
Abstract
Ion-translocating ATPases and ATP synthases (F-, V-, A-type ATPases, and several P-type ATPases and ABC-transporters) catalyze ATP hydrolysis or ATP synthesis coupled with the ion transport across the membrane. F-, V-, and A-ATPases are protein nanomachines that combine transmembrane transport of protons or sodium ions with ATP synthesis/hydrolysis by means of a rotary mechanism. These enzymes are composed of two multisubunit subcomplexes that rotate relative to each other during catalysis. Rotary ATPases phosphorylate/dephosphorylate nucleotides directly, without the generation of phosphorylated protein intermediates. F-type ATPases are found in chloroplasts, mitochondria, most eubacteria, and in few archaea. V-type ATPases are eukaryotic enzymes present in a variety of cellular membranes, including the plasma membrane, vacuoles, late endosomes, and trans-Golgi cisternae. A-type ATPases are found in archaea and some eubacteria. F- and A-ATPases have two main functions: ATP synthesis powered by the proton motive force (pmf) or, in some prokaryotes, sodium-motive force (smf) and generation of the pmf or smf at the expense of ATP hydrolysis. In prokaryotes, both functions may be vitally important, depending on the environment and the presence of other enzymes capable of pmf or smf generation. In eukaryotes, the primary and the most crucial function of F-ATPases is ATP synthesis. Eukaryotic V-ATPases function exclusively as ATP-dependent proton pumps that generate pmf necessary for the transmembrane transport of ions and metabolites and are vitally important for pH regulation. This review describes the diversity of rotary ion-translocating ATPases from different organisms and compares the structural, functional, and regulatory features of these enzymes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
ATP is one of the most important mobile carriers of metabolically available energy in a living cell. Many enzymes couple the exergonic reaction of ATP hydrolysis with vital energy-dependent biochemical processes. ATP hydrolysis also powers the transmembrane transport of ions and low-molecular-weight substances and provides energy for performing mechanical work (muscle fibers contraction, cilia beating, separation of chromosomes during division, etc.). The ATP/ADP ratio in the cell affects the intracellular concentrations of other nucleotides. Nucleoside diphosphate kinases catalyze the reaction of γ-phosphate transfer from ATP to GDP, CDP, TDP, and UDP. The resulting nucleoside triphosphates provide energy for the synthesis of proteins, nucleic acids, lipids, and some other anabolic biochemical reactions.
Rotary ion-translocating ATPases/ATP synthases are a large group of enzymes that includes both ATP “suppliers” and “consumers”. In some cases, the same enzyme can perform both functions, depending on physiological conditions. Moreover, in vitro ATP synthesis can be catalyzed even by an enzyme that does not perform this reaction in vivo. In this review, the terms ATPase and ATP synthase are used as synonyms, but the latter is used only for the enzymes that synthesize ATP in vivo.
Rotary ion-translocating ATPases are subdivided into three families: F-, V- and A-type enzymes. F-type enzymes were the first rotary ATPases discovered. They have been extensively studied since the middle of the twentieth century, and today, they are the most well-described family. F-type ATPases are found in mitochondria, chloroplasts, most eubacteria, and some archaea (together with A-ATPases). The letter “F” stands for “factor”, as it was suggested in the first papers on the mitochondrial oxidative phosphorylation to indicate protein factors necessary for the ATP synthesis during respiration.
V-ATPases have been discovered much later, in the 1980s, in the vacuoles of eukaryotic cells (hence the letter “V”); the history of this discovery is described in detail in [1]. At about the same time, it was found that ion-translocating ATPases of archaea and of some eubacteria are different from the F-type enzymes and resemble eukaryotic V-ATPases. For this reason, these enzymes are often called V-ATPases, especially those found in eubacteria (e.g., Enterococcus hirae or Thermus thermophilus). However, there are significant differences between eukaryotic V-ATPases and prokaryotic enzymes (see below), and in the early 1990s, it was proposed to classify rotary ion-translocating ATPases of archaea as a separate group of A-ATPases [2]. At present, the classification that assigns all prokaryotic enzymes of this type to the A-ATPase family is accepted by several research groups [3-6]. We find this classification convenient; it avoids unnecessary confusion, and in this review, we use the term “V-ATPases” for eukaryotic enzymes only.
Rotary ion-translocating ATPases have a common evolutionary origin [7] and share a similar structure and catalytic mechanism. This suggests that enzymes of this type had already been present in a common ancestor of bacteria, archaea, and eukaryotes [8]. Rotary ATPases and ATP synthases interconvert the two main biological “energy currencies”, ATP and the transmembrane difference of the electrochemical ion potential (Δμ~H+ or, in the case of some prokaryotes, Δμ~Na+), and play a key role in the membrane energization and regulation of intracellular nucleotide concentrations.
COMMON STRUCTURAL AND FUNCTIONAL FEATURES OF ROTARY ATPases
Subunit composition of rotary ATPases. All rotary ATPases have a similar overall structure and subunit organization. They consist of two subcomplexes: hydrophilic F1/V1/A1, which binds nucleotides and phosphate and catalyzes ATP synthesis and/or hydrolysis, and hydrophobic FO/VO/AO located in the membrane and responsible for ion transport. These two parts are connected by a central stalk and by 1 to 3 peripheral stalks [9].
All rotary ATPases share a set of similar key subunits involved in ATP synthesis/hydrolysis and transmembrane ion transfer. The main functional components of the F1 subcomplex are the α3β3 hexamer that contains the nucleotide-binding sites and the elongated subunit γ, which occupies the central cavity of the hexamer. Subunit γ together with subunit ε (δ in the mitochondrial F1) constitute the central stalk of the enzyme. The γε complex is attached to the ring of the c subunits. The translocation of ions occurs at the interface of the c-ring and the hydrophobic a subunit immersed into the membrane. This process is accompanied by the rotation of the cnγε complex (rotor) relative to the stator part of the enzyme consisting of the a subunit, the α3β3 hexamer, and the peripheral stalk. The rotation of subunit γ inside the α3β3 hexamer is coupled to the conformational changes in the catalytic sites that result in ATP synthesis/hydrolysis.
Homologs of the above-described subunits are found in all known F-ATPases (Fig. 1). The subunit composition of the chloroplast enzyme does not differ significantly from that of the bacterial FOF1 [14]. The central stalk of the mitochondrial ATP synthase contains an additional small subunit, called ε (not related to the bacterial or chloroplast ε). The peripheral stalk of the mitochondrial enzyme does not have much in common with that of bacterial/chloroplast FOF1: it consists of nine or more types of subunits that vary in organisms from different taxonomic groups (see the Table) [11, 15, 16]. Some of the peripheral stalk subunits are involved in the dimerization of the mitochondrial enzyme. Dimerization is apparently a distinctive feature of all mitochondrial F-ATPases. It has been described for enzymes from yeast [17, 18], algae [15, 16], higher plants [19], and, finally, vertebrates [20]. Two FOF1 molecules contact via the membrane regions of the peripheral stalks and form a V-shaped dimer that bends the inner mitochondrial membrane. The dimers are arranged in extended rows [21] located at the rims of mitochondria cristae and are essential for the cristae formation [22, 23]. A more detailed comparison of the structures of F-ATPases from different organisms and organelles can be found in the recent review [5].
A- and V-type ATPases have several structural features that are absent in F-ATPases. Prokaryotic A-ATPases have two peripheral stalks [12], and eukaryotic V-ATPases have three [13]. Each stalk consists of two subunits, E and G, possibly homologous to the δ and b subunits, respectively, of the peripheral stalk of bacterial/chloroplast F-ATPases [7, 24]. The central stalk of A- and V-ATPases includes additional specific subunits involved in the attachment of D subunit, a functional analog of the F-ATPase γ subunit, to the c-ring. V-ATPases contain heterooligomeric c-ring that consists of several copies of the c subunit, one c′′ subunit homologous to c, and, in the yeast enzyme, also a copy of the c′ subunit [13, 25]. The a subunit in V-ATPases (and its homolog I in A-ATPases) has a hydrophilic collar-shaped domain that is absent in F-ATPases. This domain protrudes from the membrane and is involved in the binding of two peripheral stalks. The third peripheral stalk in V-ATPases is attached to the a subunit via a separate C subunit, which is absent in the A-type enzymes [13]. It seems probable that eukaryotic V-ATPases have evolved from an ancient archaeal A-ATPase. The similarity of structural features, subunit composition, and amino acid sequences between V-type and A-type enzymes supports this hypothesis. The structure of A- and V-ATPases has been characterized in detail in several reviews (see [26-28]). A complete list of rotary ATPases subunits is given in the Table below. It should be noted once again that, despite the structural differences described above, all rotary ATPases share the same set of subunits directly involved in the substrate binding and in ion transport. This indicates that the catalytic mechanism of these enzymes is highly conserved and likely has a common evolutionary origin.

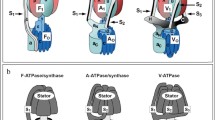
Comparison of structures of rotary ATPases: F-ATPase of thermophilic bacterium Bacillus sp. PS3 (PDB 6N2Y [10]), F-ATPase of porcine (Sus scrofa) mitochondria (PDB 6J5I [11]), A-ATPase of thermophilic bacterium Thermus thermophilus (PDB 6R0W [12]), and rat (Rattus norvegicus) V-ATPase (PDB 6VQ9, 6VQC, and 6VQI [13]). Subunits with proposed structural and/or function homology are shown with the same color. The rotor subunits are outlined with a black thick line; the dotted line shows a part of the rotor hidden behind the stator subunits.
THE CATALYTIC MECHANISM OF F1/A1/V1
In the hydrophilic F1-subcomplex, the α and β subunits (B and A in A1- and V1-subcomplexes) alternate with each other, and the nucleotide-binding sites are located on the interfaces of the αβ pairs. All rotary ATPases have three catalytic sites formed mostly by the residues of the β subunits in F-ATPases [29] and the A subunits in V- and A-ATPases [13, 30] and have an extremely conserved structure [31]. Catalysis also involves a conserved arginine residue (“arginine finger”), which belongs to the α subunit in F-ATPases and to the B subunit in A- and V-ATPases and interacts with the γ-phosphate group of ATP bound in the catalytic site [32, 33].
The substrates of the rotary ATPases are magnesium complexes of nucleotides. In the absence of magnesium, free nucleotides bind to the enzyme, but neither hydrolysis nor synthesis occurs.
