
Abstract
Anaerobic production of pyruvic acid from glucose by recombinant Escherichia coli strains with impaired fermentation ability during respiration with nitrate as an external terminal electron acceptor was studied. During nitrate respiration in a minimal salt medium lacking ammonium ions, the core E. coli strain MG1655 ∆ackA-pta, ∆poxB, ∆ldhA, ∆adhE, ∆ptsG, PLglk, PtacgalP, ∆frdAB, ∆pflB, ∆sdhAB, ∆aceEF converted glucose into pyruvic acid with a yield of 1.72 mol/mol, secreting lactic acid as the only detected byproduct. The deletion of the lldD and dld genes blocked the secretion of this byproduct. The corresponding strain lacking the respiratory L- and D-lactate dehydrogenases LldD and Dld synthesized pyruvic acid from glucose with a yield of 1.76 mol/mol, consuming the available carbohydrate substrate incompletely. Enforced ATP hydrolysis due to the action of the pyruvic acid–oxaloacetic acid–malic acid–pyruvic acid or pyruvic acid–phosphoenolpyruvate–pyruvic acid futile cycles led to a drastic increase in glucose consumption by recombinants while maintaining the levels of substrate to the target product conversion. As a result, during anaerobic nitrate respiration and enforced ATP hydrolysis pyruvic acid was produced from glucose with a yield of 1.77–1.78 mol/mol with almost exhaustive consumption of the substrate by recombinants and no or minimal byproduct formation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Pyruvic acid is both a key metabolite of living cells and an important, industrially relevant compound that can serve as a convenient precursor in the further synthesis of a wide range of high value-added substances. Organic synthesis with pyruvic acid as the starting material can produce food supplements, pharmaceuticals, and solvents, such as acetaldehyde [1, 2] and butanol [3], D/L-alanine, aromatic L-amino acids [4], 4-dihydroxy-L-phenylalanine [5], and N-acetyl-D-neuraminic acid [6]. In addition, pyruvic acid can be directly used in nutrition as a dietary supplement with many properties that are beneficial for human health [7].
Pyruvic acid is currently produced via petrochemical synthesis. However, the current strategies for sustainable development imply a gradual but steady replacement of chemical-industry products with the corresponding products of microbial biotechnology. Thus, it is of immediate interest to obtain microbial producers of pyruvic acid that can efficiently synthesize the target compound from cheap and readily available renewable raw materials (i.e., plant-biomass sugars). It should be noted that pyruvic acid acts in the cell as the main precursor metabolite for many biochemical pathways leading to various classes of organic molecules. Thus, highly efficient pyruvic-acid producers can also serve as platform strains for the further development of specialized producers of other valuable compounds. Recent years have seen remarkable progress in the engineering of processes for the biological conversion of carbohydrates into pyruvic acid with the use of various species of industrially relevant microorganisms [8]. The best characteristics were achieved with a directly engineered Escherichia coli strain, which was able to synthesize pyruvic acid from glucose with a yield of 1.53 mol/mol amounting, to 77% of the theoretical maximum [9].
Glycolysis is the key biochemical pathway supplying the cell with pyruvic acid during the utilization of the relevant substrates (primarily, glucose). It is a conservative metabolic pathway in many microorganisms, including the facultatively anaerobic bacterium E. coli, which is traditionally used in the industrial biotechnology. It is well known that glycolysis is not transcriptionally limited and is controlled primarily by the intracellular ratios of NADH/NAD+ and ATP/ADP [10, 11]. Glucose utilization during glycolysis results in the formation of two pyruvic acid molecules, two NADH molecules, and two ATP molecules. Under aerobic conditions, the NADH produced during glycolysis is reoxidized by the respiratory electron-transport chain with oxygen as the terminal electron acceptor, while pyruvic acid and ATP are involved in the anabolic biomass-formation processes. Under anaerobic conditions, a substantial portion of the glycolytically formed pyruvic acid directly or indirectly participates in fermentation reactions to maintain the intracellular redox balance, acting as an internal terminal electron acceptor for NADH oxidation.
