natural selection: “… any hypothetical ‘accelerated ageing gene’ would be disadvantageous to the individual. It is therefore difficult to see how genes for accelerated ageing could be maintained in stable equilibrium, as individuals in whom the genes were inactivated by mutation would enjoy a selection advantage” [13]; “The anomalous nature of ageing as a putative adaptation is that it is bad for the individual in which the process is exhibited. An animal that grows to maturity and thereafter reproduces indefinitely has, other things being equal, a greater Darwinian fitness than one that grows to maturity and then survives and reproduces for only a fixed period of time” [17].
The frequency variation, between one generation and the next, of a gene C, acting on the individual I (Δc), may be expressed by the formula (1):
where: S is the advantage or disadvantage (i.e., greater or smaller fitness) for I caused by C; P is the residual reproductive capacity of I at the age when C acts.
If the value of S is negative, Δc will be negative, i.e., the frequency of C will decrease. Consequently, according to this formula, any gene harmful to the individual in which it acts would be eliminated by natural selection and so aging-causing genes and any phenoptotic phenomena cannot be adaptive.
Let us now consider the supra-individual selection mechanism known as kin selection and based on inclusive fitness [18, 19]. This selective mechanism considers both the individual I1 in which the gene C is present and acts and other n individuals genetically related to I (I2, I3...) for which the action of C has any consequence for their survival capacity. In this case, Δc is described by the formula (2):
where: Sx = advantage or disadvantage for the individual Ix deriving from the action of C; Px = reproductive value of Ix at the age when C acts; rx = coefficient of relationship between Ix and I1.
If the value of the summation is positive, gene C is favored by natural selection and this can also occur in the case in which S1 is negative (that is, in the case that C is disadvantageous for the individual I1 in which it acts).
It should be noted that if C acts only on I1, the formula (2) becomes (1). Therefore, selection at the individual level is only a particular case of a more general way of describing natural selection; and the two formulas do not represent alternative theories.
Moreover, as discussed in [6], §2.2 Supra-individual selection, particular cases of group selection can be described in terms of inclusive fitness by appropriate transformations of (2), and particular population models also allow to explain the existence of genes that are harmful to the individual in which they act. An important deduction is that it is wrong to exclude a priori the possibility that genes harmful to the individual in which they act (e.g., genes determining aging or, in general, phenoptotic phenomena), are favored by natural selection.
B. Weaknesses of the theories maintaining the hypothesis of aging as a non-adaptive phenomenon. Disregarding the validity of the three aforementioned arguments against the possibility that aging is an adaptive phenomenon, there are three traditional theories that somehow try to consider the mechanisms of selection to justify aging and a varied set of theories explaining aging as the gradual accumulation of the effects of harmful agents of various types:
B.1. Mutation Accumulation theory [20, 21]. This theory starts from the fact that over the course of life the number of surviving individuals decreases and therefore a harmful gene that acts at older ages (let’s call it “t-gene” for brevity) is removed by natural selection more weakly than an equivalent gene acting at younger ages. Consequently, the noxious genes acting at greater ages, by the accumulation of their harmful effects, progressively causes the general age-related impairment defined as aging.
Already in 1988 [3], a simple mathematical model was proposed to verify if a strong load of t-genes could cause a progressive age-related increase in mortality, i.e., if it could determine a life table similar to that of an aging species. The model, proposed again in other works (e.g. [6, 22]) and never invalidated by other authors, demonstrated that t-genes do not give rise to life tables of the aforementioned type and that therefore t-genes are not a plausible cause of aging.
However, even accepting that mutation accumulation theory is an admissible hypothesis, it should explain the diversity of aging rates among the various species. It is certainly quite weak to postulate that in species with a lower aging rate the number and effects of t-genes are smaller and that in species with “negligible senescence” t-genes are insufficient to cause detectable aging.
Furthermore, there is another implicit postulate in mutation accumulation theory, namely that the frequency of mutations not eliminated by specific mechanisms cannot be further restrained by natural selection. In contradiction with this thesis:
– There is an inverse correlation between lifespan and somatic mutation rates per year [23] (clearly, considering only the mutations not eliminated by specific mechanisms). Without this inverse relationship, animals with greater longevity should have more cells with a critical level of cancer-causing mutations [24]. In fact, this demonstrates that natural selection is perfectly capable of developing efficient mechanisms to restrain mutations and consequently it appears unfounded to argue that aging is somehow caused by the accumulation of mutations that natural selection cannot oppose.
– Some studies show that individuals with genetic alterations causing higher mutation rates show no signs of accelerated aging [25], “which argues against a straightforward connection between the accumulation of mutations and aging” [24].
B.2. Antagonistic Pleiotropy theory [16, 26]. This theory postulates the existence of genes with beneficial effects in young or adult ages but harmful in later ages. As the advantage in early ages outweighs the disadvantage in later ages (also because in these later ages there are fewer survivors), these pleiotropic genes would be favored by natural selection and so their cumulative effects would determine aging.
By implication, this theory also postulates the non-existence or impossibility of existence of analogous genes that are advantageous for all ages.
Furthermore, if we want to explain the differences between the various species in aging rhythms (i.e., the rates of mortality increase), it appears necessary to postulate that the number and effects of the hypothetical genes with pleiotropic effects are proportional to aging rhythms.
Finally, wanting to explain the species with “negligible senescence,” it appears also necessary to postulate that in such species the aforementioned hypothetical genes are so few and with so limited effects as to justify the non-detectability of senescence.
Unfortunately, apart from a load of postulates that makes the theory difficult to accept as scientific, there is no evidence showing the existence of genes with the aforementioned pleiotropic effects (first postulate) nor any elements supporting the other postulates highlighted.
B.3. Disposable Soma theory [27, 28]. This theory postulates that for the organism there are limited resources, which are not better specified.
Admitting this limit, as the organism must necessarily divide insufficient resources to best meet all needs, natural selection chooses a compromise between the need to promote reproductive capacity and that of continuously repairing physiological systems, i.e., counteracting aging. The partial satisfaction of the needs of the second type progressively compromises the functionality of the organism, i.e., determines aging.
There are other more or less explicit postulates for this theory. The first is the existence of an undefined resource with limited availability. The second is that the limitation of the aforementioned resource forces a choice between reproductive potential and the ability to counteract the decay of functions.
The third is that in the comparison between species, the limitation of resources varies according to their aging rates.
The fourth is that such a limitation of resources does not exist for species with “negligible senescence.”
For this theory, apart from the plurality of postulates that makes it difficult to accept as scientific, there are no works showing the existence of the undefined resource with limited availability or an inverse correlation between reproductive capacity and aging rates.
Furthermore, the caloric restriction, which presumably would further restrict the hypothetical limited resource, should limit the duration of life, but the evidence appears to contrast this prediction (for a discussion see [6], §4.4.5 Effects of caloric restriction on lifespan).
B.4. Damage Accumulation theories [6, 29]. There are many factors that have been proposed to cause progressive damage to organisms and so aging, e.g.: cellular “wear and tear”; mechanochemical deterioration of cellular colloids; changes in specified tissues (nervous/endocrine/vascular/connective); toxic products of intestinal bacteria; accumulation of “metaplasm” or of metabolites; the action of gravity; accumulation of heavy water; increasing entropy; accumulation of chemical alterations due to DNA transcription errors; deleterious effects of oxidation; oxidative effects of free radicals on DNA/mitochondria/whole body; age-related inflammatory phenomena; age-related immunological alterations [6, 29].
For some of these theories, the starting point is a well-known association between older ages and the phenomena proposed as the cause of aging. For example, inflammatory phenomena and immunological alterations are correlated with age and are proposed as a cause of aging [30, 31]. However, the aforementioned association does not allow to exclude that the opposite is true, that is, that aging is the cause of the age-related increase in inflammatory phenomena and immunological alterations.
A similar objection can be formulated for the theories for which aging is caused by oxidation phenomena on the basis of the known association between aging and the accumulation of oxidized products. With regard to the opposite thesis that aging causes the accumulation of oxidized substances, it is opportune to report, among other things, various experimental works in which, by various manipulations, differentiated cells are brought back to the condition of embryonic or stem cells in which there is no accumulation of oxidized substances. For example, the introduction of four factors (Oct3/4, Sox2, c-Myc, and Klf4) transforms (i.e., reprograms) adult fibroblasts into induced pluripotent stem cells which have the cell marker genes and the growth properties of embryonic stem cells [32]; in humans, tetraploid cells showing the functional characteristics of embryonic stem cells were obtained by fusion of somatic cells with embryonic stem cells (“Analysis of genome-wide transcriptional activity, reporter gene activation, allele-specific gene expression, and DNA methylation showed that the somatic genome was reprogrammed to an embryonic state” [33]).
In some cases, the proposed association between aging and a specific cause appears to be contradicted by the evidence. For example, for the hypothesis that aging is influenced by a faster metabolism, considering that the needs of flight require an accelerated metabolism, this thesis is contradicted by the remarkable longevity observed among birds in comparison with mammals (“The maximum life-span in birds… is materially longer than in mammals of comparable size and activity” [29]).
In general, regarding the causal priority of aging or, on the contrary, of cell damage of various types, it is important to remember what Fossel [34] states on the basis of the results of many works: “Cells do not senesce because of wear and tear, but because they permit wear and tear to occur because of an altered pattern of gene expression.” (p. 53) “…cells do not senesce because they are damaged, but permit damage because they senesce” (p. 55).
However, in general, the Damage Accumulation theories do not explain the differences in aging rates as justified by the hypothesized harmful factor, nor do they explain the existence of species with “negligible senescence.”
C. A general argument against non-adaptive theories of aging and in support of the opposite thesis. A factor common to all the theories mentioned in the previous section is that aging is always interpreted as something contrasted by selection because inevitably harmful. Consistent with these theories, if extrinsic mortality (i.e., the basic rate of mortality distinct from the increased rate of mortality due to aging) is high, the strength of natural selection against natural aging acts less effectively and so the theoretical prediction is that aging is earlier and stronger: “The principal determinant in the evolution of longevity is predicted to be the level of extrinsic mortality. If this level is high, life expectancy in the wild is short, the force of selection attenuates fast, deleterious gene effects accumulate at earlier ages, and there is little selection for a high level of somatic maintenance. Consequently, the organism is predicted to be short lived… Conversely, if the level of extrinsic mortality is low, selection is predicted to postpone deleterious gene effects and to direct greater investment in building and maintaining durable soma” [13].