In F-ATPases, the α3β3 hexamer contains three additional (noncatalytic) nucleotide-binding sites, which are located mainly on the α subunits. The noncatalytic sites can bind ATP and other purine nucleotides and are probably involved in the regulation of the enzyme activity (see reviews [34, 35]). For A- and V-type ATPases, the very existence of noncatalytic sites is a matter of discussion. In the structures available, the B subunit does not contain a bound nucleotide [12, 13, 36]. At the same time, there are experiments demonstrating the binding of photoaffinity analogs of ATP and ADP to the B subunit of some A-ATPases, so some researchers suggest that this subunit may still bind nucleotides and play a role in the regulation of catalysis [4, 37]. The appearance of the catalytic and noncatalytic subunits in the hexamer is most likely the result of gene duplication in the common ancestor of prokaryotes even before their separation into archaea and eubacteria [38].
During the synthesis/hydrolysis of ATP, the three catalytic sites function cooperatively. At a single point in time, the sites have different affinity for the substrates and reaction products, and the catalytic events at one site trigger conformational transitions in the other two. This idea was first proposed for F-ATPase by Paul Boyer and later formed the basis for the currently accepted model of catalysis, which was called the binding change mechanism [39]. The cooperation between the sites is ensured by the asymmetric subunit of the central stalk (γ in F-ATPases and D in A- and V-ATPases), which rotates inside the catalytic hexamer and sequentially interacts with its subunits. One complete revolution of this subunit is accompanied by the synthesis or hydrolysis of three ATP molecules. Rotation of the γ subunit during catalysis was predicted from the structure of the F1-subcomplex from bovine mitochondria [29] and was then demonstrated directly in single molecules of bacterial F1 [40]. Later, the single-molecule methods for monitoring ATP hydrolysis by the F1 complex have been significantly improved and used to obtain an impressive amount of experimental data that have clarified in detail the molecular mechanism of ATP hydrolysis in F-ATPases (see, for example, reviews [41-43]).
The rotational mechanism of the A1 subcomplex has been studied mostly in the bacterial enzymes from Enterococcus hirae [44] and Thermus thermophilus [45, 46]. It has its own features but obeys the general principles described above. The ATP-dependent rotation of the central stalk has also been shown for eukaryotic V-ATPase [47]. Thus, the fundamental similarity of F-, A-, and V-ATPases is supported not only on the level of structure but also on the functional level by a common mechanism of rotational catalysis.
Another interesting topic is the substrate specificity of rotary ATPases. It has been extensively studied in isolated enzymes [48-50] or their subcomplexes [51-54], mitochondrial membranes [55], chloroplasts [56], vacuoles [57], and plasma membranes of eukaryotic [58] and prokaryotic cells [59, 60]. It seems likely that all rotary ATPases have significant GTPase and ITPase activities that are comparable to the ATPase activity (from 30 to 90% for different enzymes or their subcomplexes), and that GTP or ITP hydrolysis is coupled to the ion transport. The synthesis of GTP and ITP was shown for the F-ATPase from Escherichia coli [59]. Experiments with the F-ATPase from the thermophilic bacterium Bacillus sp. PS3 also demonstrated GTP synthesis and, albeit with very low efficiency, synthesis of UTP and CTP [60]. Thus, it seems that ion-translocating rotary ATPases can directly affect not only the ATP/ADP but also the GTP/GDP ratio in a living cell.
Some enzymes, especially archaeal ATPases, can also catalyze the hydrolysis of pyrimidine nucleoside triphosphates (UTP and/or CTP), but this reaction proceeds much slower [49, 50, 53, 54, 61, 62] and is poorly coupled with proton transport [57]. The UTPase or CTPase activity comparable to the ATPase activity was demonstrated only for few AOA1 complexes [63]. The UTPase activity of bovine V-ATPase was found to be insensitive to bafilomycin, an inhibitor of ion transport through the VO complex, which indicates a loss of coupling between VO and V1 during UTP hydrolysis [58]. A single-molecule study of the F1 subcomplex of the thermophilic bacterium Bacillus sp. PS3 showed that the γ subunit rotates upon hydrolysis of purine but not pyrimidine nucleotides [48]. Thus, rotary ATPases are unlikely to have a notable direct effect on the content of pyrimidine nucleotides in the cell.
ION TRANSPORT BY FO/AO/VO: ION SPECIFICITY AND ION/ATP RATIO
The transmembrane ion transport by rotary ATPases occurs at the interface of the c-ring and subunit a (or, in the case of V-ATPases, the membrane C-terminal domain of subunit a) adjacent to the c-ring. In A-type ATPases, the subunits homologous to c and a are called K (or L) and I, correspondingly.
The outer surface of the c-ring (K- or L-rings in A-ATPases) bears several ion-binding sites that face the lipid bilayer. The ions bind to the carboxyl groups of the aspartate or glutamate residues located in the middle of the outer transmembrane α-helices of the c/K/L subunits. Subunit a (I in A-ATPases) interacts with the c/K/L-ring and forms two aqueous half-channels that open on the opposite sides of the membrane. The half-channels are separated by a conserved arginine residue, which belongs to the a subunit. This arginine is located at the interface of subunit a and the c/K/L-ring and is necessary for the ion transport [64].
The operation principle of rotary ion-translocating complex was proposed by Vladimir P. Skulachev and Alexei N. Glagolev in 1978 to explain the mechanism of bacterial flagellum rotation [65]. Their hypothesis assumed that the ion transfer between the two shifted half-channels occurs when an oligomeric ring-shaped protein complex carrying ion-binding sites rotates in the plane of the membrane. Models describing similar mechanisms of rotational ion transport had been proposed for F-ATPases even before the structure of the FO complex was solved [66, 67] (Fig. 2). The cryo-EM structures of F-, A-, and V-ATPases obtained recently are in good agreement with this model [10, 11, 13, 14, 68, 69].

A scheme of transmembrane proton transport by rotary ATPase during ATP hydrolysis. Protons can reach the ion-binding groups of the c subunits (K or L in A-ATPases) only through the half-channels formed by the a subunit (I in A-ATPases) and the c-ring (K- or L-ring in A-ATPases). Hydrolysis of ATP causes the rotation of the rotor subcomplex, including the c-ring (K- or L-ring in A-ATPases). The highly conserved arginine residue of the a/I subunit at the a-c/K/L-ring interface is positively charged and prevents proton leakage between the half-channels.
Most prokaryotic A- and F-type ATPases, as well as all known F- and V-ATPases of eukaryotes, translocate hydrogen ions. Until the early 1980s, the bioenergetics of ATP synthesis/hydrolysis was generally believed to be associated exclusively with proton transport. In 1984, Vladimir P. Skulachev suggested an idea of sodium bioenergetics, assuming the possibility of ATP synthesis powered by the energy of Δμ~Na+ [70, 71]. This hypothesis was later confirmed by the discovery in some eubacteria and archaea of F- and A-ATPases that couple ATP synthesis/hydrolysis to the transmembrane transport of sodium ions (see reviews [72, 73]). They may function as ATP synthases or ATP-dependent Na+ pumps that control the concentration of sodium in the cytoplasm. In terms of evolution, Na+-translocating rotary ATPases are most likely the ancestors of the H+-translocating ATPases [74]. It seems quite probable that ancient prokaryotes had Na+-translocating ATP synthases that used Δμ~Na+ generated during the synthesis of acetate from carbon dioxide and molecular hydrogen [75].
Na+-translocating ATPases are found in many marine and pathogenic prokaryotes, where they might play an important role in maintaining low intracellular sodium concentration. These enzymes are also found in several species of anaerobic eubacteria and archaea, in which they synthesize ATP using Δμ~Na+ [73]. Some prokaryotes possess a special type of Na+-translocating F-ATPase that presumably works only as an ATP-dependent ion pump and extrudes excessive sodium out of the cell. A distinctive feature of this enzyme is the lack of subunit δ that is compensated by the additional domain of the b subunit. Such F-ATPases were named N-ATPases; it was also found that organisms that have such N-ATPase always have one more rotary proton-translocating ATPase [76].
It should also be noted that some N-ATPases can be actually H+-translocating enzymes. There is experimental evidence suggesting that the N-ATPase of the pathogenic bacterium Burkholderia pseudomallei transports protons, not sodium ions [77].
The ion specificity of rotary ATPases is determined by the primary structure of the c/K/L and a/I subunits. Noteworthy, all rotary ATPases are theoretically capable of translocating protons; for several Na+-dependent ATPases, the proton transport has been demonstrated experimentally at low sodium concentration [78-80]. Under physiological conditions, however, the concentration of sodium ions exceeds the concentration of protons by about six orders of magnitude. Thus, H+-transporting ATPases have developed an extremely high H+/Na+ selectivity during evolution. Apparently, this selectivity is provided by the hydrophobic environment of the ion-binding carboxyl group. In turn, in sodium-transporting enzymes, the proton selectivity is lower because of the polar groups at the ion-binding site. These polar groups belong to the protein backbone or side chains of glutamine, serine, threonine, and tyrosine residues and, together with the ion-binding carboxyl group, coordinate the sodium ion. In some cases, ion coordination also involves a water molecule [81, 82]. Molecular dynamics simulations and mutagenesis experiments have confirmed that the ion selectivity is determined by the arrangement and the ratio of polar and hydrophobic groups in the ion-binding site [83, 84]. Some ATPases, e.g., A-ATPase of the methanogenic archaea Methanosarcina acetivorans, are capable of both H+ and Na+ transport under physiological conditions [85].
The number of ions translocated by a rotary ATPase per 1 ATP molecule (the H+/ATP or Na+/ATP ratio) is a key parameter of cell bioenergetics. The relationship between this parameter and enzyme function is discussed in the corresponding section below. The number of ATP molecules synthesized or hydrolyzed during one complete revolution of the rotor is the same for all rotary ATPases and equals 3 (the number of catalytic sites). In turn, the number of ions transported across the membrane during one revolution corresponds to the number of conserved ion-binding glutamate/aspartate carboxyl groups in the c-ring. This value is species-specific and depends on the stoichiometry of the c/K/L-ring, on the number of the ion-binding carboxyl groups per c/K/L subunit (Fig. 3), and on the coupling efficiency, i.e., the probability of “slipping”, when the enzyme transports ions across the membrane but no synthesis/hydrolysis of ATP occurs.

Types of c/K/L subunits of rotary ATPases. The number of subunits in the c/K/L-ring is given in brackets. Examples of organisms that have an ATPase with the corresponding type of c/K/L subunits are also indicated. Ion-binding glutamate/aspartate residues are denoted as E/D. The simplest c subunit (E. coli-like, top left) is typical for F-ATPases.
The number of ion-translocating groups in the c-ring varies from 8 (F-ATPase of bovine mitochondria [86], A-ATPase of Methanococcus jannaschii [87]) to 17 (N-ATPase of pathogenic bacterium B. pseudomallei [77]). Eukaryotic V-ATPases with known c-ring stoichiometry (yeast and mammalian enzymes) contain 10 ion-translocating groups each [13, 25].