In E. coli, the key enzymes responsible for the anaerobic dissimilation of pyruvic acid include lactate dehydrogenase LdhA (EC 1.1.1.28), acetate kinase AckA (EC 2.7.2.1), phosphotransacetylase Pta (EC 2.3.1.8), bifunctional alcohol/alhehyde dehydrogenase AdhE (EC 1.1.1.1/1.2.1.3), catalyzing the formation of the main mixed-acid fermentation products, i.e., lactic acid, acetic acid, and ethanol, as well as pyruvate formate-lyase PflB (EC 2.3.1.54), which converts pyruvic acid into formic acid and acetyl-CoA. The inactivation of these enzymes is an obligatory requirement to enable efficient anaerobic production of pyruvic acid by recombinant E. coli strains. However, E. coli strains deficient in mixed-acid fermentation pathways are incapable of anaerobic growth [12]. Moreover, the residual ability of corresponding strains to reoxidize NADH is rather low, while their need for ATP is dramatically decreased. As a result, such mutants are almost incapable of utilizing glucose under anaerobic conditions [13]. At the same time, anaerobic processes are more preferable than aerobic processes for the biosynthesis of valuable products, because they have lower capital and operating costs and fewer issues related to mass transfer and contamination [14]. Since E. coli is a facultatively anaerobic bacterium, the anaerobic production of pyruvic acid from glucose by recombinants devoid of fermentation pathways can be achieved with a dual-phase fermentation process, comprising an aerobic phase for biomass accumulation followed by an anaerobic production phase. NAD+ regeneration under anoxic conditions can be ensured in this case via anaerobic respiration with an external electron acceptor, such as DMSO [15], nitrate [16], or even an electrode functioning in the anode mode [17].
Indeed, it was previously demonstrated that a directly engineered E. coli strain possessing a modified system of glucose transport and phosphorylation, devoid of the main mixed-acid fermentation pathways and the ability to perform fumarate-succinate interconversion and also uncapable to form acetyl-CoA from the corresponding three-carbon precursor, in the presence of sodium nitrate in the medium anaerobically converts glucose into pyruvic acid with a rather high molar yield [18]. The only byproduct secreted by the strain, in addition to pyruvic acid, was lactic acid, which most likely formed resulting from the involvement of the target-product in reactions of alternative anaerobic respiration with an internal electron acceptor [19, 20]. At the same time, despite the effecient reoxidation of the glycolytically formed NADH during anaerobic respiration with both external and internal electron acceptors the strain did not completely consume the available glucose. The key factor limiting glucose consumption by the recombinant strain under anoxic conditions appeared to be excessive intracellular ATP levels.
The goal of the work was to improve the anaerobic production of pyruvic acid from glucose by recombinant Escherichia coli strains with impaired fermentation ability via enforced ATP hydrolysis.
MATERIALS AND METHODS
Reagents. The restriction endonuclease BglII, Т4 DNA ligase, Taq DNA polymerase (Thermo Scientific, Lithuania), and high-fidelity DNA polymerase Kapa HiFi (Roche, Switzerland) were used in the work. Polymerase chain reaction (PCR) products were purified via agarose gel electrophoresis and further isolated from gel with a QIAquick Gel Extraction Kit (Qiagen, United States). Table 1 presents the oligonucleotides used in the work (Evrogen, Russia). The culture media components, salts, and other reagents were produced by Panreac (Spain) and Sigma (United States).
Bacterial strains, plasmids, and media. Table 2 lists the bacterial strains and plasmids used in the work. The E. coli strain K-12 MG1655 (VKPM B-6195), the previously constructed strain E. coli MG1655 ∆ackA-pta, ∆poxB, ∆ldhA, ∆adhE, ∆ptsG, PLglk, PtacgalP [21], possessing a modified system of glucose transport and phosphorylation and lacking the main mixed-acid fermentation pathways designated as PA4, and its derivative, the strain PA4FPSA [18], with an additionally blocked fumarate–succinate interconversion and incapable of pyruvic acid convertion to acetyl-CoA, were used as the starting strains for the construction of all of the recombinants obtained in the work.