The first theory that, using a precise theoretical model, proposed aging as an adaptive phenomenon in terms of supra-individual selection predicted the exact opposite, namely that, other factors being equal, with higher levels of extrinsic mortality aging would be delayed causing greater lifespan and that the opposite would have occurred with lower levels of extrinsic mortality (“Methuselah effect” [3]) A similar prediction was proposed in other following theoretical models in which aging was proposed as an adaptive phenomenon favored by natural selection at supra-individual level [35-37]. This seemingly paradoxical theoretical prediction can be explained in non-mathematical terms by a simple reasoning. If aging, in terms of natural selection, is positive under certain ecological conditions, but is restrained by the disadvantages caused by a shorter lifespan, for a species there will be an optimal value of the mean duration of life where advantages and disadvantages of aging are in balance. The mean duration of life in a species is the consequence of the combined action of aging (intrinsic mortality) and extrinsic mortality. If extrinsic mortality is higher, intrinsic mortality must be lower to have the aforementioned optimal life span value. On the contrary, if extrinsic mortality is lower, intrinsic mortality must be higher, that is, faster aging is required, if the aforementioned optimal value is to be achieved.
Therefore, we have two opposite theoretical predictions in adaptive and non-adaptive theories of aging. However, in a work based on observations in the wild [14], the inverse correlation between extrinsic mortality and aging rhythms was described, in accordance with the prediction of adaptive theories of aging and in opposition to what was predicted by the non-adaptive hypotheses of aging. This was underlined in other papers [6, 38, 39], but, until today, there is no work proposing an alternative interpretation of the aforementioned data that could be compatible with the non-adaptive theories of aging.
Part II. The existence of aging mechanisms in accordance with the thesis of aging as a programmed phenomenon. Modern science is fundamentally and firmly based on experimental evidence. Even the apparently better theoretical arguments and the theories that seem compatible with the evidence cannot be considered as scientific theories in their own right in absence of clear and indisputable experimental proofs and confirmations. Therefore, although what has been stated up to now could appear to support the thesis of aging as a programmed phenomenon (i.e., adaptive and within the concept of phenoptosis) against the opposite thesis, the first hypothesis must be considered of undefined validity without the support of univocal experimental evidence that confirms it and at the same time falsifies the opposite thesis of aging as a non-programmed phenomenon. It should be emphasized that this concept also applies to the opposite general idea of aging as a non-adaptive phenomenon and to the various distinct theories that fall under this thesis, sometimes presented as a sure valid scientific explanation of ageing (e.g., “The three theories [B.1, B.2, and B.3 in Part I] provide complementary explanations of why ageing occurs” [13]).
In this regard, there is a possible fundamental discriminating element between the theories that interpret aging as a harmful phenomenon insufficiently opposed by selection and the opposing theories that interpret aging as an adaptive phenomenon: for the second thesis, specific mechanisms, genetically determined and regulated, which determine and modulate aging, are absolutely necessary.
The hypothetical existence of such mechanisms, if described, demonstrated and confirmed by valid and various experiments, would be the indispensable and fundamental proof for the thesis of aging as an adaptive phenomenon, i.e., as a particular type of phenoptosis. In fact, the possible existence of mechanisms that somehow progressively compromise survival capacities would be proof of aging as an adaptive and programmed phenomenon. On the contrary, these mechanisms would require rational and valid justifications if the opposite thesis of aging as a non-adaptive phenomenon can still be regarded as plausible.
However, it is possible to describe a complex and sophisticated mechanism that appears to explain how aging is determined. This mechanism, described in two recent works [40, 41], is defined as “subtelomere–telomere theory of aging”. It derives from a previous theory, defined as “telomere theory of aging”, and is the result of decades of work by many researchers, most of whom were not in some way conditioned by the desire to describe the molecular and cellular mechanisms underlying aging as a programmed phenomenon. Indeed, researchers who have greatly contributed, and continue to contribute, to the description of these supposed aging mechanisms are often supporters of the opposite thesis that maintains the inexistence of such mechanisms. For example, Leonard Hayflick, the researcher who first demonstrated the limitations in cellular duplication [42], previously considered as non-existent but which are fundamental for the mechanisms of aging, has also long been a staunch supporter of the thesis of aging as a non-adaptive phenomenon [11].
In this work, instead of repeating unnecessarily all that was stated in the two works cited above [40, 41], the three fundamental phases of the development of the subtelomere–telomere theory will be outlined, together with some main features of the proposed mechanism, in particular those that appear incompatible with the thesis of aging as a non-adaptive and non-programmed phenomenon.
Phase I. Telomere theory. In 1961, a pivotal work showed that normal non-tumor cells were limited in their duplication capacities [42]. Regarding this limit, ten years later Olovnikov observed that DNA duplicating enzyme did not replicate a small terminal part of the molecule: so, DNA was shortened at each duplication and this could be the rational explanation of the limits in the duplication capacities [43].
Shortly thereafter, the same Author pointed out that to justify the greater duplication capacities, or the absence of duplication limits for cell duplication in stem and germline cells, respectively, an enzyme was needed to compensate partially or entirely, after the duplication, for the non-replicated portion of the DNA molecule [44]. This enzyme (afterwards called telomerase) was isolated 12 years later [45].
This series of discoveries led to a hypothesis that seemed to explain aging. The progressive shortening of the terminal parts of the DNA molecules, the telomeres, could justify the progressive cellular alterations, including the triggering of cell senescence (see below), and the consequent impairment of the organism in general, namely aging. However, this theory, which can be defined as “telomere theory of aging”, was contradicted by some experimental facts:
1) In the comparison between various species, there was no correlation between telomere length and longevity [46]. As strong example, humans show much shorter telomeres than mice and hamster but much longer longevity [47]. Furthermore, between the cells of a cloned animal, obtained from the somatic cell of a donor, and the donor cells, the telomere lengths were different in the first germ cell of each organism, but the longevity appeared the same [48, 49]. These and other experimental evidences of the same type (see also [34], pp. 59-61) were against the plausibility of telomere theory.
2) In cell cultures that divided synchronously, cell senescence was not triggered when a certain critical number of duplications was reached (i.e., when a critical telomere shortening was reached), but appeared as a phenomenon of progressively increasing probability correlated with the shortening of the telomere but not determined by critical levels of telomere shortening [50].
3) Cells showing no duplication and turnover in the organism (perennial cells; e.g., most of the neurons), and so without any telomere shortening, aged as other cells with turnover.
Consequently, telomere theory appeared not evaluable as a valid explanation of aging and could at best explain some features of this phenomenon.
Phase II. Subtelomere–telomere theory in its first formulation. An attempt to overcome the deficiencies of the telomere theory was then proposed by assuming a fundamental role of the portion of the DNA molecule adjacent to the telomere, namely the subtelomere [34, 51], and this proposal was defined as the “subtelomere–telomere theory” [6, 40, 52].
This theory is mainly based on two phenomena:
– In yeast, genes inserted in a subtelomeric position are repressed under particular conditions [53];
– Also in yeast, which is a unicellular species where telomerase is always active in wild strains and therefore the telomere does not shorten at each duplication, each cell divides into two cells, one called mother cell and the other daughter cell. In the cells of the mother line, at each duplication there is an accumulation on the subtelomere of particular molecules, extrachromosomal ribosomal DNA circles (ERCs), with progressive inhibition of the subtelomere determining the inhibition of numberless cellular functions and an increasing risk of cell senescence, which in yeast leads to apoptosis, i.e., the death of the individual [54]. Furthermore, in yeast tlc1Δ mutants where telomerase is inactive, in the cells of the daughter line in which there is no accumulation of ERCs, in correlation with telomere shortening at each duplication there are cellular alterations indistinguishable from those of mother lineage cells [55].
These experimental results, and others not reported here for the sake of brevity, led to the proposition that there was “… a heterochromatin ‘hood’ that covers the telomere and a variable length of the subtelomeric chromosome… As the telomere shortens, the hood slides further down the chromosome (the heterochromatin hood remains invariant in size and simply moves with the shortening terminus) …the result is an alteration of transcription from portions of the chromosome immediately adjacent to the telomeric complex, usually causing transcriptional silencing, although the control is doubtless more complex than merely telomere effect through propinquity... These silenced genes may in turn modulate other, more distant genes (or set of genes). There is some direct evidence for such modulation in the subtelomere…” [34], p. 50.
In short, the subtelomere–telomere theory proposed:
(i) the existence of a telomeric hood with a size defined in the first cell of the organism and modeled on the length of the telomere and not on the basis of a predefined length;
(ii) the invariance of the size of the hood at each duplication;
(iii) that this hood, in many multicellular species like ours, in relation to the shortening of the telomere at each duplication (which happens if the telomerase is inactive), slipped on the subtelomere and progressively inhibited more and more particular hypothetical sequences, defined as “r” sequences. This mechanism was also valid in the cells of the daughter line in yeast tlc1Δ mutants, while in the cells of the mother line of wild strains the inhibition of the subtelomere was caused by the progressive accumulation of ERCs;
(iv) that the repression of the hypothetical subtelomeric “r” sequences caused a different modulation of other sequences regulated by them and that this caused a different modulation of many other cellular sequences with progressive general alteration of the cellular functions;
(v) that among these alterations there was also a progressive increasing vulnerability to the activation of cell senescence, a “fundamental cellular program” [56] characterized by many alterations of cellular functions, including cellular secretions (senescence-associated secretory phenotype, SASP), which are harmful to other cells and to the organs and tissues to which the cells belong [57] and are “associated with inflammation and malignancy” [58]. It should be noted that, in yeast, cell senescence induces apoptosis of the cell and so the immediate death of the individual. On the contrary, in species with multicellular individuals, cell senescence causes resistance to apoptosis, but the accumulation of senescent cells reduces the general efficiency of the organism, causing or aggravating conditions of disease and risk of death, so much so that the elimination of senescent cells is a method actively studied to combat various types of disease and even aging [52].