The c subunit in its simplest form (e.g., in the F-ATPases of E. coli or mitochondria) is a hairpin of two transmembrane α-helices and contains a single ion-binding Glu or Asp residue located in the middle part of the C-terminal α-helix. The two helices are connected by a loop that is formed by polar residues and interacts with other subunits of the rotor. Most of the studied F-ATPases and many A-type enzymes have the c/K/L subunits of this type [88, 89]. However, in all groups of rotary ATPases, there are examples of gene duplications that resulted in the appearance of subunits containing two or more α-helical hairpins [90]. In some cases, duplication was also accompanied by the loss of the ion-binding group in one of the hairpins. The most diverse are the K/L subunits of A-ATPases. A typical variant is a single hairpin [88, 91]. Also common is a two-hairpin subunit that contains two (methanogens) or one (Pyrococcus and Thermococcus) ion-binding sites [92, 93]. The K/L subunit of the M. jannaschii enzyme consists of three hairpins and has two ion-binding sites [87]. Finally, the genome sequence of the methanogen Methanopyrus kandleri suggests the existence of a giant c subunit composed of 13 hairpins that form a monomeric ring with 13 ion-binding carboxyl groups [94].
In V-ATPases, the heteromeric cc′′-ring (cc′c′′-ring in yeast) is composed of subunits that have two α-helical hairpins each, but only one of the hairpins retains the ion-binding residue [90]. The heteromeric ring is also found in F-ATPases: the c-ring of Na+-translocating F-ATPase of Acetobacterium woodii contains nine copies of the “usual” hairpin c subunit composed of two α-helices and a single copy of the double-hairpin c subunit that has only one ion-binding site [95, 96].
For any particular rotary ATPase, the stoichiometry of the c-ring is constant and determined by the amino acid sequence of c subunit. It was demonstrated that mutations in the c subunit of F-ATPase could alter the ring stoichiometry [97, 98].
FUNCTIONS OF F-TYPE AND A-TYPE ATPases
All rotary ATPases establish a certain balance between the ATP, ADP, GTP, GDP, inorganic phosphate, and magnesium ion concentrations, pH, and transmembrane electrochemical potential difference of the coupling ion (Δμ~H+ or Δμ~Na+; for simplicity, only the proton transfer will be discussed below, although the logic remains the same for sodium-translocating enzymes). This balance is quantitatively determined by the H+/ATP ratio specific for a particular enzyme, which, as mentioned above, depends on the number of ion-binding carboxyl groups in the c-ring.
The Gibbs free energy of ATP synthesis in a cell under physiological conditions is about 55 kJ/mol [99]. The amount of energy released during transmembrane transfer of a single proton down the electrochemical gradient depends on the magnitude of Δμ~H+. In the coupling membranes of prokaryotes, mitochondria, and chloroplasts, this value is usually about 10-22 kJ/mol, corresponding to the protonmotive force (pmf) of approximately 100-220 mV. Therefore, depending on the Δμ~H+ magnitude, from 2.5 to 5.5 protons must be transported across the membrane to synthesize one ATP molecule. Indeed, the H+/ATP ratio of mammalian mitochondrial F-ATPase, as inferred from the enzyme structure, is 8/3 ≈ 2.7, and Δμ~H+ across the inner mitochondrial membrane is about 18-22 kJ/mol (pmf ≈ 180-220 mV) [100]. A higher H+/ATP ratio in rotary ATPases that have a larger number of ion-binding sites in the c-ring presumably allows ATP synthesis at lower Δμ~H+ level.
The direction of the reaction catalyzed by rotary ATPases depends on the presence and activity of other Δμ~H+ generators. Mitochondrial ATP synthase starts hydrolyzing ATP under ischemic conditions when the respiratory chain activity decreases. In chloroplasts, ATP hydrolysis starts when the light intensity is low. In prokaryotic cells, Δμ~H+ can be generated by the respiratory or photosynthetic electron transport chains, bacteriorhodopsins, metabolite transporters, etc. If the activity of these enzymes is decreased, the ATPase hydrolyzes ATP and pumps protons out of the cell, acting as a Δμ~H+ generator itself. In prokaryotes, this process may be of great importance since Δμ~H+ is necessary for the transport of ions and small molecules across the membrane, flagellum rotation, etc. However, such activity may quickly deplete the intracellular ATP pool if the latter is not replenished from other sources. Therefore, it is not surprising that many F-ATPases are regulated by several mechanisms suppressing their ATPase activity when the ATP/ADP ratio drops and Δμ~H+ decreases [14, 35, 101]. Some of these mechanisms are also known for A-ATPases (see below).
It is often implied that the most crucial function of rotary ATPases is ATP synthesis at the expense of Δμ~H+ generated by other enzymes. In prokaryotes lacking such enzymes, rotary ATPase works exclusively as a proton pump, generating Δμ~H+ and elevating the cytoplasm pH. The latter function may be vitally important for acidophilic prokaryotes.
As noted above, A-ATPases are typical for archaea, while F-type ATPases prevail in eubacteria. However, many eubacterial species have acquired A-ATPase operons during evolution, and in some cases have lost F-ATPase operons. We analyzed 711 fully sequenced genomes of prokaryotic species (83 archaea and 628 eubacteria) – a set used in the latest version of the COG (Clusters of Orthologous Groups of proteins) database [102]. Our analysis showed that the A-ATPase operon had been acquired independently by different groups of eubacteria through the horizontal gene transfer. Among the species analyzed, operons encoding rotary ATPases are distributed as follows (Fig. 4): (i) all 83 archaeal genomes contain A-ATPase genes, and one species (Methanosarcina acetivorans) also contains the operon encoding the N-type F-ATPase; (ii) out of 628 eubacteria, 488 have only a single F-ATPase operon, 19 have only a single A-ATPase operon, 47 have both A- and F-type operons (among them, 4 F-ATPases appear to be of N-type), and, finally, several eubacterial species possess more than one operon encoding an ATPase of a particular type; (iii) 12 of 628 eubacteria completely lack rotary ion-translocating ATPases; these organisms are plant or insect endosymbionts.

Distribution of various types of rotary ATPase operons in prokaryotic organisms from the COG database. This database is built using genomes of 83 archaea and 628 eubacteria and provides an adequate coverage of the entire phylogenetic diversity of known prokaryotes. See the text for quantitative data.
These data indicate that in eubacteria, F- and A-ATPases are functionally interchangeable. Moreover, in some cases, the presence of more than one rotary ATPase (presumably with different coupling ion, ion/ATP ratio, or regulatory features) is likely to provide an evolutionary advantage.
Besides ATP synthesis and ATP-driven generation of Δμ~H+, eukaryotic F-ATPases perform several other functions. It was noted above that the dimerization of mitochondrial ATP synthase is necessary for the formation of cristae. Another important, albeit debated, function of this enzyme is its participation in the generation of mitochondrial permeability transition pore (mPTP) ‒ a protein complex that forms a channel in the mitochondrial membrane and renders it permeable for molecules up to 1.5 kDa. In mammalian mitochondria, mPTP formation occurs under stress conditions (e.g., in response to increasing concentration of calcium ions or reactive oxygen species) and results in the dissipation of Δμ~H+, swelling of mitochondria, release of mitochondrial proteins into the cytoplasm, and triggering of programmed cell death [103]. Several researchers suggest that mPTP formation involves mitochondrial ATP synthase [104]. However, experiments with mitochondria that lacked ATP synthase subunits that are presumably involved in mPTP formation contradicted this hypothesis [105].
V-ATPase FUNCTIONS
F- and A-type ATPases in living cells can perform both ATP synthesis and ATP-driven Δμ~H+ generation. The direction of the reaction depends on the species and on the environment. V-ATPases in vivo are only capable of transmembrane proton transport at the expense of ATP hydrolysis. However, yeast V-ATPase was able to catalyze ATP synthesis in vitro when Δμ~H+ was generated by pyrophosphatase artificially introduced into the same membrane [106]. This suggests that V-ATPase is capable of catalyzing both ATP synthesis and hydrolysis. But in vivo the synthase activity is not observed since most types of cellular membranes where V-ATPases reside lack other Δμ~H+ generators, and the Δμ~H+ magnitude on the plasma membrane of an eukaryotic cell is insufficient for ATP synthesis.
The physiological functions of V-ATPases are numerous and have been discussed in detail in many reviews (see e.g. [1, 107, 108]). Below, we will limit ourselves to only a brief outline of this topic.
Most often, V-ATPase works in tandem with other membrane transporters and acts as a motor that generates Δμ~H+, which is then converted into various types of work necessary for the vital functions of the cell and the organism. In most cases, it powers the transport of ions and low-molecular-weight compounds across the membrane (Fig. 5).

V-ATPase hydrolyses ATP and generates Δμ~H+ (both the electric potential difference and the concentration gradient). Various membrane transporters (symporters, antiporters, carriers, and channels) use the energy of Δμ~H+ for translocation of ions (Me+, metal cations; A–, organic acid anions; Cl–, etc.) and low-molecular compounds (C, neurotransmitters, peptides, etc.).
One of the most important V-ATPase functions is the acidification of cellular compartments and, in some tissues, of the extracellular medium. In this case, the electrical component of Δμ~H+ generated by V-ATPase is compensated by the transport of other ions (e.g., Cl– symport), making it possible to maintain a larger ΔpH and to achieve deeper acidification of the compartment interior. V-ATPase is involved in the acidification of the Golgi apparatus trans-cisternae and subsequent protein sorting [109] and provides the energy for the retrograde protein transport [28]. V-ATPase also lowers the pH inside the lysosomes and endosomes [107] and plays an important role in endocytosis (including clathrin-mediated endocytosis) by acidifying the content of the imported vesicles [110]. Proton transport from the cytoplasm of osteoclasts to the extracellular space by V-ATPase is necessary for normal bone tissue resorption and for maintenance of a balance between bone formation and degradation. Increased activity of V-ATPase causes osteopetrosis, while diminished activity may lead to osteoporosis [111]. In the epithelial cells of renal tubules, V-ATPase acidifies the urine. If this process is compromised, the inability of distal tubules to maintain pH gradient leads to an excessive loss of potassium and sodium ions with the urine and the emergence of distal renal tubular acidosis [112].