The bacteria were cultured in reach LB, SOB, SOC media and the minimal М9 medium [22], with the addition of ampicillin (100 µg/mL) or chloramphenicol (30 µg/mL) if necessary. The media were also supplemented with sodium acetate to enable the aerobic growth of the ∆aceEF PA4FPSA strain and its derivatives.
Recombinant strain construction. All chromosome modifications were performed as described previously [23].
To inactivate the lldD and dld genes, linear DNA fragments, containing chloramphenicol resistance marker (cat gene), were obtained by PCR using the primer pairs P1 and P2 and P3 and P4 and the pMW118-(λattL-Cm-λattR) plasmid [24] as a template. The obtained DNA fragments were individually integrated into the chromosome of the E. coli MG1655 strain carrying the pKD46 helper plasmid. The correspondence between the assumed and experimentally obtained structures of the chromosomes of the selected strains with the individually inactivated lldD and dld genes was confirmed by PCR with pairs of the locus-specific primers P5 and P6 and P7 and P8.
The DNA fragment to replace the native regulatory region of the ppsA gene with the artificial genetic element PL-SDφ10 containing the phage lambda PL promoter and the efficient ribosome binding site of φ10 gene from the T7 phage was constructed in several stages. At the first stage, a DNA fragment containing the BglII recognition site, PL promoter, the SD sequence of φ10 gene from T7 phage, and 36 nucleotides complementary to the 5'–end of coding region of the ppsA gene was obtained by PCR.
The fragment was obtained in two steps. As the first step, the DNA fragment containing the BglII recognition site, PL promoter, and part of the SD sequence of φ10 gene from T7 phage was obtained using primers P9 and P10 and genomic DNA of phage lambda as a template. The obtained PCR product served as a template for subsequent PCR with the primers P9 and P11. Primer P11 contained a region complementary to the 3'-end of the PL promoter, SD sequence of φ10 gene from T7 phage, and the first 36 nucleotides of the ppsA gene open reading frame. At the same time, the second step of DNA-fragment construction was performed. A DNA fragment containing the BglII recognition site, chloramphenicol resistance marker (cat gene), and 36 nucleotides homologous to the DNA region located immediately upstream of the coding region of the ppsA gene was obtained by PCR with the P12 and P13 primers and the pMW118-(λattL-Cm-λattR) plasmid as a template. The obtained DNA fragments were treated with the BglII restriction endonuclease and ligated with T4 DNA ligase. The ligation product was amplified using primers P11 and P13. The obtained PCR product was integrated into the chromosome of the E. coli MG1655 strain carrying the helper plasmid pKD46. The correspondence between the desired and experimentally obtained nucleotide sequence of the new regulatory element introduced upstream of the coding region of ppsA gene was confirmed by sequencing using primers P14 and P15.
The corresponding individual genetic modifications were further introduced into the chromosomes of the target recombinants via P1-dependent transduction [22]. The marker flanked by the phage lambda att-sites was removed from the chromosomes of the target strains using the pMWts-Int/Xis plasmid as described previously [25]. The strains were transformed with the plasmids according to the standard protocols.
Strain cultivation. The recombinant strains were grown overnight in M9 medium containing 2 g/L of glucose at 37°C. Overnight cultures were diluted ten times by the addition of 45 mL of M9 medium containing 10 g/L of yeast extract to 5 mL of the culture. The resulting cultures were grown in 750-mL flasks at 37°C on a rotary shaker at 250 rpm for 8 h. The cell suspensions were centrifuged for 15 min at 2000 g at 4°C. The cell pellets were resuspended in 15 mL of modified M9 medium lacking ammonium ions and containing 10 g/L of glucose and 50 mM of sodium nitrate to an OD600 of ~10. The cultures were further incubated under anaerobic conditions for 24 h in 15-mL screw-cap tubes at 37°C on the rotary shaker at 250 rpm. When strains containing the pMW-derived plasmids were grown, the media also contained 100 µg/mL of ampicillin (OOO Sintez, Russia). To ensure the functional activity of pyruvate carboxylase in strains carrying the Bacillus subtilis pycA gene, NaHCO3 was added to the media used at the last stage of culturing to a final concentration of 10 g/L.