The subtelomere–telomere theory made it possible to overcome the first two of the aforementioned objections for the telomere theory (for the third objection, see below):
– For the first: The hypothesis that the size of the hood was defined in the first cell of the organism and did not change when the telomere shortened implied that telomere length in the first cell was not important while the subsequent telomere shortening at each duplication was critical for the efficiency of the cells and so of the organism. Therefore, a relationship between initial telomere length and longevity was no longer expected and this was in agreement with the evidence of no correlation between these two values.
– For the second: As subtelomere repression was progressive and from this derived a progressive increasing vulnerability to cell senescence, this explained why the activation of cell senescence was not conditioned by a critical telomere length but was related to the progressive shortening of the telomere.
The subtelomere–telomere theory also gave an answer to another possible objection. There are many telomeres in each cell (e.g., for our species, as there are 2 telomeres for each DNA molecule, 2 copies of DNA molecule for each chromosome, and 23 chromosomes, therefore: no. of telomeres = 2 × 2 × 23 = 92) and telomeres do not have the same initial length (i.e., in the first cell of the organism) also among chromosomes of the same cell: “…telomere lengths within the same cell are heterogeneous and certain chromosome arms typically have either short or long telomeres” [59]. Furthermore, the telomere length differs between individuals of the same species and these differences are inherited from the parents [60].
Any mechanism based on the initial absolute length of the telomeres would have led to a different regulation depending on the different lengths and this would have been a probable source of disharmony. On the contrary, assuming a hood that in the first cell of the organism was modeled on the specific length of each telomere, for telomeres of different length the same shortening (in terms of lost bp pairs), would have determined the same degree of subtelomeric repression.
However, the subtelomere–telomere theory, although it appeared rationally coherent and with predictions in agreement with the evidence, had a big vulnerable point. In fact, it hypothesized the existence of particular telomeric sequences, the “r” sequences, of which there were indirect theoretical clues but no direct evidence. Consequently, the theory could be evaluated as possible but was not proven by direct evidence.
Phase III. Subtelomere–telomere theory and TERRA sequences. However, while the “r” sequences were hypothesized without any direct evidence for their existence, sequences with the characteristics hypothesized for them were already an active object of study by brilliant researchers that did not appear in any way to have the purpose of filling the aforementioned gap in the subtelomere–telomere theory.
As early as 1990, two subtelomeric sequences were described, named TelBam3.4 and TelSau2.0 [61], with conserved regions (1.6 kb and 1.3 kb long, respectively) precisely described in [62].
These sequences, defined as TElomeric Repeat-containing RNA (TERRA; here, for brevity, also “T-sequences”): (i) are not coding for protein production; (ii) however, are subject to transcription, producing RNA sequences (here, for brevity, defined as “T-transcripts”); (iii) have been described for our species, but also for mouse, Zebrafish, plants and yeast (for the references, see [40].
About T-sequences:
– In mammals, the transcription, by the action of the enzyme RNA polymerase II, starts from a promoter in the subtelomeric DNA and proceeds toward the telomeric repeated motifs including some of them in the transcription [63-65]. The transcription of T-sequences starts from subtelomeric promoters located on at least two-thirds of chromosome ends [62, 66, 67];
– “The first human subtelomeric promoters that were identified comprise CpG dinucleotide-rich DNA islands shared among multiple chromosome ends... These CpG islands are characterized by the presence of the so-called 61-29-37 repeats located directly upstream of TERRA Transcription Start Site (TSS) and at ~1 kb from the telomeric tract…” [68];
– T-sequences are a general feature in eukaryotic cells and “are emerging as new key players in several important biological processes” [68]. It has been highlighted that “…TERRA is evolutionarily conserved in vertebrates” [69] and this implies that they certainly have a very important function from ancestral times;
– “TERRA read coverage was high within subtelomeric regions of nearly all chromosomes (Chr), most prominently Chr. 2, 9, 13, 18, and the sex chromosomes, with targets being as much as tens of kilobases away from the telomeric repeat... TERRA also bound within internal chromosomal regions and within genes, where it favored introns… TERRA binds chromatin targets throughout the genome… TERRA binds both in cis at telomeres and in trans within or near genes.” [70];
– There are “…significant changes in expression of TERRA targets relative to non-targets after TERRA depletion…, indicating that TERRA target genes were more likely to be affected by TERRA depletion… Interestingly, subtelomeric target genes were consistently downregulated… Internal target genes could either be up- or down-regulated… In the mouse ES [embryonic stem] cell genome, we identified thousands of cis and trans chromatin binding sites” [70];
– T-transcripts bind to many loci outside telomeres where noncoding DNA sequences appear to have important regulatory functions about gene expression [70, 71];
– “The vast majority of TERRA-binding sites were found outside of telomeres, mostly in distal intergenic and intronic regions of the genome where TERRA regulates gene expression” [68].
– Physical exercise was shown to increase TERRA levels in biopsies of skeletal muscle from healthy young subjects and this is in accordance with the idea that physical activity protects against aging [67].
About T-sequences and telomere protection:
– in embryonic stem cells of mice, depletion of T-transcripts is related to reduced telomere protection [70, 71];
– T-sequences knockdown is related to alterations in the capping function and a loss of telomeric integrity was shown after TERRA knockdown [70];
– T-transcripts antagonize ATRX (a particular protein that is related to alpha thalassemia mental retardation X-related syndrome) and are important for telomere protection: “TERRA and ATRX share hundreds of target genes and are functionally antagonistic at these loci: Whereas TERRA activates, ATRX represses gene expression. At telomeres, TERRA competes with telomeric DNA for ATRX binding, suppresses ATRX localization, and ensures telomeric stability” [70];
– the inhibition of T-sequences transcription activates the mechanisms for DNA damage response at telomeres [72];
– the deletion of the 20q locus determines a strong decrease in the TERRA levels and a consequent massive DNA damage response, which appears to be a “demonstration in any organism of the essential role of TERRA in the maintenance of telomeres” [73].
The characteristics of the T-sequences appear to correspond precisely to what subtelomere–telomere theory hypothesizes for the “r” sequences. In fact, they:
– are positioned in the subtelomere;
– are inhibited in relation to the shortening of the telomere with respect to the initial length;
– have transcripts that perform regulatory functions for other regulatory sequences both adjacent and non-adjacent in the DNA molecule in which they are present, but also in other DNA molecules of the same cell;
– influence innumerable cellular functions in various ways;
– are essential for telomere stability and therefore also to reduce the probability of cell senescence activation;
– are widespread and evolutionarily stable and therefore surely perform a function of great importance.
In short, the T-sequences are the “r” sequences transformed from hypothesis into reality. Regarding the functional significance of the T-sequences, for any possible explanation alternative to that expressed by the subtelomere–telomere theory, it would be necessary to provide a justification of why sequences of such great importance for the functioning of the whole cell are in the most vulnerable position to inhibition in case of telomere shortening. On the contrary, for the subtelomere–telomere theory, the T-sequences and the consequences of their vulnerable position are fundamental part of a gradual mechanism of self-destruction of the organism and are completely compatible with the hypothesis of aging as a form of phenoptosis according to the definition proposed by Skulachev [1].
EPIGENETIC MODIFICATIONS IN AGING
About epigenetic modifications in aging, there are a number of facts to consider:
– There is a strong correlation between age and epigenetic modifications of DNA, which also depend on the type of cell and tissue [74, 75]. In particular, among the age-related epigenetic modifications, cytosine-5 methylation within CpG dinucleotides, defined as DNA methylation, is the most studied [76, 77];
– Epigenetic modifications caused by DNA methylation are practically zero for embryonic cells and for induced pluripotent stem cells (iPSCs), while they grow in relation to the number of cellular duplications [76, 77]. It should be emphasized that these epigenetic changes are reversible as shown by the fact that the transformation of adult somatic cells into iPSCs brings these changes back to values practically equal to zero as for embryonic cells [76];
– CpG sequences that show DNA methylation in relation to age [78-81] are limited to specific DNA parts, in particular DNA stretches where CpG nucleotides are about 1 per 10 bp, defined as CpG islands (CGIs). They constitute only 2% of the entire DNA [74], and often coincide with the transcription start sites of genes [82]. The methylation of these CGIs appears correlated with the silencing of the promoters present in them [83], and vice versa the demethylation restores promoter expression [84];
– Age-related DNA methylation of CGIs in some cases is hypomethylation and in others hypermethylation [80, 81, 85, 86];
– For our species, these types of age-related DNA methylation have been proposed as an indicator for assessing age. The most reliable indicator [76] shows a correlation with age equal to 0.96 and an error of 3.6 years;
– Similar epigenetic modifications are documented for mammals in general. In a study on 128 mammal species (with maximum longevity between 3.8 and 211 years and an analogous great variety of adult weight) a similar indicator was proposed with correlation greater than 0.96 and median relative error of less than 3.5% [77];
– In general, CGIs appear evolutionarily conserved to such an extent as to allow the definition of the aforementioned reliable index that is valid for mammals in general [77];
– In addition to DNA methylation, there are other age-related epigenetic changes (e.g., “reduced bulk levels of the core histones, altered patterns of histone posttranslational modifications… replacement of canonical histones with histone variants, and altered noncoding RNA expression” [87], histone methylation, nucleosome remodeling, reduction of heterochromatin, changes in histone marks [88, 89]). However, with regard to these epigenetic changes, no reliable indicator has been proposed such as the two mentioned above.