Besides maintaining pH homeostasis, V-ATPase is also necessary for the transmembrane transport of ions and low-molecular-weight compounds. In S. cerevisiae, V-ATPase is required for normal functioning of Na+/H+-exchangers involved in the yeast response to hyperosmotic and oxidative stresses [113]. In plant cells, V-ATPase is involved in the salt stress response and participates in the vacuolar accumulation of metal ions (including toxic metals) and inorganic and organic acids [114]. In synaptic vesicles, V-ATPase generates Δμ~H+ used by specific membrane transporters that exchange H+ and neurotransmitters and allows to increase the intravesicular concentration of the latter more than 1000-fold as compared to that in the cytoplasm [1].
V-ATPase is also an essential element of several cell signaling pathways. Suppression of its activity affects the crosstalk of lysosomes with the mTORC1 complex – one of the most important regulators of amino acid metabolism. V-ATPase-mediated acidification of lysosomal content is a necessary step in the mTORC1 activation in response to the increase in the concentration of amino acids [115]. V-ATPase also participates in the regulation of glucose metabolism. A decrease in the intracellular glucose level leads to the dissociation of V1 and VO subcomplexes, resulting in the enzyme inactivation (see the section below for details).
REGULATION OF THE ACTIVITY OF ROTARY ATPases
The activity of rotary ATPases is regulated by modulating the expression levels of the corresponding genes. Eukaryotes might have several isoforms for some subunits of rotary ATPases so that the enzyme can be adjusted to the needs of different tissues and cell organelles. Finally, the activity of rotary ATPases can be regulated by post-translational modifications, primarily by protein phosphorylation. Each of these regulatory levels is a large and complex topic and lies beyond the scope of this review. Below, we will discuss in detail only general regulatory mechanisms determined by the amino acid sequence of the enzyme and not associated with its individual isoforms.
As mentioned above, the regulatory features of rotary ATPases largely depend on the mode of enzyme operation. Some ATPases should be able to switch promptly between ATP synthase and hydrolase activities; others work exclusively as ATP-dependent proton (or sodium) pumps. The first group includes F-ATPases of mitochondria, chloroplasts, and photosynthetic and aerobic prokaryotes. The ATPase activity of these enzymes is typically suppressed at low ATP/ADP ratios and/or when Δμ~H+ drops below the level required for ATP synthesis. Re-activation of the enzyme occurs when Δμ~H+ rises above this level. Several mechanisms of such regulation have been described (see review [116]). All F-ATPases studied so far can be noncompetitively inhibited by the MgADP complex (ADP inhibition). This inhibition occurs when MgADP binds at the catalytic site in the absence of inorganic phosphate and is different from simple inhibition by the reaction product [35]. The ATPase activity of many prokaryotic and chloroplast enzymes is also suppressed by conformational changes of the ε subunit [117]. Chloroplast ATP synthase has a unique redox regulation that results in the enzyme inhibition during the nighttime. A pair of cysteine residues in the γ subunit get oxidized and form a disulfide bond in the dark, causing suppression of the ATPase activity [101]. In mitochondria and some α-proteobacteria, if the membrane is de-energized, the hydrolytic activity of F-ATPase is repressed by the binding of special small inhibitory proteins (IF1 and ζ, respectively) [118, 119]. Several additional regulatory mechanisms preventing ATP hydrolysis have been described for prokaryotic F-ATPases (see ref. [35] for more detail).
The regulation of A-ATPases has been studied less comprehensively. However, at least one of the regulatory mechanisms described above, ADP inhibition, was also found in A-ATPases. Therefore, it is probable that ADP inhibition is one of the most ancient regulatory mechanisms of rotary ATPases. F-ATPases of some organisms, in which the synthase activity prevails (e.g., Paracoccus denitrificans, some bacilli species, and chloroplasts), are so susceptible to ADP inhibition that their ATPase activity is negligibly small without additional stimulation, resulting in the release of the inhibitory ADP. In this case, the ATPase activity can be detected after a brief membrane energization or in the presence of certain activators, such as detergents, alcohols, sulfite, and some other oxyanions.
On the other hand, E. coli F-ATPase, which works as an ATP-dependent Δμ~H+ generator when the bacterium dwells anaerobically in the human intestine, hydrolyzes ATP at a high rate even under de-energized conditions and in the absence of stimulating compounds mentioned above. These compounds activate E. coli enzyme to a lesser extent than F-ATPases of chloroplasts and bacilli [35], indicating, in our opinion, that ADP inhibition of the E. coli enzyme is less pronounced. For A-ATPases, various degrees of ADP inhibition have also been shown. The ATPase activities of A-type ATP synthases from two species, Th. thermophilus and Methanosarcina mazei Gö1, are strongly inhibited by ADP [120-122]. At the same time, the A-ATPase of E. hirae, an enzyme that carries out ATP-dependent pumping of sodium ions from the cell, did not demonstrate any signs of ADP inhibition [123]. Several amino acid residues modulating the strength of ADP inhibition in the F- and A-type enzymes were identified by mutagenesis experiments [122, 124, 125].
ADP also inhibits eukaryotic V-ATPases. Clearly, ADP may act as a competitive inhibitor, preventing ATP binding at the enzyme’s catalytic site [126]. However, it was demonstrated that ADP can also inhibit V-ATPases’ activity in a noncompetitive manner [127-129], possibly via a mechanism similar to that in F- and A-ATPases.
The activity of A-ATPases is regulated by one more mechanism associated with the F subunit. The latter is a small protein that, together with the D subunit, belongs to the rotor part of the enzyme and has a flexible elongated domain that can interact with the catalytic hexamer. This interaction was shown to promote the ATPase activity of Th. thermophilus and M. mazei Gö1 A1 subcomplexes [130, 131]. The conformational changes of the F subunit regulatory domain are presumably nucleotide-dependent [132]. The F subunit-dependent activation was also shown for E. hirae A1, but this activation was noticeably weaker [133]. To some extent, the F subunit of A-ATPases resembles the ε subunit of bacterial F-ATPases. The latter is also a part of the rotor and contains flexible regulatory domain, which can adopt an extended conformation, interact with the catalytic hexamer, and affect ATP hydrolysis. However, subunit ε inhibits the ATPase activity of F-ATPases ([117], but see also [134]), and the sequence homology of the F-ATPase subunit ε and A-ATPase subunit F is not evident. Some authors suggest an evolutionary relationship between the F subunit and the globular domain of the F-ATPase γ subunit [135]. In V-ATPases, the regulatory role of the F subunit has not been studied. In the structural models published, this subunit is always in a compact conformation and does not seem to interact with the catalytic hexamer [13, 136].
The activity of V-ATPases is mainly regulated by the reversible dissociation of the V1 and VO subcomplexes. This regulatory mechanism is specific for V-ATPases and has been demonstrated for the enzymes from insects, mammals, and baker’s yeast [137]. A key player in this process is the C subunit. In the full-size VOV1, this subunit is required for the attachment of the peripheral stalks to VO, and dissociation of this subunit destabilizes the enzyme complex [138]. After dissociation, V1 loses its ATPase activity. The inactivation is accompanied by conformational rearrangements of the H subunit [139, 140]. Free VO, in turn, loses the ability to transport protons due to the conformational changes in the cytoplasmic domain of subunit a. The latter interacts with the d subunit and, thereby, stops proton translocation [141, 142]. One of the main factors causing VOV1 dissociation in vivo is a decrease in glucose concentration [113]. The close relationship between glucose metabolism and dissociation/association of V-ATPase is controlled by several signaling pathways [143]. Moreover, V-ATPase can interact directly with two enzymes involved in glycolysis, aldolase and phosphofructokinase. When the glucose concentration is high, these interactions cause association of V1 and VO, followed by re-activation of V-ATPase [144, 145]. Coordination of the V-ATPase activity and glycolysis is presumably required to preserve ATP upon starvation and to prevent cytoplasm acidification during intense anaerobic glycolysis [143].
It should be noted that in F-ATPases the nucleotide-binding hydrophilic subcomplex F1 is tightly bound to the ion-translocating membrane subcomplex FO. The separation of the two subcomplexes requires severe treatment with chaotropic agents in the absence of magnesium ions. Therefore, dissociation of F-ATPases in vivo seems extremely unlikely.
ATP hydrolysis by V-ATPases is also suppressed in a redox-dependent manner due to the formation of a disulfide bond between two conserved cysteine residues in subunit A. One of these residues belongs to the catalytic site. This regulatory mechanism of V-ATPase has been repeatedly demonstrated in vitro for the mammalian and fungal enzymes [146-150]. In yeast, indirect evidence of the physiological significance of this mechanism has also been obtained [151]. In vitro oxidation of sulfhydryl groups in the catalytic site was demonstrated to inhibit the activity of plant V-ATPase [152]. However, in vivo experiments in Arabidopsis thaliana failed to reveal any significant effect of this regulatory mechanism in plant cells [153]. We did not find any in vivo evidence of the redox-dependent regulation of the activity of A-ATPases, although it is known that oxidants may induce cross-link formation between the subunits of the catalytic hexamer of A-ATPase and decrease the enzyme activity [63].
CONCLUSION
Rotary ion-translocating ATPases play a key role in the bioenergetics of most living organisms. These enzymes have a common evolutionary origin and a similar catalytic mechanism, but their regulatory features are remarkably diverse and depend on the physiological functions of these ATPases in the cell. An imbalance in the regulation of these enzymes might cause numerous pathologies; therefore, research in this area is of fundamental interest and is also important for developing new therapies. Distinctive features of rotary ATPases of pathogenic microorganisms make these enzymes promising targets for new specific inhibitors that will suppress microbial activity without affecting similar enzymes in human cells [154].
References
Beyenbach, K. W. (2006) The V-type H+ ATPase: molecular structure and function, physiological roles and regulation, J. Exp. Biol., 209, 577-589, https://doi.org/10.1242/jeb.02014.
Ihara, K., Abe, T., Sugimura, K. I., and Mukohata, Y. (1992) Halobacterial A-ATP synthase in relation to V-ATPase, J. Exp. Biol., 172, 475-485.
Müller, V., and Grüber, G. (2003) ATP synthases: structure, function and evolution of unique energy converters, Cell. Mol. Life Sci., 60, 474-494, https://doi.org/10.1007/s000180300040.
Grüber, G., and Marshansky, V. (2008) New insights into structure-function relationships between archeal ATP synthase (A1A0) and vacuolar type ATPase (V1V0), BioEssays, 30, 1096-1099, https://doi.org/10.1002/bies.20827.
Kühlbrandt, W. (2019) Structure and mechanisms of F-type ATP synthases, Ann. Rev. Biochem., 88, 515-549, https://doi.org/10.1146/annurev-biochem-013118-110903.
Hilario, E., and Gogarten, J. P. (1998) The prokaryote-to-eukaryote transition reflected in the evolution of the V/F/A-ATPase catalytic and proteolipid subunits, J. Mol. Evol., 46, 703-715, https://doi.org/10.1007/pl00006351.