The cell suspensions collected for the analysis were centrifuged at 10 000 g for 10 min. The obtained supernatants were used to determine the concentrations of secreted metabolites and residual glucose. All experiments were performed at least in triplicate.
Analytical techniques. The concentrations of the organic acids in the culture liquids freed from the biomass by centrifugation were determined via high-performance liquid chromatography (HPLC) with the Waters HPLC system (United States). The Rezex ROA-Organic Acid H+ (8%) ion-exclusion column (300 × 7.8 mm, 8 µm, Phenomenex, United States) was used with detection at 210 nm. An aqueous solution of sulfuric acid (2.5 mM) was used as a mobile phase with a flow rate of 0.5 mL/min. To measure the glucose concentrations, the system was equipped with a Waters 2414 refractive index detector and a Spherisorb-NH2 column (4.6 × 250 mm, 5 μм, Waters, United States). An acetonitrile/water mixture (vol/vol 75/25) served as a mobile phase at a flow rate of 1.0 mL/min.
RESULTS AND DISCUSSION
The previously constructed E. coli strain PA4FPSA was used as the core pyruvic-acid producer. In this strain, the main mixed-acid fermentation pathways, which are responsible for anaerobic pyruvate dissimilation and lead to the formation of acetic and lactic acids, as well as ethanol, were inactivated via the deletion of the ackA, pta, poxB, ldhA, and adhE genes, which encode the enzymes catalyzing the corresponding reactions. The anaerobic respiration with fumarate as the internal electron acceptor was prevented primarily due to the deletion of the frdAB genes, which encode the components of fumarate reductase (EC 1.3.5.4). It is well-known that succinate dehydrogenase (EC 1.3.5.1) can functionally replace fumarate reductase in E. coli strains with inactivated frdAB genes [26]. Thus, the sdhAB genes, which encode the components of succinate dehydrogenase, were also deleted in this strain. The involvement of pyruvic acid, through the intermediate formation of acetyl-CoA, in the reaction of the tricarboxylic acid cycle (TCA), which is activated upon respiration, was prevented resulting from the deletion of the pflB and aceEF genes, which encode pyruvate formate-lyase and the components of the pyruvate dehydrogenase enzymatic complex (EC 1.2.4.1/2.3.1.12/1.8.1.4). In addition, the system of glucose transport and phosphorylation was modified to consume ATP for phosphorylation of the carbohydrate substrate instead of phosphoenolpyruvate (PEP) for a potential reduction of the inhibition of the initial and terminal stages of glycolysis by the ATP generated at its intermediate stages. This was achieved via inactivation of the ptsG gene, which encodes the glucose permease of the PEP-dependent phosphotransferase system with the overexpression of the galP and glk genes encoding the H+-galactose symporter and ATP-dependent glucokinase (EC 2.7.1.2). As a result, after aerobic growth, the PA4FPSA strain was able to convert glucose anaerobically to pyruvic acid upon respiration with nitrate as an external electron acceptor with a yield of ~1.7 mol/mol [18].
The main detected byproduct synthesized by the strain was lactic acid. The results of enantiomeric analysis of the lactic acid secreted by the strain indicated that the respiratory lactate dehydrogenases LldD and Dld (EC 1.1.5.12) could be responsible for the formation of this compound. Normally, the corresponding enzymes catalyze the quinone-dependent conversion of L-lactic acid and D-lactic acid into pyruvic acid [19]. However, under anaerobic conditions, their inverted quinol-dependent action, which resulted in the formation of the corresponding reduced product from pyruvic acid, could also contribute to the maintenance of the intracellular redox balance in the strain with impaired fermentation ability.