Regarding the correlation between epigenetic modifications and T-sequences, we have:
– The subtelomeric CGIs “promote transcription of TERRA molecules.” [62];
– “Subtelomeric DNA methylation is… decreased in conjunction with telomere shortening in Terc–/– mice.” [90]
– In mice, subtelomere methylation is related to telomere shortening [91]. “Furthermore, the abrogation of master epigenetic regulators, such as histone methyltransferases and DNA methyltransferases, correlates with loss of telomere-length control, and telomere shortening to a critical length affects the epigenetic status of telomeres and subtelomeres.” [91];
– “Both healthy controls and sarcoidosis patients showed that long telomeres (>9.4 kb) decrease and short telomeres (<4.4 kb) increase with aging, accompanying relative increases of long telomeres with subtelomeric hypermethylation and short telomeres with subtelomeric hypomethylation. This suggested that the aging-related telomere shortening is associated with the surrounding subtelomeric hypomethylation.” [92]
– In humans, for leukocytes “…shorter telomeres are associated with decreased methylation levels of multiple cytosine sites located within 4 Mb of telomeres… significant enrichment of positively associated methylated CpG sites in subtelomeric loci (within 4 Mb of the telomere) (p < 0.01)” [93]. Telomere shortening modifies gene expression and increase gravity and risk of age-related diseases [93].
This indicates that with longer telomeres there is greater demethylation of the T-sequences and with shorter telomeres there is greater methylation of the T-sequences, which should mean less and greater repression of the T-sequences, respectively.
As for the effects that T-sequences (presumably first level regulators) have on other regulatory sequences (presumably second level regulators), an important clue is what is reported (i) for the cells in relation to the number of duplications; and (ii) for the cells in the state of cell senescence where there should be the maximum repression of the T-sequences:
– For mesenchymal stem cells (MSCs) “expansion of MSC has a very consistent impact on DNA-methylation profiles;” “517 CpG sites were consistently differentially methylated in early versus late passages” [94];
– Cell senescence in MSCs is associated with histone marks of aging such as trimethylation at specific targets and DNA methylation in specific CGIs [94];
– In aged MSCs, in some CpG sites there is hypomethylation and in others hypermethylation: “almost one third of the CpG sites reveal age-associated changes on DNA methylation, of which 60% become hypomethylated and 40% hypermethylated upon aging.” [95]
These data indicate that there is a link between repression of T-sequences and a series of epigenetic DNA modifications and that so these epigenetic modifications, certainly age-related, are not primary or correlated with hypothetical random factors but are secondary to the activity more or less repressed of the T-sequences.
Consequently, when aging is rightly described as an epigenetic phenomenon, this should be integrated by highlighting the dependence of this phenomenon on the regulation (i.e., the degree of repression) of the T-sequences.
The fact that aging is an epigenetic phenomenon should not surprise or lead us to believe that this is an exception in the general organization of the organism, or worse still that it is something determined by random factors.
When it was discovered that the synthesis of proteins depended on specific sections of DNA with protein-coding capabilities (defined as genes), it was initially believed that DNA was mainly composed of genes. The subsequent discovery that a very large part of the DNA was not protein-coding even led to the belief that this was “junk” DNA [96]. Subsequently, it was observed that species with enormous difference in the degree of complexity had a similar number of genes (e.g., “The scientific community was astonished that the number of human genes is equal to that of a rather unsophisticated nematode.” [97]) and The Human Genome and ENCODE Projects showed that “the protein-coding potential of the mammalian genome is extremely limited… Although only 2% of the genome is coding, >90% is transcribed. This transcriptional activity largely produces long noncoding RNAs (lncRNA), the functions of which have remained mostly unknown.” [70]
All this implies that most of the “program” that defines the functions and the development of an organism is not in that small part of the DNA encoding protein sequences but in the remaining part of the DNA, which is transcribed but does not encode proteins and which regulates, through mechanisms of repression/activation, up-regulation/down-regulation of non-protein-coding or protein-coding sections of the DNA, every characteristic of an organism both at the cellular level and in the development and functioning of the organism.
Consequently, it is likely that every function of the cell and of the whole organism is usually obtained through epigenetic modifications (i.e., regulations) and it should not surprise that aging is also an effect of epigenetic modifications. It should also be noted that defining aging as an epigenetic phenomenon appears hardly compatible with the interpretation of aging as a result of the casual accumulation of damage of various kinds.
CELL SENESCENCE
A cell in the condition defined as cell senescence is a cell in which a specific cellular program, a “fundamental cellular program” [56], has been activated and not a generic way of naming an old cell. Cell senescence is triggered in normal cells by various factors [56], such as telomere shortening [98], and is characterized by:
i) replicative senescence, i.e., blockage of cell replication capacities [99, 100];
ii) specific alterations of cell functions [100-102], with big transcriptional modifications (“Senescence-related chromatin remodelling leads to profound transcriptional changes” [103]);
iii) specific alterations of extracellular secretions (senescence-associated secretory phenotype, or SASP) [58, 104];
iv) resistance to apoptosis [102, 105].
By identifying senescent cells with the expression of p16Ink4a, the absolute number of these cells and their fraction on the total number of cells increase with age [106, 107], and their number is clearly related to aging manifestations and age-related diseases [108, 109].
The selective elimination of senescent cells improves and reduces manifestations of aging and age-related diseases [109, 110]. So, their selective elimination by appropriate drugs or associations of drugs, defined as senolytics, is an important and actual therapeutic goal [110, 111], a topic deepened in [52].
It is evident that senescent cells are harmful to the individual in which they accumulate and strongly contribute to the manifestations of aging and age-related diseases. For the subtelomere–telomere theory they constitute a fundamental part of the damage resulting from telomere shortening and the consequent subtelomere repression, which, among other things, increase the probability of activation of the cell senescence program. For theories that interpret aging as a non-adaptive and non-programmed phenomenon, it is essential to justify cell senescence in a different way from that of being part of a mechanism of self-destruction (i.e., aging).
The only alternative explanation proposed for cell senescence is that this phenomenon, as it would somehow thwart cell proliferation, would constitute a general defense against cancer [112, 113]. Moreover, considering the damage caused by cell senescence, it has been considered an evolutionary trade-off between its harm and the necessity of opposing cancerous proliferation, i.e., a good example of antagonistic pleiotropy [114].
There are many facts and arguments that counter this proposal (deepened in [52] and [6], §5.4 Limits in cell duplication capacities and other effects of telomere–subtelomere–telomerase system explained as a general defense against cancer), e.g.:
– Cell senescence, as part of the alterations defined as SASP determine the secretion of “myriad factors associated with inflammation and malignancy” [58];
– In mice, the selective elimination of senescent cells determined, in addition to an increased lifespan and fewer age-dependent alterations, also a delay in the progression of cancer [107];
– In humans, various studies have shown a relation between short telomeres and cancer risk [115, 116];
– In normal mice, induced telomerase expression delayed aging and increased longevity but did not increase cancer risk [117];
– In yeast, cell senescence causes immediate apoptosis, i.e., death [118], and this cannot have any anti-cancer significance in a single-celled species;
– An accurate review has shown that, for 175 animal species studied in the wild, there is a progressive age-related increase in mortality that significantly reduces the average life span [15]. However, for no species cancer mortality is documented to significantly influence a mortality increase in the wild. It appears illogical to propose that cell senescence is an effective defense against cancer as it reduces cell duplication capacities at ages when the increase in mortality drastically reduces the number of surviving individuals and cancer still does not significantly affect this number of survivors. This argument was already proposed on the basis of data obtained from the study of a human population in the wild: “This completely disproves the hypothesis that the reduction of cell duplication capacities would be a defense against cancer: it would be like arguing that a defense against a deadly disease has the effect of mass killing before the disease begins to kill” [119];
– However, “If cellular senescence is designed to cut off cancerous cell lines, why would senescent cells remain alive and toxic? They could, instead, be programmed to be good citizens and dismantle themselves via apoptosis to facilitate recycling of proteins and nutrients. The fact that senescent cells emit poisons is completely consonant with the theory that cellular senescence is a form of programmed organismal death” [120].
In short, cell senescence, an important part of the mechanisms of aging in the context of subtelomere–telomere theory and of the general hypothesis of programmed adaptive aging, finds no justification for accepting the idea that aging is a non-adaptive phenomenon.
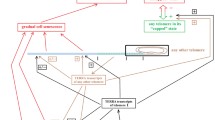
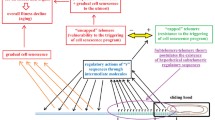
GRADUAL CELL SENESCENCE
The subtelomere–telomere theory maintains that in relation to telomere shortening there is a progressive inhibition of the subtelomere, i.e., of the T-sequences. This causes: (i) a progressive increase in the possibility of activation of cell senescence; and (ii) a progressive alteration of cellular functions, phenomena defined as “gradual cell senescence” that must be distinguished from cell senescence [121].
There is evidence supporting the existence of gradual cell senescence as a phenomenon distinct from cell senescence:
– In vitro, mesenchymal stem cells (MSCs) show gradual changes in mRNA expression with “a consistent pattern of alterations in the global gene expression… These changes are not restricted to later passages, but are continuously acquired with increasing passages.” [122]. Furthermore, in proportion to the number of duplications, MSCs show gradual changes in DNA methylation (hypomethylation in some points and hypermethylation in others) whose magnitude can be used to calculate the number of duplications [123-125];
– In a work about the effects of telomere shortening, the authors state: “Our results demonstrate that the expression of a subset of subtelomeric genes is dependent on the length of telomeres and that widespread changes in gene expression are induced by telomere shortening long before telomeres become rate-limiting for division or before short telomeres initiate DNA damage signaling. These changes include up-regulation and down-regulation of gene expression levels” [126].
The study of gradual cell senescence must avoid the possible confusion due to the overlap, in a culture, between the effects of gradual cell senescence in some cells and those of cell senescence in others. This difficulty is entirely avoided in cultures of yeast, a unicellular organism where the cell senescence of an individual leads to apoptosis, i.e., death [127, 128]. In yeast, in the cells of the mother line of wild strains (in which telomerase is always active and therefore the telomeres do not shorten at each duplication [129]), the repression of the subtelomere is caused by the accumulation of ERCs and, in relation to the number of duplications, increasing functional alterations and susceptibility to cell senescence are observed [127, 128]. Furthermore, in yeast mutant tlc1Δ strains, in which telomerase is inactive, the telomere shortens at each duplication, and the repression of the subtelomere is likely caused by the sliding of the subtelomeric hood on the telomere: in the cells of the daughter line, in which there is no accumulation of ERCs, there is a transcriptome similar to that of the mother line cells with an equal number of previous duplications and so with similar functional alterations [55].