Mulkidjanian, A. Y., Makarova, K. S., Galperin, M. Y., and Koonin, E. V. (2007) Inventing the dynamo machine: the evolution of the F-type and V-type ATPases, Nat. Rev. Microbiol., 5, 892-899, https://doi.org/10.1038/nrmicro1767.
Gogarten, J. P., and Taiz, L. (1992) Evolution of proton pumping ATPases: rooting the tree of life, Photosynth. Res., 33, 137-146, https://doi.org/10.1007/BF00039176.
Kühlbrandt, W., and Davies, K. M. (2016) Rotary ATPases: a new twist to an ancient machine, Trends Biochem. Sci., 41, 106-116, https://doi.org/10.1016/j.tibs.2015.10.006.
Guo, H., Suzuki, T., and Rubinstein, J. L. (2019) Structure of a bacterial ATP synthase, eLife, 8, https://doi.org/10.7554/eLife.43128.
Gu, J., Zhang, L., Zong, S., Guo, R., Liu, T., et al. (2019) Cryo-EM structure of the mammalian ATP synthase tetramer bound with inhibitory protein IF1, Science, 364, 1068- 1075, https://doi.org/10.1126/science.aaw4852.
Zhou, L., and Sazanov, L. A. (2019) Structure and conformational plasticity of the intact Thermus thermophilus V/A-type ATPase, Science, 365, https://doi.org/10.1126/science.aaw9144.
Abbas, Y. M., Wu, D., Bueler, S. A., Robinson, C. V., and Rubinstein, J. L. (2020) Structure of V-ATPase from the mammalian brain, Science, 367, 1240-1246, https://doi.org/10.1126/science.aaz2924.
Hahn, A., Vonck, J., Mills, D. J., Meier, T., and Kühlbrandt, W. (2018) Structure, mechanism, and regulation of the chloroplast ATP synthase, Science, 360, https://doi.org/10.1126/science.aat4318.
Murphy, B. J., Klusch, N., Langer, J., Mills, D. J., Yildiz, Ö., and Kühlbrandt, W. (2019) Rotary substates of mitochondrial ATP synthase reveal the basis of flexible F1-FO coupling, Science, 364, https://doi.org/10.1126/science.aaw9128.
Mühleip, A., McComas, S. E., and Amunts, A. (2019) Structure of a mitochondrial ATP synthase with bound native cardiolipin, eLife, 8, https://doi.org/10.7554/eLife.51179.
Arnold, I., Pfeiffer, K., Neupert, W., Stuart, R. A., and Schägger, H. (1998) Yeast mitochondrial F1F0-ATP synthase exists as a dimer: identification of three dimer-specific subunits, EMBO J., 17, 7170-7178, https://doi.org/10.1093/emboj/17.24.7170.
Guo, H., Bueler, S. A., and Rubinstein, J. L. (2017) Atomic model for the dimeric FO region of mitochondrial ATP synthase, Science, 358, 936-940, https://doi.org/10.1126/science.aao4815.
Eubel, H., Jänsch, L., and Braun, H.-P. (2003) New insights into the respiratory chain of plant mitochondria. Supercomplexes and a unique composition of complex II, Plant Physiol., 133, 274-286, https://doi.org/10.1104/pp.103.024620.
Strauss, M., Hofhaus, G., Schröder, R. R., and Kühlbrandt, W. (2008) Dimer ribbons of ATP synthase shape the inner mitochondrial membrane, EMBO J., 27, 1154-1160, https://doi.org/10.1038/emboj.2008.35.
Blum, T. B., Hahn, A., Meier, T., Davies, K. M., and Kühlbrandt, W. (2019) Dimers of mitochondrial ATP synthase induce membrane curvature and self-assemble into rows, Proc. Natl. Acad. Sci. USA, 116, 4250-4255, https://doi.org/10.1073/pnas.1816556116.
Paumard, P., Vaillier, J., Coulary, B., Schaeffer, J., Soubannier, V., Mueller, D. M., et al. (2002) The ATP synthase is involved in generating mitochondrial cristae morphology, EMBO J., 21, 221-230, https://doi.org/10.1093/emboj/21.3.221.
Davies, K. M., Anselmi, C., Wittig, I., Faraldo-Gómez, J. D., and Kühlbrandt, W. (2012) Structure of the yeast F1F0-ATP synthase dimer and its role in shaping the mitochondrial cristae, Proc. Natl. Acad. Sci. USA, 109, 13602-13607, https://doi.org/10.1073/pnas.1204593109.
Muench, S. P., Trinick, J., and Harrison, M. A. (2011) Structural divergence of the rotary ATPases, Quart. Rev. Bioph., 44, 311-356, https://doi.org/10.1017/S0033583510000338.
Mazhab-Jafari, M. T., Rohou, A., Schmidt, C., Bueler, S. A., Benlekbir, S., Robinson, C. V., et al. (2016) Atomic model for the membrane-embedded VO motor of a eukaryotic V-ATPase, Nature, 539, 118-122, https://doi.org/10.1038/nature19828.
Grüber, G., Manimekalai, M. S. S., Mayer, F., and Müller, V. (2014) ATP synthases from archaea: the beauty of a molecular motor, Biochim. Biophys. Acta, 1837, 940-952, https://doi.org/10.1016/j.bbabio.2014.03.004.
Harrison, M. A., and Muench, S. P. (2018) The vacuolar ATPase – a nano-scale motor that drives cell biology, in Membrane Protein Complexes: Structure and Function (Harris, J. R., and Boekema, E. J., eds.) Springer Singapore, Singapore, p. 409-459, https://doi.org/10.1007/978-981-10-7757-9_14.
Vasanthakumar, T., and Rubinstein, J. L. (2020) Structure and roles of V-type ATPases, Trends Biochem. Sci., 45, 295-307, https://doi.org/10.1016/j.tibs.2019.12.007.
Abrahams, J. P., Leslie, A. G. W., Lutter, R., and Walker, J. E. (1994) Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria, Nature, 370, 621-628.
Arai, S., Saijo, S., Suzuki, K., Mizutani, K., Kakinuma, Y., Ishizuka-Katsura, Y., et al. (2013) Rotation mechanism of Enterococcus hirae V1-ATPase based on asymmetric crystal structures, Nature, 493, 703-707, https://doi.org/10.1038/nature11778.
Schäfer, G., Engelhard, M., and Müller, V. (1999) Bioenergetics of the Archaea, Microbiol. Mol. Biol. Rev., 63, 570-620, https://doi.org/10.1128/mmbr.63.3.570-620.1999.
Kumar, A., Manimekalai, M. S. S., Balakrishna, A. M., Jeyakanthan, J., and Grüber, G. (2010) Nucleotide binding states of subunit A of the A-ATP synthase and the implication of P-loop switch in evolution, J. Mol. Biol., 396, 301-320, https://doi.org/10.1016/j.jmb.2009.11.046.
Komoriya, Y., Ariga, T., Iino, R., Imamura, H., Okuno, D., and Noji, H. (2012) Principal role of the arginine finger in rotary catalysis of F1-ATPase, J. Biol. Chem., 287, 15134-15142, https://doi.org/10.1074/jbc.M111.328153.
Malyan, A. N. (2013) Noncatalytic nucleotide binding sites: properties and mechanism of involvement in ATP synthase activity regulation, Biochemistry (Moscow), 78, 1512-1523, https://doi.org/10.1134/S0006297913130099.
Lapashina, A. S., and Feniouk, B. A. (2018) ADP-inhibition of H+-FOF1-ATP synthase, Biochemistry (Moscow), 83, 1141-1160, https://doi.org/10.1134/S0006297918100012.
Suzuki, K., Mizutani, K., Maruyama, S., Shimono, K., Imai, F. L., Muneyuki, E., et al. (2016) Crystal structures of the ATP-binding and ADP-release dwells of the V1 rotary motor, Nat. Commun., 7, 13235, https://doi.org/10.1038/ncomms13235.
Schäfer, I. B., Bailer, S. M., Düser, M. G., Börsch, M., Bernal, R. A., et al. (2006) Crystal structure of the archaeal A1AO ATP synthase subunit B from Methanosarcina mazei Gö1: implications of nucleotide-binding differences in the major A1AO subunits A and B, J. Mol. Biol., 358, 725-740, https://doi.org/10.1016/j.jmb.2006.02.057.
Gogarten, J. P., Kibak, H., Dittrich, P., Taiz, L., Bowman, E. J., Bowman, B. J., et al. (1989) Evolution of the vacuolar H+-ATPase: implications for the origin of eukaryotes, Proc. Natl. Acad. Sci. USA, 86, 6661-6665, https://doi.org/10.1073/pnas.86.17.6661.
Boyer, P. D. (1997) The ATP synthase – a splendid molecular machine, Annu. Rev. Biochem., 66, 717-749.
Noji, H., Yasuda, R., Yoshida, M., and Kinosita, K. (1997) Direct observation of the rotation of F1-ATPase, Nature, 386, 299-302, https://doi.org/10.1038/386299a0.
Okuno, D., Iino, R., and Noji, H. (2011) Rotation and structure of FOF1-ATP synthase, J. Biochem., 149, 655-664, https://doi.org/10.1093/jb/mvr049.
Junge, W., and Nelson, N. (2015) ATP synthase, Annu. Rev. Biochem., 84, 631-657, https://doi.org/10.1146/annurev-biochem-060614-034124.
Noji, H., Ueno, H., and McMillan, D. G. G. (2017) Catalytic robustness and torque generation of the F1-ATPase, Biophys. Rev., 9, 103-118, https://doi.org/10.1007/s12551-017-0262-x.
Iida, T., Minagawa, Y., Ueno, H., Kawai, F., Murata, T., and Iino, R. (2019) Single-molecule analysis reveals rotational substeps and chemo-mechanical coupling scheme of V-ATPase, J. Biol. Chem., 294, 17017-17030, https://doi.org/10.1074/jbc.RA119.008947.
Furuike, S., Nakano, M., Adachi, K., Noji, H., Kinosita, K., and Yokoyama, K. (2011) Resolving stepping rotation in Thermus thermophilus H(+)-ATPase/synthase with an essentially drag-free probe, Nat. Commun., 2, 233, https://doi.org/10.1038/ncomms1215.
Imamura, H., Takeda, M., Funamoto, S., Shimabukuro, K., Yoshida, M., and Yokoyama, K. (2005) Rotation scheme of V1-motor is different from that of F1-motor, Proc. Natl. Acad. Sci. USA, 102, 17929-17933.