The phenomenon of gradual cell senescence could be interpreted as an effect of random degenerative factors completely outside of a hypothetical aging program. However:
– The functional alterations of MSCs related to the previous number of duplications can be canceled by reprogramming them to induced pluripotent stem cells (iPSCs) [130].
– These iPSCs, regardless of the age of the donor and the source of the cell, showed a rejuvenated profile [130]. Furthermore, for induced MSCs (iMSCs) “…DNA methylation, related to age, was completely erased, and iMSCs reacquired senescence-associated DNA methylation during culture in vitro” [95].
– It is possible to obtain from iPSCs induced MSCs with fewer epigenetic changes and better cellular functions [131].
It should be considered that gradual cell senescence appears to be the consequence of the repression of particular sequences, the T-sequences, in a subtelomeric position that is critically subject to this repression. As gradual cell senescence reduces the functional efficiency of the cell (and of the organism if the cell is part of multicellular organism), the phenomenon is not justifiable in terms of individual selection and it seems necessary to hypothesize its advantage in terms of supra-individual selection in the context of a more general phenoptotic program.
LIMITS IN CELL DUPLICATION AND ATROPHIC SYNDROME
Somatic cells are largely subject to cell turnover. In fact, cells that continuously die from necrosis (caused by traumatic events, infection, or inflammation) and from apoptosis or other types of programmed cell death ([6], §6.1.2 Alterations of cell turnover), are continuously replaced. For some cell types the turnover is very slow (“…bone …has a turnover time of about ten years in humans…” [132]; if “dying myocytes were not constantly replaced, the entire organ would disappear in ≈4.5 years” [133]) while for other cell types the turnover is very fast (e.g., in “the intestinal epithelium …cells are replaced every three to six days” [132]).
For some types of cells (most types of neurons, including retina photoreceptors) there is no turnover and they are defined as perennial cells, but they depend for their vitality on other cells that are subject to turnover [134].
One might believe that, in particular for cell types with fast turnover, the progressive shortening of telomeres is the critical element which could be at the basis of aging. However, some studies have revealed a more complex situation which is well explainable:
– A review [135] investigated in humans the reduction of telomere length from newborns to centenarians. For some tissues with cell types showing no or very low turnover (e.g., cerebral tissue and cardiac muscle), the reduction in telomere length, measured in base pairs (bp) lost per year, was undetectable;
– The same review highlighted that: (i) for some cell types the reduction of telomere length was critical (e.g., for hepatocytes there was a shortening rate of 120 bp/year and the mean telomere length decreased from 13.7 ± 2.5 kbp in newborns to 8.7 ± 1.4 in centenarians); and (ii) for many tissues the reduction rates per year had intermediate values, mostly within 20-60 bp per year;
– Another study showed that in four types of cells or tissues with quite different rates of cell turnover (skeletal muscle, leukocytes, skin, and subcutaneous fat), the rates of telomere shortening were similar [136];
– Telomere lengths appeared to be similar in different tissues and organs of the human fetus [137];
– Telomere lengths in stem cells of hematic cell types showing high turnover are shorter than that of stem cells of other cell types with low turnover [136].
These results allowed to hypothesize that, in the growth period, the stem progenitor cells for each cell type undergo an expansion (i.e., proliferation) that is proportional to the subsequent rhythms of cell turnover (e.g., modest proliferation for stem cells of skeletal muscle that shows slow turnover and massive expansion for stem cells of hematopoietic cells that show high turnover). Telomere length of each type of stem cells was reduced in proportion to the degree of expansion, while in later life stages, telomere lengths were reduced with similar rhythms for different cell types [136].
Moreover, mesenchymal stem cells, in proportion to their expansion, i.e., the number of duplications, showed consistent epigenetic changes [94];
These facts, which certainly involve a set of sophisticated regulations, together with what has been outlined for cell senescence and gradual cell senescence, allow us to provide a general interpretation of aging:
– As telomerase in stem cells is not perfectly active as in embryonic cells, there is always a certain probability that a stem cell passes to the condition of cell senescence in which there is the block of duplication capacities [50]. Consequently, with the progressive reduction of the number of stem cells capable of duplicating themselves, the cell renewal capacities are gradually slowed down and reduced;
– Apart from the decline in cell turnover capacity due to the reduction of the number of stem cells, for the various cell types there is a progressive increase in the number of cells in which (i) for some of them the partial shortening of telomeres causes the alterations of the gradual cell senescence; and (ii) for others, the activation of cell senescence determines altered cellular functions and abnormal secretions (SASP), which affect the functioning of other cells. (For these reasons, the goal of selectively eliminating cells altered by cell senescence to counteract some manifestations of aging and age-related diseases has been proposed and is the subject of multiple studies [52]);
– As regards perennial cells (most of the neurons, retina photoreceptors included, and eye lens fibers) they depend on specialized cells subject to turnover (particular types of gliocytes including retina pigmented cells, and lens epithelium cells, respectively). These trophic cells are subject to the aging phenomena indicated for other cells with turnover and their decline determines the aging of perennial cells [134, 138, 139].
These phenomena cause a progressive alteration of all tissues and organs that has been described as “atrophic syndrome” and is characterized by:
“a) reduced mean cell duplication capacity and slackened cell turnover;
b) reduced number of cells (atrophy);
c) substitution of missing specific cells with nonspecific cells;
d) hypertrophy of the remaining specific cells;
e) altered functions of cells with shortened telomeres or definitively in noncycling state;
f) alterations of the surrounding milieu and of the cells depending on the functionality of the senescent or missing cells;
g) vulnerability to cancer because of dysfunctional telomere-induced instability…” [140].
THE FIXED SIZE OF THE TELOMERIC HETEROCHROMATIN HOOD
The telomeric heterochromatin hood is discussed in [41], in particular highlighting the characteristics required by the subtelomere–telomere theory for the telomeric hood and its function, its possible structure (RAP1, TIN2, TRF1, TRF2, TPP1, and POT1 proteins that form a chain of shelterin complexes) and the questions that still need an explanation.
It is important to underline an essential point. The subtelomere–telomere theory requires that the cap must have for each telomere a size determined in the first cell of the organism according to the length of the telomere and that this size must not vary in subsequent cell duplications even when the telomere is shortened. If the theory is true, a possible prediction is that the cellular amount of shelterin proteins should not be related to the total length of the telomeres but should be constant. On the contrary, if the size of the hood shrinks, there should be a reduction in the amount of shelterin proteins related to the shortening of the telomeres.
However, the abundance and stoichiometry of the proteins that constitute the shelterin complexes were shown to be “similar in primary and transformed cells and… not correlated with telomere length” [141].
CONCLUSION
Among living beings, the term of life is usually determined or influenced by one of various well-known forms of programmed death [7], subsequently included under the unifying necessary neologism and concept of phenoptosis [1].
Exceptions to this rule are organisms that divide into two completely equal offspring individuals (e.g., bacteria) and species that show no increase in age-related mortality (species with “Negligible Senescence” [7]).
In this general context, we should not be surprised that aging, manifested in our species and in many other species familiar to us, is a further form of phenoptosis. Moreover, the existence of species with negligible senescence demonstrates that aging is not an obligatory and common fate for all species, even when there are related species where aging is common [7].
The acceptance of the idea that aging can be a form of phenoptosis immediately leads to the objection of the need to demonstrate the existence of specific, genetically determined and modulated mechanisms that cause aging. The above shows that the existence of such mechanisms appears sufficiently documented to make this hypothesis very plausible and worthy of further investigation. Also, for the opposite thesis that aging is the cumulative progressive effect of heterogeneous damages, various inconsistencies of this thesis and some characteristics of the phenomena proposed as part of the aging mechanisms should be justified in a valid way.
However, in general, the resistance to the idea of aging as an adaptive and programmed phenomenon and the tenacious defense of the opposite hypothesis, even underestimating arguments and empirical data that highlight various inconsistencies, has deeper roots.
Modern biology is firmly based on Darwinism, that is, evolution by natural selection and all that follows from it. Darwinism in its original (but not exclusive!) form proposed by Charles Darwin is based on individual selection. Theoretical arguments and empirical data strongly lead us to believe that Darwinism must be extended to consider also mechanisms of supra-individual selection, which was not excluded by Darwin himself.
If we accept this idea, overcoming an unduly narrower conception of Darwinism, many things become more easily explained. In this context, the concept of phenoptosis, of which aging is a form, appears completely natural and represents a pivotal signal of innovation in the context of Darwinism.
References
Skulachev, V. P. (1997) Aging is a specific biological function rather than the result of a disorder in complex living systems: biochemical evidence in support of Weismann’s hypothesis, Biochemistry (Moscow), 62, 1191-1195.
Skulachev, V. P. (1999) Phenoptosis: programmed death of an organism, Biochemistry (Moscow), 64, 1418-1426.
Libertini, G. (1988) An adaptive theory of the increasing mortality with increasing chronological age in populations in the wild, J. Theor. Biol., 132, 145-162, https://doi.org/10.1016/s0022-5193(88)80153-x.
Skulachev, V. P. (2002) Programmed death phenomena: from organelle to organism, Ann. N. Y. Acad. Sci., 959, 214-237, https://doi.org/10.1111/j.1749-6632.2002.tb02095.x.
Libertini, G. (2012) Classification of phenoptotic phenomena, Biochemistry (Moscow), 77, 707-715, https://doi.org/10.1134/S0006297912070024.
Libertini, G., Corbi, G., Conti, V., Shubernetskaya, O., and Ferrara, N. (2021) Evolutionary Gerontology and Geriatrics – Why and How We Age, in Advances in Studies of Aging and Health, 2, Switzerland, Springer, https://doi.org/10.1007/978-3-030-73774-0.
Finch, C. E. (1990) Longevity, Senescence, and the Genome, University of Chicago Press, Chicago.
Darwin, C. R. (1869) Origin of Species, 5th Edn., John Murray, London.
Darwin, C. R. (1871) The Descent of Man, and Selection in Relation to Sex, John Murray, London.
Kirkwood, T. B. (2005) Understanding the odd science of aging, Cell, 120, P437-P447, https://doi.org/10.1016/j.cell.2005.01.027.
Hayflick, L. (2000) The future of ageing, Nature, 408, 267-269, https://doi.org/10.1038/35041709.
Vaupel, J. W., Baudisch, A., Dölling, M., Roach, D. A., and Gampe, J. (2004) The case for negative senescence, Theor. Popul. Biol., 65, 339-351, https://doi.org/10.1016/j.tpb.2003.12.003.
Kirkwood, T. B., and Austad, S. N. (2000) Why do we age? Nature, 408, 233-238, https://doi.org/10.1038/35041682.
Ricklefs, R. E. (1998) Evolutionary theories of aging: confirmation of a fundamental prediction, with implications for the genetic basis and evolution of life span, Am. Nat., 152, 24-44, https://doi.org/10.1086/286147.
Nussey, D. H., Froy, H., Lemaitre, J. F., Gaillard, J. M., and Austad, S. N. (2013) Senescence in natural populations of animals: widespread evidence and its implications for bio-gerontology, Ageing Res. Rev., 12, 214-225, https://doi.org/10.1016/j.arr.2012.07.004.
Williams, G. C. (1957) Pleiotropy, natural selection and the evolution of senescence, Evolution, 11, 398-411, https://doi.org/10.2307/2406060.
Kirkwood, T. B., and Melov, S. (2011) On the programmed/non-programmed nature of ageing within the life history, Curr. Biol., 21, R701-707, https://doi.org/10.1016/j.cub.2011.07.020.
Hamilton, W. D. (1964) The genetical evolution of social behaviour. II, J. Theor. Biol., 7, 1-52, https://doi.org/10.1016/0022-5193(64)90039-6.
Trivers, R. L. (1971) The evolution of reciprocal altruism, Quart. Rev. Biol., 46, 35-57, https://doi.org/10.1086/406755.
Medawar, P. B. (1952) An Unsolved Problem in Biology, H. K. Lewis, London. Reprinted in The Uniqueness of the Individual (Medawar, P. B., 1957) Methuen, London.
Mueller, L. D. (1987) Evolution of accelerated senescence in laboratory populations of Drosophila, Proc. Natl. Acad. Sci. USA, 84, 1974-1977, https://doi.org/10.1073/pnas.84.7.1974.
Libertini, G., Rengo, G., and Ferrara, N. (2017) Aging and aging theories, J. Geront. Geriatr., 65, 59-77.
Cagan, A., Baez-Ortega, A., Brzozowska, N., Abascal, F., Coorens, T. H. H., et al. (2022) Somatic mutation rates scale with lifespan across mammals, Nature, 604, 517-524, https://doi.org/10.1038/s41586-022-04618-z.
Gorelick, A. N., and Naxerova, K. (2022) Mutational clocks tick differently across species, Nature, 604, 435-436, https://doi.org/10.1038/d41586-022-00976-w.
Robinson, P. S., Coorens, T. H. H., Palles, C., Mitchell, E., Abascal, F., et al. (2021) Increased somatic mutation burdens in normal human cells due to defective DNA polymerases, Nat. Genet., 53, 1434-1442, https://doi.org/10.1038/s41588-021-00930-y.
Rose, M. R. (1991) Evolutionary Biology of Aging, Oxford University Press, Oxford (UK).
Kirkwood, T. B. (1977) Evolution of ageing, Nature, 270, 301-304, https://doi.org/10.1038/270301a0.
Kirkwood, T. B., and Holliday, R. (1979) The evolution of ageing and longevity, Proc. R. Soc. Lond. B Biol. Sci., 205, 531-546, https://doi.org/10.1098/rspb.1979.0083.
Comfort, A. (1979) The Biology of Senescence, Livingstone, London.
Fülöp, T., Witkowski, J. M., Pawelec, G., Alan, C., and Larbi, A. (2014) On the immunological theory of aging, Interdiscip. Top. Gerontol., 39, 163-176, https://doi.org/10.1159/000358904.
Franceschi, C., Garagnani, P., Morsiani, C., Conte, M., Santoro, A., et al. (2018) The continuum of aging and age-related diseases: common mechanisms but different rates, Front. Med. (Lausanne), 5, 61, https://doi.org/10.3389/fmed.2018.00061.
Takahashi, K., and Yamanaka, S. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors, Cell, 126, 663-676, https://doi.org/10.1016/j.cell.2006.07.024.
Cowan, C. A., Atienza, J., Melton, D. A., and Eggan, K. (2005) Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells, Science, 309, 1369-1373, https://doi.org/10.1126/science.1116447.
Fossel, M. B. (2004) Cells, Aging and Human Disease, Oxford University Press, New York.
Travis, J. M. (2004) The evolution of programmed death in a spatially structured population, J. Gerontol. A Biol. Sci. Med. Sci., 59, 301-305, https://doi.org/10.1093/gerona/59.4.b301.
Martins, A. C. (2011) Change and aging senescence as an adaptation, PLoS One, 6, e24328, https://doi.org/10.1371/journal.pone.0024328.
Mitteldorf, J., and Martins, A. C. (2014) Programmed life span in the context of evolvability, Am. Nat., 184, 289-302, https://doi.org/10.1086/677387.
Libertini, G. (2006) Evolutionary explanations of the “actuarial senescence in the wild” and of the “state of senility”, ScientificWorldJournal, 6, 1086-1108, https://doi.org/10.1100/tsw.2006.209.
Libertini, G. (2008) Empirical evidence for various evolutionary hypotheses on species demonstrating increasing mortality with increasing chronological age in the wild, ScientificWorldJournal, 8, 182-193, https://doi.org/10.1100/tsw.2008.36.
Libertini, G., Corbi, G., and Ferrara, N. (2020) Importance and meaning of TERRA sequences for aging mechanisms, Biochemistry (Moscow), 85, 1505-1517, https://doi.org/10.1134/S0006297920120044.
Libertini, G., Shubernetskaya, O., Corbi, G., and Ferrara, N. (2021) Is evidence supporting the subtelomere–telomere theory of aging? Biochemistry (Moscow), 86, 1766-1781, https://doi.org/10.1134/S0006297921120026.
Hayflick, L., and Moorhead, P. S. (1961) The serial cultivation of human diploid cell strains, Exp. Cell Res., 25, 585-621, https://doi.org/10.1016/0014-4827(61)90192-6.
Olovnikov, A. M. (1971) Principle of marginotomy in template synthesis of polynucleotides, Doklady Biochem., 201, 394-397.
Olovnikov, A. M. (1973) A theory of marginotomy: the incomplete copying of template margin in enzyme synthesis of polynucleotides and biological significance of the problem, J. Theor. Biol., 41, 181-190, https://doi.org/10.1016/0022-5193(73)90198-7.
Greider, C. W., and Blackburn, E. H. (1985) Identification of a specific telomere terminal transferase activity in Tetrahymena extracts, Cell, 43 (2 Pt 1), 405-413, https://doi.org/10.1016/0092-8674(85)90170-9.
Gorbunova, V., Bozzella, M. J., and Seluanov, A. (2008) Rodents for comparative aging studies: from mice to beavers, Age (Dordr.), 30, 111-119, https://doi.org/10.1007/s11357-008-9053-4.
Slijepcevic, P., and Hande, M. P. (1999) Chinese hamster telomeres are comparable in size to mouse telomeres, Cytogenet. Cell Genet., 85, 196-199, https://doi.org/10.1159/000015292.
Kubota, C., Yamakuchi, H., Todoroki, J., Mizoshita, K., Tabara, N., et al. (2000) Six cloned calves produced from adult fibroblast cells after long-term culture, Proc. Natl. Acad. Sci. USA, 97, 990-995, https://doi.org/10.1073/pnas.97.3.990.
Lanza, R. P., Cibelli, J. B., Faber, D., Sweeney, R. W., Henderson, B., et al. (2001) Cloned cattle can be healthy and normal, Science, 294, 1893-1894, https://doi.org/10.1126/science.1063440.
Blackburn, E. H. (2000) Telomere states and cell fates, Nature, 408, 53-56, https://doi.org/10.1038/35040500.
Libertini, G., and Ferrara, N. (2016) Possible interventions to modify aging, Biochemistry (Moscow), 81, 1413-1428, https://doi.org/10.1134/S0006297916120038.
Libertini, G., Ferrara, N., Rengo, G., and Corbi, G. (2018) Elimination of senescent cells: prospects according to the subtelomere–telomere theory, Biochemistry (Moscow), 83, 1477-1488, https://doi.org/10.1134/S0006297918120064.
Gottschling, D. E., Aparicio, O. M., Billington, B. L., and Zakian, V. A. (1990) Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription, Cell, 63, 751-762, https://doi.org/10.1016/0092-8674(90)90141-z.
Sinclair, D. A., and Guarente, L. (1997) Extrachromosomal rDNA circles – a cause of aging in yeast, Cell, 91, 1033-1042, https://doi.org/10.1016/s0092-8674(00)80493-6.
Lesur, I., and Campbell, J. L. (2004) The transcriptome of prematurely aging yeast cells is similar to that of telomerase-deficient cells, Mol. Biol. Cell, 15, 1297-1312, https://doi.org/10.1091/mbc.e03-10-0742.
Ben-Porath, I., and Weinberg, R. (2005) The signals and pathways activating cellular senescence, Int. J. Biochem. Cell Biol., 37, 961-976, https://doi.org/10.1016/j.biocel.2004.10.013.
Campisi, J., and d’Adda di Fagagna, F. (2007) Cellular senescence: when bad things happen to good cells, Nat. Rev. Mol. Cell. Biol., 8, 729-740, https://doi.org/10.1038/nrm2233.
Coppé, J. -P., Patil, C. K., Rodier, F., Sun, Y., Muñoz, D. P., et al. (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor, PLoS Biol., 6, 2853-2868, https://doi.org/10.1371/journal.pbio.0060301.