Hirata, T., Iwamoto-Kihara, A., Sun-Wada, G.-H., Okajima, T., Wada, Y., and Futai, M. (2003) Subunit rotation of vacuolar-type proton pumping ATPase: relative rotation of the G and C subunits, J. Biol. Chem., 278, 23714-23719, https://doi.org/10.1074/jbc.M302756200.
Noji, H., Bald, D., Yasuda, R., Itoh, H., Yoshida, M., and Kinosita, K. (2001) Purine but not pyrimidine nucleotides support rotation of F(1)-ATPase, J. Biol. Chem., 276, 25480-25486, https://doi.org/10.1074/jbc.M102200200.
Pisa, K. Y., Huber, H., Thomm, M., and Müller, V. (2007) A sodium ion-dependent A1AO ATP synthase from the hyperthermophilic archaeon Pyrococcus furiosus: A1AO ATPase of Pyrococcus furiosus, FEBS J., 274, 3928-3938, https://doi.org/10.1111/j.1742-4658.2007.05925.x.
Yokoyama, K., Akabane, Y., Ishii, N., and Yoshida, M. (1994) Isolation of prokaryotic VOV1-ATPase from a thermophilic eubacterium Thermus thermophilus, J. Biol. Chem., 269, 12248-12253.
Yoshida, M., Sone, N., Hirata, H., and Kagawa, Y. (1975) A highly stable adenosine triphosphatase from a thermophilic bacterium. Purification, properties, and reconstitution, J. Biol. Chem., 250, 7910-7916.
Senior, A. E., Lee, R. S., al-Shawi, M. K., and Weber, J. (1992) Catalytic properties of Escherichia coli F1-ATPase depleted of endogenous nucleotides, Arch. Biochem. Biophys., 297, 340-344.
Iida, T., Hoaki, T., Kamino, K., Inatomi, K., Kamagata, Y., and Maruyama, T. (1996) Vacuolar-type ATPase in a hyperthermophilic archaeum, Thermococcus sp., Biochem. Biophys. Res. Commun., 229, 559-564, https://doi.org/10.1006/bbrc.1996.1843.
Konishi, J., Wakagi, T., Oshima, T., and Yoshida, M. (1987) Purification and properties of the ATPase solubilized from membranes of an acidothermophilic archaebacterium, Sulfolobus acidocaldarius, J. Biochem., 102, 1379-1387, https://doi.org/10.1093/oxfordjournals.jbchem.a122184.
Pedersen, P. L. (1976) ATP-dependent reactions catalyzed by inner membrane vesicles of rat liver mitochondria. Kinetics, substrate specificity, and bicarbonate sensitivity, J. Biol. Chem., 251, 934-940.
Vambutas, V. K., and Racker, E. (1965) Partial resolution of the enzymes catalyzing photophosphorylation. I. Stimulation of photophosphorylation by a preparation of a latent, Ca++-dependent adenosine triphosphatase from chloroplasts, J. Biol. Chem., 240, 2660-2667.
Struve, I., and Lüttge, U. (1987) Characteristics of MgATP2–-dependent electrogenic proton transport in tonoplast vesicles of the facultative crassulacean-acid-metabolism plant Mesembryanthemum crystallinum L., Planta, 170, 111-120, https://doi.org/10.1007/BF00392387.
Pacheco, G., Lippo de Bécemberg, I., Gonzalez de Alfonzo, R., and Alfonzo, M. J. (1996) Biochemical characterization of a V-ATPase of tracheal smooth muscle plasma membrane fraction, Biochim. Biophys. Acta, 1282, 182-192, https://doi.org/10.1016/0005-2736(96)00038-7.
Perlin, D. S., Latchney, L. R., Wise, J. G., and Senior, A. E. (1984) Specificity of the proton adenosine triphosphatase of Escherichia coli for adenine, guanine, and inosine nucleotides in catalysts and binding, Biochemistry, 23, 4998-5003, https://doi.org/10.1021/bi00316a026.
Suzuki, T., Wakabayashi, C., Tanaka, K., Feniouk, B. A., and Yoshida, M. (2011) Modulation of nucleotide specificity of thermophilic FOF1-ATP synthase by epsilon-subunit, J. Biol. Chem., 286, 16807-16813, https://doi.org/10.1074/jbc.M110.209965.
D’Auzac, J. (1977) ATPase membranaire de vacuoles lysosomales: les lutoides du latex d’Hevea brasiliensis, Phytochemistry, 16, 1881-1885, https://doi.org/10.1016/0031-9422(77)80088-5.
Gräf, R., Harvey, W. R., and Wieczorek, H. (1996) Purification and properties of a Cytosolic V1-ATPase, J. Biol. Chem., 271, 20908-20913, https://doi.org/10.1074/jbc.271.34.20908.
Mayer, F., Lim, J. K., Langer, J. D., Kang, S. G., and Mueller, V. (2015) Na+ transport by the A(1)A(O)-ATP synthase purified from Thermococcus onnurineus and reconstituted into liposomes, J. Biol. Chem., 290, 6994-7002, https://doi.org/10.1074/jbc.M114.616862.
Valiyaveetil, F. I., and Fillingame, R. H. (1997) On the role of Arg-210 and Glu-219 of subunit a in proton translocation by the Escherichia coli FOF1-ATP synthase, J. Biol. Chem., 272, 32635-32641, https://doi.org/10.1074/jbc.272.51.32635.
Glagolev, A. N., and Skulachev, V. P. (1978) The proton pump is a molecular engine of motile bacteria, Nature, 272, 280-282, https://doi.org/10.1038/272280a0.
Junge, W., Lill, H., and Engelbrecht, S. (1997) ATP synthase: an electrochemical transducer with rotatory mechanics, Trends Biochem. Sci., 22, 420-423, https://doi.org/10.1016/s0968-0004(97)01129-8.
Vik, S. B., and Antonio, B. J. (1994) A mechanism of proton translocation by F1FO ATP synthases suggested by double mutants of the a subunit, J. Biol. Chem., 269, 30364-30369.
Srivastava, A. P., Luo, M., Zhou, W., Symersky, J., Bai, D., Chambers, M. G., et al. (2018) High-resolution cryo-EM analysis of the yeast ATP synthase in a lipid membrane, Science, 360, https://doi.org/10.1126/science.aas9699.
Murata, T., Yamato, I., Kakinuma, Y., Leslie, A. G. W., and Walker, J. E. (2005) Structure of the rotor of the V-Type Na+-ATPase from Enterococcus hirae, Science, 308, 654-659, https://doi.org/10.1126/science.1110064.
Skulachev, V. P. (1984) Membrane bioenergetics – should we build the bridge across the river or alongside of it? Trends Biochem. Sci., 9, 182-185, https://doi.org/10.1016/0968-0004(84)90134-8.
Skulachev, V. P. (1985) Membrane-linked energy transductions. Bioenergetic functions of sodium: H+ is not unique as a coupling ion, Eur. J. Biochem., 151, 199-208, https://doi.org/10.1111/j.1432-1033.1985.tb09088.x.
Dimroth, P., and Cook, G. M. (2004) Bacterial Na+- or H+-coupled ATP synthases operating at low electrochemical potential, Adv. Microb. Physiol., 49, 175-218.
Mulkidjanian, A. Y., Dibrov, P., and Galperin, M. Y. (2008) The past and present of sodium energetics: may the sodium-motive force be with you, Biochim. Biophys. Acta, 1777, 985-992, https://doi.org/10.1016/j.bbabio.2008.04.028.
Mulkidjanian, A. Y., Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2008) Evolutionary primacy of sodium bioenergetics, Biol. Direct., 3, 13, https://doi.org/10.1186/1745-6150-3-13.
Poehlein, A., Schmidt, S., Kaster, A.-K., Goenrich, M., Vollmers, J., et al. (2012) An ancient pathway combining carbon dioxide fixation with the generation and utilization of a sodium ion gradient for ATP synthesis, PLoS One, 7, e33439, https://doi.org/10.1371/journal.pone.0033439.
Dibrova, D. V., Galperin, M. Y., and Mulkidjanian, A. Y. (2010) Characterization of the N-ATPase, a distinct, laterally transferred Na+-translocating form of the bacterial F-type membrane ATPase, Bioinformatics, 26, 1473-1476, https://doi.org/10.1093/bioinformatics/btq234.
Schulz, S., WiIkes, M., Mills, D. J., Kuhlbrandt, W., and Meier, T. (2017) Molecular archifecture of the N-type ATPase rotor ring from Barkholderia pseudomallei, EMBO Rep., 18, 526-535, https://doi.org/10.15252/embr.201643374.
Laubinger, W., and Dimroth, P. (1989) The sodium ion translocating adenosine triphosphatase of Propionigenium modestum pumps protons at low sodium ion concentrations, Biochemistry, 28, 7194-7198, https://doi.org/10.1021/bi00444a010.
Neumann, S., Matthey, U., and Kaim, G. (1998) Purification and properties of the F1Fo ATPase of Ilyobacter tartaricus, a sodium ion pump, J. Bacteriol., 180, 3312-3316, https://doi.org/10.1128/JB.180.13.3312-3316.1998.
McMillan, D. G. G., Ferguson, S. A., Dey, D., Schröder, K., Aung, H. L., Carbone, V., et al. (2011) A1Ao-ATP synthase of Methanobrevibacter ruminantium couples sodium ions for ATP synthesis under physiological conditions, J. Biol. Chem., 286, 39882-39892, https://doi.org/10.1074/jbc.M111.281675.
Murata, T., Yamato, I., Kakinuma, Y., Shirouzu, M., Walker, J. E., Yokoyama, S., et al. (2008) Ion binding and selectivity of the rotor ring of the Na+-transporting V-ATPase, Proc. Natl. Acad. Sci. USA, 105, 8607-8611, https://doi.org/10.1073/pnas.0800992105.
Meier, T., Krah, A., Bond, P. J., Pogoryelov, D., Diederichs, K., and Faraldo-Gómez, J. D. (2009) Complete ion-coordination structure in the rotor ring of Na+-dependent F-ATP synthases, J. Mol. Biol., 391, 498-507, https://doi.org/10.1016/j.jmb.2009.05.082.
Krah, A., Pogoryelov, D., Langer, J. D., Bond, P. J., Meier, T., and Faraldo-Gómez, J. D. (2010) Structural and energetic basis for H+ versus Na+ binding selectivity in ATP synthase FO rotors, Biochim. Biophys. Acta, 1797, 763-772, https://doi.org/10.1016/j.bbabio.2010.04.014.
Leone, V., Pogoryelov, D., Meier, T., and Faraldo-Gómez, J. D. (2015) On the principle of ion selectivity in Na+/H+-coupled membrane proteins: experimental and theoretical studies of an ATP synthase rotor, Proc. Natl. Acad. Sci. USA, 112, 1057-1066, https://doi.org/10.1073/pnas.1421202112.