Londoño-Vallejo, J. A., DerSarkissian, H., Cazes, L., and Thomas, G. (2001) Differences in telomere length between homologous chromosomes in humans, Nucleic Acids Res., 29, 3164-3171, https://doi.org/10.1093/nar/29.15.3164.
Hjelmborg, J. B., Dalgård, C., Möller, S., Steenstrup, T., Kimura, M., et al. (2015) The heritability of leucocyte telomere length dynamics, J. Med. Genet., 52, 297-302, https://doi.org/10.1136/jmedgenet-2014-102736.
Brown, W. R., MacKinnon, P. J., Villasanté, A., Spurr, N., Buckle, V. J., et al. (1990) Structure and polymorphism of human telomere-associated DNA, Cell, 63, 119-132, https://doi.org/10.1016/0092-8674(90)90293-n.
Nergadze, S. G., Farnung, B. O., Wischnewski, H., Khoriauli, L., Vitelli, V., et al. (2009) CpG-island promoters drive transcription of human telomeres, RNA, 15, 2186-2194, https://doi.org/10.1261/rna.1748309.
Azzalin, C. M., Reichenbach, P., Khoriauli, L., Giulotto, E., and Lingner, J. (2007) Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends, Science, 318, 798-801, https://doi.org/10.1126/science.1147182.
Schoeftner, S., and Blasco, M. A. (2008) Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II, Nat. Cell Biol., 10, 228-236, https://doi.org/10.1038/ncb1685.
Feuerhahn, S., Iglesias, N., Panza, A., Porro, A., and Lingner, J. (2010) TERRA biogenesis, turnover and implications for function, FEBS Lett., 584, 3812-3818, https://doi.org/10.1016/j.febslet.2010.07.032.
Porro, A., Feuerhahn, S., Delafontaine, J., Riethman, H., Rougemont, J., et al. (2014) Functional characterization of the TERRA transcriptome at damaged telomeres, Nat. Commun., 5, 5379, https://doi.org/10.1038/ncomms6379.
Diman, A., Boros, J., Poulain, F., Rodriguez, J., Purnelle, M., et al. (2016) Nuclear respiratory factor 1 and endurance exercise promote human telomere transcription, Sci. Adv., 2, e1600031, https://doi.org/10.1126/sciadv.1600031.
Diman, A., and Decottignies, A. (2018) Genomic origin and nuclear localization of TERRA telomeric repeat-containing RNA: from Darkness to Dawn, FEBS J., 285, 1389-1398, https://doi.org/10.1111/febs.14363.
Azzalin, C. M., and Lingner, J. (2008) Telomeres: the silence is broken, Cell Cycle, 7, 1161-1165, https://doi.org/10.4161/cc.7.9.5836.
Chu, H. -P., Cifuentes-Rojas, C., Kesner, B., Aeby, E., Lee, H.-G., et al. (2017) TERRA RNA antagonizes ATRX and protects telomeres, Cell, 170, 86-101, https://doi.org/10.1016/j.cell.2017.06.017.
Chu, H. -P., Froberg, J. E., Kesner, B., Oh, H. J., Ji, F., et al. (2017) PAR-TERRA directs homologous sex chromosome pairing, Nat. Struct. Mol. Biol., 24, 620-631, https://doi.org/10.1038/nsmb.3432.
Bettin, N., Oss Pegorar, C., and Cusanelli, E. (2019) The emerging roles of TERRA in telomere maintenance and genome stability, Cells, 8, 246, https://doi.org/10.3390/cells8030246.
Montero, J. J., Lopez de Silanes, I., Grana, O., and Blasco, M. A. (2016) Telomeric RNAs are essential to maintain telomeres, Nat. Commun., 7, 12534, https://doi.org/10.1038/ncomms12534.
Illingworth, R., Kerr, A., Desousa, D., Jørgensen, H., Ellis, P., et al. (2008) A novel CpG island set identifies tissue-specific methylation at developmental gene loci, PLoS Biol., 6, e22, https://doi.org/10.1371/journal.pbio.0060022.
Bernstein, B. E., Stamatoyannopoulos, J. A., Costello, J. F., Ren, B., Milosavljevic, A., et al. (2010) The NIH roadmap epigenomics mapping consortium, Nat. Biotechnol., 28, 1045-1048, https://doi.org/10.1038/nbt1010-1045.
Horvath, S. (2013) DNA methylation age of human tissues and cell types, Genome Biol., 14, 3156, doi: https://doi.org/10.1186/gb-2013-14-10-r115. Erratum in: Horvath, S. (2015) Erratum to: DNA methylation age of human tissues and cell types, Genome Biol., 16, 96, doi: 10.1186/s13059-015-0649-6.
Mammalian Methylation Consortium (2021) Universal DNA methylation age across mammalian tissues, bioRxiv, https://doi.org/10.1101/2021.01.18.426733.
Rakyan, V. K., Down, T. A., Maslau, S., Andrew, T., Yang, T. P., et al. (2010) Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains, Genome Res., 20, 434-439, https://doi.org/10.1101/gr.103101.109.
Teschendorff, A. E., Menon, U., Gentry-Maharaj, A., Ramus, S. J., Weisenberger, D. J., et al. (2010) Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer, Genome Res., 20, 440-446, https://doi.org/10.1101/gr.103606.109.
Horvath, S., Zhang, Y., Langfelder, P., Kahn, R., Boks, M., et al. (2012) Aging effects on DNA methylation modules in human brain and blood tissue, Genome Biol., 13, R97, https://doi.org/10.1186/gb-2012-13-10-r97.
Bell, J. T., Tsai, P. C., Yang, T. P., Pidsley, R., Nisbet, J., et al. (2012) Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population, PLoS Genet., 8, e1002629, https://doi.org/10.1371/journal.gen.1002629.
Bird, A. (2002) DNA methylation patterns and epigenetic memory, Genes Dev., 16, 6-21, https://doi.org/10.1101/gad.947102.
Stein, R., Razin, A., and Cedar, H. (1982) In vitro methylation of the hamster adenine phosphoribosyltransferase gene inhibits its expression in mouse L cells, Proc. Natl. Acad. Sci. USA, 79, 3418-3422, https://doi.org/10.1073/pnas.79.11.3418.
Hansen, R. S., and Gartler, S. M. (1990) 5-Azacytidine-induced reactivation of the human X chromosome-linked PGK1 gene is associated with a large region of cytosine demethylation in the 5′ CpG island, Proc. Natl. Acad. Sci. USA, 87, 4174-4178, https://doi.org/10.1073/pnas.87.11.4174.
Bollati, V., Schwartz, J., Wright, R., Litonjua, A., Tarantini, L., et al. (2009) Decline in genomic DNA methylation through aging in a cohort of elderly subjects, Mech. Ageing Dev., 130, 234-239, https://doi.org/10.1016/j.mad.2008.12.003.
Christensen, B. C., Houseman, E. A., Marsit, C. J., Zheng, S., Wrensch, M. R., et al. (2009) Aging and environmental exposures alter tissue specific DNA methylation dependent upon CpG island context, PLoS Genet., 5, e1000602, https://doi.org/10.1371/journal.pgen.1000602.
Pal, S., and Tyler, J. K. (2016) Epigenetics and aging, Sci. Adv., 2, e1600584, https://doi.org/10.1126/sciadv.1600584.
Greer, E. L., and Shi, Y. (2012) Histone methylation: a dynamic mark in health, disease and inheritance, Nat. Rev. Genet., 13, 343-357, https://doi.org/10.1038/nrg31730.
Booth, L. N., and Brunet, A. (2016) The aging epigenome, Mol. Cell, 62, 728-744, https://doi.org/10.1016/j.molcel.2016.05.013.
Benetti, R., García-Cao, M., and Blasco, M. A. (2007) Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres, Nat. Genet., 39, 243-250, https://doi.org/10.1038/ng1952.
Blasco, M. A. (2007) The epigenetic regulation of mammalian telomeres, Nat. Rev. Genet., 8, 299-309, https://doi.org/10.1038/nrg2047.
Maeda, T., Guan, J. Z., Higuchi, Y., Oyama, J., and Makino, N. (2009) Aging-related alterations of subtelomeric methylation in sarcoidosis patients, J. Gerontol. A Biol. Sci. Med. Sci., 64, 752-760, https://doi.org/10.1093/gerona/glp049.
Buxton, J. L., Suderman, M., Pappas, J. J., Borghol, N., McArdle, W., et al. (2014) Human leukocyte telomere length is associated with DNA methylation levels in multiple subtelomeric and imprinted loci, Sci. Rep., 4, 4954, https://doi.org/10.1038/srep04954.
Schellenberg, A., Lin, Q., Schüler, H., Koch, C. M., Joussen, S., et al. (2011) Replicative senescence of mesenchymal stem cells causes DNA-methylation changes which correlate with repressive histone marks, Aging (Albany NY), 3, 873-888, https://doi.org/10.18632/aging.100391.
Zhou, X., Hong, Y., Zhang, H., and Li, X. (2020) Mesenchymal stem cell senescence and rejuvenation: current status and challenges, Front. Cell Dev. Biol., 8, 364, https://doi.org/10.3389/fcell.2020.00364.
Ohno, S. (1972) So much “junk” DNA in our genome, in Evolution of Genetic Systems (Smith, H. H., ed.) Gordon and Breach, New York, pp. 366-370.
Moraes, F., and Góes, A. (2016) A decade of human genome project conclusion: Scientific diffusion about our genome knowledge, Biochem. Mol. Biol. Educ., 44, 215-223, https://doi.org/10.1002/bmb.20952.
D’Adda di Fagagna, F., Reaper, P. M., Clay-Farrace, L., Fiegler, H., Carr, P., et al. (2003) A DNA damage checkpoint response in telomere-initiated senescence, Nature, 426, 194-198, https://doi.org/10.1038/nature02118.
Cristofalo, V. J., and Pignolo, R. J. (1993) Replicative senescence of human fibroblast-like cells in culture, Physiol. Rev., 73, 617-638, https://doi.org/10.1152/physrev.1993.73.3.617.