Schlegel, K., Leone, V., Faraldo-Gómez, J. D., and Müller, V. (2012) Promiscuous archaeal ATP synthase concurrently coupled to Na+ and H+ translocation, Proc. Natl. Acad. Sci. USA, 109, 947-952, https://doi.org/10.1073/pnas.1115796109.
Watt, I. N., Montgomery, M. G., Runswick, M. J., Leslie, A. G. W., and Walker, J. E. (2010) Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria, Proc. Natl. Acad. Sci. USA, 107, 16823-16827, https://doi.org/10.1073/pnas.1011099107.
Ruppert, C., Kavermann, H., Wimmers, S., Schmid, R., Kellermann, J., Lottspeich, F., et al. (1999) The proteolipid of the A1AO ATP synthase from Methanococcus jannaschii has six predicted transmembrane helices but only two proton-translocating carboxyl groups, J. Biol. Chem., 274, 25281-25284, https://doi.org/10.1074/jbc.274.36.25281.
Wilms, R., Freiberg, C., Wegerle, E., Meier, I., Mayer, F., and Müller, V. (1996) Subunit structure and organization of the genes of the A1AO ATPase from the Archaeon Methanosarcina mazei Gö1, J. Biol. Chem., 271, 18843-18852, https://doi.org/10.1074/jbc.271.31.18843.
Steinert, K., Wagner, V., Kroth-Pancic, P. G., and Bickel-Sandkötter, S. (1997) Characterization and subunit structure of the ATP synthase of the halophilic archaeon Haloferax volcanii and organization of the ATP synthase genes, J. Biol. Chem., 272, 6261-6269, https://doi.org/10.1074/jbc.272.10.6261.
Kibak, H., Taiz, L., Starke, T., Bernasconi, P., and Gogarten, J. P. (1992) Evolution of structure and function of V-ATPases, J. Bioenerg. Biomembr., 24, 415-424, https://doi.org/10.1007/BF00762534.
Ihara, K., Watanabe, S., Sugimura, K.-I., Katagiri, I., and Mukohata, Y. (1997) Identification of proteolipid from an extremely halophilic archaeon Halobacterium salinarumas an N, N′-dicyclohexyl-carbodiimide binding subunit of ATP synthase, Arch. Biochem. Biophys., 341, 267-272, https://doi.org/10.1006/abbi.1997.9972.
Vonck, J., Pisa, K. Y., Morgner, N., Brutschy, B., and Müller, V. (2009) Three-dimensional structure of A1AO ATP synthase from the hyperthermophilic archaeon Pyrococcus furiosus by electron microscopy, J. Biol. Chem., 284, 10110-10119, https://doi.org/10.1074/jbc.M808498200.
Mayer, F., Leone, V., Langer, J. D., Faraldo-Gómez, J. D., and Müller, V. (2012) A c subunit with four transmembrane helices and one ion Na+-binding site in an archaeal ATP synthase: implications for c ring function and structure, J. Biol. Chem., 287, 39327-39337, https://doi.org/10.1074/jbc.M112.411223.
Slesarev, A. I., Mezhevaya, K. V., Makarova, K. S., Polushin, N. N., Shcherbinina, O. V., et al. (2002) The complete genome of hyperthermophile Methanopyrus kandleri AV19 and monophyly of archaeal methanogens, Proc. Natl. Acad. Sci. USA, 99, 4644-4649, https://doi.org/10.1073/pnas.032671499.
Müller, V., Aufurth, S., and Rahlfs, S. (2001) The Na+ cycle in Acetobacterium woodii: identification and characterization of a Na+ translocating F1F0-ATPase with a mixed oligomer of 8 and 16 kDa proteolipids, Biochim. Biophys. Acta Bioenergetics, 1505, 108-120, https://doi.org/10.1016/S0005-2728(00)00281-4.
Matthies, D., Zhou, W., Klyszejko, A. L., Anselmi, C., Yildiz, Ö., Brandt, K., et al. (2014) High-resolution structure and mechanism of an F/V-hybrid rotor ring in a Na+-coupled ATP synthase, Nat. Commun., 5, 5286, https://doi.org/10.1038/ncomms6286.
Pogoryelov, D., Klyszejko, A. L., Krasnoselska, G. O., Heller, E.-M., Leone, V., et al. (2012) Engineering rotor ring stoichiometries in the ATP synthase, Proc. Natl. Acad. Sci. USA, 109, 1599-1608, https://doi.org/10.1073/pnas.1120027109.
Preiss, L., Klyszejko, A. L., Hicks, D. B., Liu, J., Fackelmayer, O. J., et al. (2013) The c-ring stoichiometry of ATP synthase is adapted to cell physiological requirements of alkaliphilic Bacillus pseudofirmus OF4, Proc. Natl. Acad. Sci. USA, 110, 7874-7879, https://doi.org/10.1073/pnas.1303333110.
Veech, R. L., King, M. T., Pawlosky, R., Bradshaw, P. C., and Curtis, W. (2019) Relationship between inorganic ion distribution, resting membrane potential, and the ΔG’ of ATP hydrolysis: a new paradigm, FASEB J., 33, 13126-13130, https://doi.org/10.1096/fj.201901942R.
Hüttemann, M., Lee, I., Pecinova, A., Pecina, P., Przyklenk, K., and Doan, J. W. (2008) Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease, J. Bioener. Biomembr., 40, 445-456, https://doi.org/10.1007/s10863-008-9169-3.
Hisabori, T., Konno, H., Ichimura, H., Strotmann, H., and Bald, D. (2002) Molecular devices of chloroplast F1-ATP synthase for the regulation, Biochim. Biophys. Acta Bioenergetics, 1555, 140-146, https://doi.org/10.1016/S0005-2728(02)00269-4.
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015) Expanded microbial genome coverage and improved protein family annotation in the COG database, Nucleic Acids Res., 43, 261-269, https://doi.org/10.1093/nar/gku1223.
Zorov, D. B., Juhaszova, M., and Sollott, S. J. (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release, Physiol. Rev., 94, 909-950, https://doi.org/10.1152/physrev.00026.2013.
Giorgio, V., von Stockum, S., Antoniel, M., Fabbro, A., Fogolari, F., Forte, M., et al. (2013) Dimers of mitochondrial ATP synthase form the permeability transition pore, Proc. Natl. Acad. Sci. USA, 110, 5887-5892, https://doi.org/10.1073/pnas.1217823110.
Carroll, J., He, J., Ding, S., Fearnley, I. M., and Walker, J. E. (2019) Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase, Proc. Natl. Acad. Sci. USA, 116, 12816-12821, https://doi.org/10.1073/pnas.1904005116.
Hirata, T., Nakamura, N., Omote, H., Wada, Y., and Futai, M. (2000) Regulation and reversibility of vacuolar H+-ATPase, J. Biol. Chem., 275, 386-389, https://doi.org/10.1074/jbc.275.1.386.
Forgac, M. (2007) Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology, Nat. Rev. Mol. Cell Biol., 8, 917-929, https://doi.org/10.1038/nrm2272.
McGuire, C., Stransky, L., Cotter, K., and Forgac, M. (2017) Regulation of V-ATPase activity, Front. Biosci., 22, 609-622, https://doi.org/10.2741/4506.
Huang, C., and Chang, A. (2011) pH-dependent cargo sorting from the Golgi, J. Biol. Chem., 286, 10058-10065, https://doi.org/10.1074/jbc.M110.197889.
Kozik, P., Hodson, N. A., Sahlender, D. A., Simecek, N., Soromani, C., Wu, J., et al. (2013) A human genome-wide screen for regulators of clathrin-coated vesicle formation reveals an unexpected role for the V-ATPase, Nat. Cell Biol., 15, 50-60, https://doi.org/10.1038/ncb2652.
Futai, M., Sun-Wada, G.-H., Wada, Y., Matsumoto, N., and Nakanishi-Matsui, M. (2019) Vacuolar-type ATPase: a proton pump to lysosomal trafficking, Proc. Japan Acad. Series B Physic. Biol. Sci., 95, 261-277, https://doi.org/10.2183/pjab.95.018.
Finberg, K. E., Wagner, C. A., Bailey, M. A., Paunescu, T. G., Breton, S., Brown, D., et al. (2005) The B1-subunit of the H+ ATPase is required for maximal urinary acidification, Proc. Natl. Acad. Sci. USA, 102, 13616-13621, https://doi.org/10.1073/pnas.0506769102.
Cotter, K., Stransky, L., McGuire, C., and Forgac, M. (2015) Recent insights into the structure, regulation, and function of the V-ATPases, Trends Biochem. Sci., 40, 611-622, https://doi.org/10.1016/j.tibs.2015.08.005.
Almeida, D. M., Oliveira, M. M., and Saibo, N. J. M. (2017) Regulation of Na+ and K+ homeostasis in plants: towards improved salt stress tolerance in crop plants, Genet. Mol. Biol., 40, 326-345, https://doi.org/10.1590/1678-4685-GMB-2016-0106.
Zoncu, R., Bar-Peled, L., Efeyan, A., Wang, S., Sancak, Y., and Sabatini, D. M. (2011) mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase, Science, 334, 678-683, https://doi.org/10.1126/science.1207056.
Feniouk, B. A., and Yoshida, M. (2008) Regulatory mechanisms of proton-translocating FOF1-ATP synthase, Results Problems Cell Differ., 45, 279-308, https://doi.org/10.1007/400_2007_043.
Feniouk, B. A., Suzuki, T., and Yoshida, M. (2006) The role of subunit epsilon in the catalysis and regulation of FOF1-ATP synthase, Biochim. Biophys. Acta, 1757, 326-338, https://doi.org/10.1016/j.bbabio.2006.03.022.
Gledhill, J. R., Montgomery, M. G., Leslie, A. G. W., and Walker, J. E. (2007) How the regulatory protein, IF1, inhibits F1-ATPase from bovine mitochondria, Proc. Natl. Acad. Sci. USA, 104, 15671-15676, https://doi.org/10.1073/pnas.0707326104.
Morales-Ríos, E., de la Rosa-Morales, F., Mendoza-Hernández, G., Rodríguez-Zavala, J. S., Celis, H., et al. (2010) A novel 11-kDa inhibitory subunit in the F1FO ATP synthase of Paracoccus denitrificans and related alpha-proteobacteria, FASEB J., 24, 599-608, https://doi.org/10.1096/fj.09-137356.