Kwon, S. M., Hong, S. M., Lee, Y. K., Min, S., and Yoon, G. (2019) Metabolic features and regulation in cell senescence, BMB Rep., 52, 5-12, https://doi.org/10.5483/BMBRep.2019.52.1.291.
Shelton, D. N., Chang, E., Whittier, P. S., Choi, D., and Funk, W. D. (1999) Microarray analysis of replicative senescence, Curr. Biol., 9, 939-945, https://doi.org/10.1016/s0960-9822(99)80420-5.
Kirkland, J. L., and Tchkonia, T. (2017) Cellular senescence: a translational perspective, EBioMedicine, 21, 21-28, https://doi.org/10.1016/j.ebiom.2017.04.013.
Van Deursen, J. M. (2014) The role of senescent cells in ageing, Nature, 509, 439-446, https://doi.org/10.1038/nature13193.
Rodier, F., Coppé, J. P., Patil, C. K., Hoeijmakers, W. A., Muñoz, D. P., et al. (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion, Nat. Cell. Biol., 11, 973-979, https://doi.org/10.1038/ncb1909.
Wang, E. (1995) Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved, Cancer Res., 55, 2284-2292.
Krishnamurthy, J., Torrice, C., Ramsey, M. R., Kovalev, G. I., Al-Regaiey, K., et al. (2004) Ink4a/Arf expression is a biomarker of aging, J. Clin. Invest., 114, 1299-1307, https://doi.org/10.1172/JCI22475.
Baker, D. J., Childs, B. G., Durik, M., Wijers, M. E., Sieben, C. J., et al. (2016) Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan, Nature, 530, 184-189, https://doi.org/10.1038/nature16932.
Baker, D. J., Jeganathan, K. B., Cameron, J. D., Thompson, M., Juneja, S., et al. (2004) BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice, Nat. Genet., 36, 744-749, https://doi.org/10.1038/ng1382.
Baker, D. J., Perez-Terzic, C., Jin, F., Pitel, K. S., Niederländer, N. J., et al. (2008) Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency, Nat. Cell. Biol., 10, 825-836, https://doi.org/10.1038/ncb1744.
Chang, J., Wang, Y., Shao, L., Laberge, R.-M., Demaria, M., et al. (2016) Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice, Nat. Med., 22, 78-83, https://doi.org/10.1038/nm.4010.
Fuhrmann-Stroissnigg, H., Ling, Y. Y., Zhao, J., McGowan, S. J., Zhu, Y., et al. (2017) Identification of HSP90 inhibitors as a novel class of senolytics, Nat. Commun., 8, 422, https://doi.org/10.1038/s41467-017-00314-z.
Campisi, J. (2003) Cancer and ageing: rival demons? Nat. Rev. Cancer, 3, 339-349, https://doi.org/10.1038/nrc1073.
Wright, W. E., and Shay, J. W. (2005) Telomere biology in aging and cancer, J. Am. Geriatr. Soc., 53, S292-S294, https://doi.org/10.1111/j.1532-5415.2005.53492.x.
Campisi, J. (2000) Cancer, aging and cellular senescence, In vivo, 14, 183-188.
Wu, X., Amos, C. I., Zhu, Y., Zhao, H., Grossman, B. H., et al. (2003) Telomere dysfunction: a potential cancer predisposition factor, J. Natl. Cancer Inst., 95, 1211-128, https://doi.org/10.1093/jnci/djg011.
Ma, H., Zhou, Z., Wei, S., Liu, Z., Pooley, K. A., et al. (2011) Shortened telomere length is associated with increased risk of cancer: a meta-analysis, PLoS One, 6, e20466, https://doi.org/10.1371/journal.pone.0020466.
Bernardes de Jesus, B., Vera, E., Schneeberger, K., Tejera, A. M., Ayuso, E., et al. (2012) Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer, EMBO Mol. Med., 4, 691-704, https://doi.org/10.1002/emmm.201200245.
Laun, P., Bruschi, C. V., Dickinson, J. R., Rinnerthaler, M., Heeren, G., et al. (2007) Yeast mother cell-specific ageing, genetic (in)stability, and the somatic mutation theory of ageing, Nucleic Acids Res., 35, 7514-7526, https://doi.org/10.1093/nar/gkm919.
Libertini, G. (2013) Evidence for aging theories from the study of a hunter-gatherer people (Ache of Paraguay), Biochemistry (Moscow), 78, 1023-1032, https://doi.org/10.1134/S0006297913090083.
Mitteldorf, J. (2013) Telomere biology: cancer firewall or aging clock? Biochemistry (Moscow), 78, 1054-1060, https://doi.org/10.1134/S0006297913090125.
Libertini, G. (2015) Non-programmed versus programmed aging paradigm, Curr Aging Sci, 8, 56-68, https://doi.org/10.2174/1874609808666150422111623.
Wagner, W., Horn, P., Castoldi, M., Diehlmann, A., Bork, S., et al. (2008) Replicative senescence of mesenchymal stem cells: a continuous and organized process, PLoS One, 3, e2213, https://doi.org/10.1371/journal.pone.0002213.
Koch, C. M. (2012) Monitoring of cellular senescence by DNA-methylation at specific CpG sites, Aging Cell, 11, 366-369, https://doi.org/10.1111/j.1474-9726.2011.00784.x.
Schellenberg, A. (2014) Proof of principle: quality control of therapeutic cell preparations using senescence-associated DNA-methylation changes, BMC Res. Notes, 7, 254, https://doi.org/10.1186/1756-0500-7-254.
Fernandez-Rebollo, E. (2020) Senescence-associated metabolomic phenotype in primary and iPSC-derived mesenchymal stromal cells, Stem Cell Rep., 14, 201-209, https://doi.org/10.1016/j.stemcr.2019.12.012.
Robin, J. D., Ludlow, A. T., Batten, K., Magdinier, F., Stadler, G., et al. (2014) Telomere position effect: regulation of gene expression with progressive telomere shortening over long distances, Genes Dev., 28, 2464-2476, https://doi.org/10.1101/gad.251041.114.
Laun, P., Pichova, A., Madeo, F., Fuchs, J., Ellinger, A., et al. (2001) Aged mother cells of Saccharomyces cerevisiae show markers of oxidative stress and apoptosis, Mol. Microbiol., 39, 1166-1173, https://doi.org/10.1111/j.1365-2958.2001.02317.x.
Herker, E., Jungwirth, H., Lehmann, K. A., Maldener, C., Fröhlich, K. U., et al. (2004) Chronological aging leads to apoptosis in yeast, J. Cell Biol., 164, 501-507, https://doi.org/10.1083/jcb.200310014.
D’Mello, N. P., and Jazwinski, S. M. (1991) Telomere length constancy during aging of Saccharomyces cerevisiae, J. Bacteriol., 173, 6709-6713, https://doi.org/10.1128/jb.173.21.6709-6713.1991.
Spitzhorn, L. S. (2019) Human iPSC-derived MSCs (iMSCs) from aged individuals acquire a rejuvenation signature, Stem Cell Res. Ther., 10, 100, https://doi.org/10.1186/s13287-019-1209-x.
Hynes, K. (2013) Mesenchymal stem cells from iPS cells facilitate periodontal regeneration, J. Dent. Res., 92, 833-839, https://doi.org/10.1177/0022034513498258.
Alberts, B., Bray, D., Hopkin, K., Johnson, A., Lewis, J., et al. (2014) Essential Cell Biology, 4th Edn., Garland Science, New York.
Anversa, P., Kajstura, J., Leri, A., and Bolli, R. (2006) Life and death of cardiac stem cells: a paradigm shift in cardiac biology, Circulation, 113, 1451-1463, https://doi.org/10.1161/CIRCULATIONAHA.105.595181.
Libertini, G., and Ferrara, N. (2016) Aging of perennial cells and organ parts according to the programmed aging paradigm, Age (Dordr.), 38, 35, https://doi.org/10.1007/s11357-016-9895-0.
Takubo, K., Aida, J., Izumiyama-Shimomura, N., Ishikawa, N., Sawabe, M., et al. (2010) Changes of telomere length with aging, Geriatr. Gerontol. Int., 10, S197-S206, https://doi.org/10.1111/j.1447-0594.2010.00605.x.
Daniali, L., Benetos, A., Susser, E., Kark, J. D., Labat, C., et al. (2013) Telomeres shorten at equivalent rates in somatic tissues of adults, Nat. Commun., 4, 1597, https://doi.org/10.1038/ncomms2602.
Okuda, K., Bardeguez, A., Gardner, J. P., Rodriguez, P., Ganesh, V., et al. (2002) Telomere length in the newborn, Pediatr. Res., 52, 377-381, https://doi.org/10.1203/00006450-200209000-00012.
Libertini, G. (2009) The role of telomere-telomerase system in age-related fitness decline, a tameable process, in Telomeres: Function, Shortening and Lengthening (Mancini, L., ed.) Nova Science Publ., New York, pp. 77-132.
Flanary, B. (2009) Telomeres: Function, Shortening, and Lengthening, in Telomeres: Function, Shortening and Lengthening (Mancini, L., ed.) Nova Science Publ. Inc., New York, pp. 379-386.
Libertini, G. (2014) Programmed aging paradigm: how we get old, Biochemistry (Moscow), 79, 1004-1016, https://doi.org/10.1134/S0006297914100034.
Takai, K. K., Hooper, S., Blackwood, S., Gandhi, R., and de Lange, T. (2010) In vivo stoichiometry of shelterin components, J. Biol. Chem., 285, 1457-1467, https://doi.org/10.1074/jbc.M109.038026.
Author information
Authors and Affiliations
Contributions
The first author took the lead in writing the manuscript. All authors discussed the results, provided critical feedback, and contributed to the final manuscript.
Corresponding author
Ethics declarations
The authors declare no conflicts of interest in financial or any other area. This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Libertini, G., Corbi, G., Shubernetskaya, O. et al. Is Human Aging a Form of Phenoptosis?. Biochemistry Moscow 87, 1446–1464 (2022). https://doi.org/10.1134/S0006297922120033
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297922120033