Yokoyama, K., Muneyuki, E., Amano, T., Mizutani, S., Yoshida, M., Ishida, M., et al. (1998) V-ATPase of Thermus thermophilus is inactivated during ATP hydrolysis but can synthesize ATP, J. Biol. Chem., 273, 20504-20510, https://doi.org/10.1074/jbc.273.32.20504.
Nakano, M., Imamura, H., Toei, M., Tamakoshi, M., Yoshida, M., and Yokoyama, K. (2008) ATP hydrolysis and synthesis of a rotary motor V-ATPase from Thermus thermophilus, J. Biol. Chem., 283, 20789-20796, https://doi.org/10.1074/jbc.M801276200.
Singh, D., and Grüber, G. (2018) Crystallographic and enzymatic insights into the mechanisms of Mg-ADP inhibition in the A1 complex of the A1AO ATP synthase, J. Struct. Biol., 201, 26-35, https://doi.org/10.1016/j.jsb.2017.10.008.
Kishikawa, J.-I., Nakanishi, A., Furuike, S., Tamakoshi, M., and Yokoyama, K. (2014) Molecular basis of ADP inhibition of vacuolar (V)-type ATPase/synthase, J. Biol. Chem., 289, 403-412, https://doi.org/10.1074/jbc.M113.523498.
Lapashina, A. S., Prikhodko, A. S., Shugaeva, T. E., and Feniouk, B. A. (2019) Residue 249 in subunit beta regulates ADP inhibition and its phosphate modulation in Escherichia coli ATP synthase, Biochim. Biophys. Acta Bioenerg., 1860, 181-188, https://doi.org/10.1016/j.bbabio.2018.12.003.
Lapashina, A. S., and Feniouk, B. A. (2019) Mutation Q259L in subunit beta in Bacillus subtilis ATP synthase attenuates ADP-inhibition and decreases fitness in mixed cultures, Biochem. Biophys. Res. Commun., 509, 102-107, https://doi.org/10.1016/j.bbrc.2018.12.075.
David, P., and Baron, R. (1994) The catalytic cycle of the vacuolar H+-ATPase. Comparison of proton transport in kidney- and osteoclast-derived vesicles, J. Biol. Chem., 269, 30158- 30163.
Moriyama, Y., and Nelson, N. (1987) Nucleotide binding sites and chemical modification of the chromaffin granule proton ATPase, J. Biol. Chem., 262, 14723-14729.
Webster, L. C., Pérez-Castiñeira, J. R., Atkins, G. L., and Apps, D. K. (1995) Allosteric regulation of proton translocation by a vacuolar adenosinetriphosphatase, Eur. J. Biochem., 232, 586-595, https://doi.org/10.1111/j.1432-1033.1995.586zz.x.
Vasilyeva, E., and Forgac, M. (1998) Interaction of the clathrin-coated vesicle V-ATPase with ADP and sodium azide, J. Biol. Chem., 273, 23823-23829, https://doi.org/10.1074/jbc.273.37.23823.
Kishikawa, J.-I., Seino, A., Nakanishi, A., Tirtom, N. E., Noji, H., Yokoyama, K., et al. (2014) F-subunit reinforces torque generation in V-ATPase, Eur. Biophys. J., 43, 415-422, https://doi.org/10.1007/s00249-014-0973-x.
Singh, D., Sielaff, H., Sundararaman, L., Bhushan, S., and Grüber, G. (2016) The stimulating role of subunit F in ATPase activity inside the A1-complex of the Methanosarcina mazei Gö1 A1AO ATP synthase, Biochim. Biophys. Acta, 1857, 177-187, https://doi.org/10.1016/j.bbabio.2015.12.003.
Singh, D., Sielaff, H., Börsch, M., and Grüber, G. (2017) Conformational dynamics of the rotary subunit F in the A3B3DF complex of Methanosarcina mazei Gö1 A-ATP synthase monitored by single-molecule FRET, FEBS Lett., 591, 854-862, https://doi.org/10.1002/1873-3468.12605.
Saijo, S., Arai, S., Hossain, K. M. M., Yamato, I., Suzuki, K., Kakinuma, Y., et al. (2011) Crystal structure of the central axis DF complex of the prokaryotic V-ATPase, Proc. Natl. Acad. Sci. USA, 108, 19955-19960, https://doi.org/10.1073/pnas.1108810108.
Akanuma, G., Tagana, T., Sawada, M., Suzuki, S., Shimada, T., Tanaka, K., et al. (2019) C-terminal regulatory domain of the ε subunit of FOF1 ATP synthase enhances the ATP-dependent H+ pumping that is involved in the maintenance of cellular membrane potential in Bacillus subtilis, Microbiol. Open, 8, e00815, https://doi.org/10.1002/mbo3.815.
Kishikawa, J.-I., Ibuki, T., Nakamura, S., Nakanishi, A., Minamino, T., Miyata, T., et al. (2013) Common evolutionary origin for the rotor domain of rotary ATPases and flagellar protein export apparatus, PLoS One, 8, e64695, https://doi.org/10.1371/journal.pone.0064695.
Balakrishna, A. M., Basak, S., Manimekalai, M. S. S., and Grüber, G. (2015) Crystal structure of subunits D and F in complex gives insight into energy transmission of the eukaryotic V-ATPase from Saccharomyces cerevisiae, J. Biol. Chem., 290, 3183-3186, https://doi.org/10.1074/jbc.M114.622688.
Kane, P. M. (2012) Targeting reversible disassembly as a mechanism of controlling V-ATPase activity, Curr. Protein Peptide Sci., 13, 117-123, https://doi.org/10.2174/138920312800493142.
Tabke, K., Albertmelcher, A., Vitavska, O., Huss, M., Schmitz, H.-P., and Wieczorek, H. (2014) Reversible disassembly of the yeast V-ATPase revisited under in vivo conditions, Biochem. J., 462, 185-197, https://doi.org/10.1042/BJ20131293.
Beltrán, C., and Nelson, N. (1992) The membrane sector of vacuolar H(+)-ATPase by itself is impermeable to protons, Acta Physiol. Scandinavica Suppl., 607, 41-47.
Zhang, J., Myers, M., and Forgac, M. (1992) Characterization of the VO domain of the coated vesicle (H+)-ATPase, J. Biol. Chem., 267, 9773-9778.
Qi, J., and Forgac, M. (2008) Function and subunit interactions of the N-terminal domain of subunit a (Vph1p) of the yeast V-ATPase, J. Biol. Chem., 283, 19274-19282, https://doi.org/10.1074/jbc.M802442200.
Couoh-Cardel, S., Milgrom, E., and Wilkens, S. (2015) Affinity purification and structural features of the yeast vacuolar ATPase VO membrane sector, J. Biol. Chem., 290, 27959-27971, https://doi.org/10.1074/jbc.M115.662494.
Hayek, S. R., Rane, H. S., and Parra, K. J. (2019) Reciprocal regulation of V-ATPase and glycolytic pathway elements in health and disease, Front. Physiol., 10, 127, https://doi.org/10.3389/fphys.2019.00127.
Lu, M., Ammar, D., Ives, H., Albrecht, F., and Gluck, S. L. (2007) Physical interaction between aldolase and vacuolar H+-ATPase is essential for the assembly and activity of the proton pump, J. Biol. Chem., 282, 24495-24503, https://doi.org/10.1074/jbc.M702598200.
Chan, C.-Y., and Parra, K. J. (2014) Yeast phosphofructokinase-1 subunit Pfk2p is necessary for pH homeostasis and glucose-dependent vacuolar ATPase reassembly, J. Biol. Chem., 289, 19448-19457, https://doi.org/10.1074/jbc.M114.569855.
Feng, Y., and Forgac, M. (1992) A novel mechanism for regulation of vacuolar acidification, J. Biol. Chem., 267, 19769-19772.
Feng, Y., and Forgac, M. (1994) Inhibition of vacuolar H+-ATPase by disulfide bond formation between cysteine 254 and cysteine 532 in subunit A, J. Biol.Chem., 269, 13224-13230.
Forgac, M. (1999) The vacuolar H+-ATPase of clathrin-coated vesicles is reversibly inhibited by S-nitrosoglutathione, J. Biol. Chem., 274, 1301-1305, https://doi.org/10.1074/jbc.274.3.1301.
Dschida, W. J., and Bowman, B. J. (1995) The vacuolar ATPase: sulfite stabilization and the mechanism of nitrate inactivation, J. Biol. Chem., 270, 1557-1563, https://doi.org/10.1074/jbc.270.4.1557.
Liu, Q., Leng, X. H., Newman, P. R., Vasilyeva, E., Kane, P. M., and Forgac, M. (1997) Site-directed mutagenesis of the yeast V-ATPase A subunit, J. Biol. Chem., 272, 11750-11756, https://doi.org/10.1074/jbc.272.18.11750.
Oluwatosin, Y. E., and Kane, P. M. (1997) Mutations in the CYS4 gene provide evidence for regulation of the yeast vacuolar H+-ATPase by oxidation and reduction in vivo, J. Biol. Chem., 272, 28149-28157, https://doi.org/10.1074/jbc.272.44.28149.
Hager, A., and Lanz, C. (1989) Essential sulfhydryl groups in the catalytic center of the tonoplast H+-ATPase from coleoptiles of Zea mays L. as demonstrated by the biotin-streptavidin-peroxidase system, Planta, 180, 116-212, https://doi.org/10.1007/BF02411417.
Seidel, T., Scholl, S., Krebs, M., Rienmüller, F., Marten, I., Hedrich, R., et al. (2012) Regulation of the V-type ATPase by redox modulation, Biochem. J., 448, 243-251, https://doi.org/10.1042/BJ20120976.
Hards, K., and Cook, G. M. (2018) Targeting bacterial energetics to produce new antimicrobials, Drug Resist. Updates: Rev.Comm. Antimicrob. Anticancer Chemother., 36, 1-12, https://doi.org/10.1016/j.drup.2017.11.001.
Acknowledgements
The authors are grateful to Vladimir P. Skulachev for the unique and inspiring atmosphere at the Belozersky Institute of Physico-Chemical Biology of Lomonosov Moscow State University (MSU). The authors also highly appreciate his effort in founding the MSU Faculty of Bioengineering and Bioinformatics, without which this work would never have been written.
Funding
This work was supported by the Russian Science Foundation (project no. 20-14-00268).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflict of interest. This article does not contain a description of studies with the involvement of humans or animal subjects.
Rights and permissions
About this article

Cite this article
Zubareva, V.M., Lapashina, A.S., Shugaeva, T.E. et al. Rotary Ion-Translocating ATPases/ATP Synthases: Diversity, Similarities, and Differences. Biochemistry Moscow 85, 1613–1630 (2020). https://doi.org/10.1134/S0006297920120135
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297920120135