In addition to the LldD and Dld lactate dehydrogenases, D-alanine dehydrogenase DadA (EC 1.4.5.-) may participate in the alternative anaerobic respiration with pyruvic acid as an internal electron acceptor [20]. To exclude the potential contribution of the corresponding enzyme to the maintenance of the intracellular redox balance, the minimal M9 medium without ammonium ions was used in the present work at the final stage of culturing to characterize the anaerobic glucose utilization and pyruvic acid biosynthesis by the PA4FPSA strain and its derivatives during dual-phase fermentation.
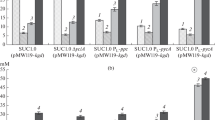
In the presence of nitrate ions and the absence of ammonium ions in the medium, the PA4FPSA strain demonstrated characteristics similar to those described previously, i.e., it consumed about 23.6 mM of glucose and synthesized ~40.8 mM (Fig. 1) of pyruvic acid with a yield of 1.72 mol/mol (Table 3). As expected, alanine was not observed among the products of anaerobic glucose utilization secreted by the strain, and lactic acid was the only byproduct (Fig. 1). The observed synthesis of lactic acid by the strain in the presence of sodium nitrate was apparently due to the high potential (~0.19 V) of the corresponding respiratory reactions utilizing the internal electron acceptor, which was only two times lower than the corresponding value (~0.42 V) for respiration involving Nar-system nitrate reductases and exceeded the value for respiration with fumarate (~0.03 V) and even DMSO (~0.16 V) as the external electron acceptors [19].

Concentrations (mM) of pyruvic acid (1) and lactic acid (2) secreted by the PA4FPSA strain and its derivatives PA4FPSAL, PA4FPSAD, and PA4FPSALD during the anaerobic glucose utilization (3) upon respiration with sodium nitrate as an external electron acceptor.
It is well known that under the conditions of nitrate respiration the expression of at least one of the quinone-dependent lactate dehydrogenases, LldD, is induced in E. coli cells [27]. Thus, the lldD gene was initially deleted in the PA4FPSA strain to prevent the undesirable production of lactic acid. The corresponding derivative PA4FPSAL synthesized pyruvic acid from glucose with almost the same efficiency as the parental strain (Fig. 1 and Table 3), but it still secreted considerable amounts of lactic acid (Fig. 1). Moreover, despite the inactivation of the L-lactate dehydrogenase LldD, the yield of lactic acid formed by the strain during anaerobic glucose utilization did not decrease (Table 3). This could indicate that the D-lactate dehydrogenase Dld was the main enzyme responsible for lactic acid production by the PA4FPSA and PA4FPSAL strains. To test this hypothesis, the dld gene was deleted in the PA4FPSA strain. When D-lactate dehydrogenase was inactivated, the anaerobic production of pyruvic acid from glucose by the corresponding PA4FPSAD strain did not change as compared to the PA4FPSA strain, while the lactic acid secretion dropped markedly (Fig. 1 and Table 3). However, the dld– mutant did not completely cease lactic acid production. Thus, despite the preferential production of lactic acid by D-lactate dehydrogenase in the PA4FPSA and PA4FPSAL strains, L-lactate dehydrogenase was able to compensate partially for the loss of activity of the alternative enzyme in the PA4FPSAD strain. For this reason, the genes encoding both lactate dehydrogenases were further concomitantly deleted in the PA4FPSA strain.
The PA4FPSALD strain with the inactivated respiratory lactate dehydrogenases LldD and Dld did not secrete any detectable quantities of lactic acid as a result of anaerobic glucose utilization. At the same time, it produced pyruvic acid (~42.1 mM) at a slightly higher level than the initial PA4FPSA strain did (Fig. 1), and the corresponding carboxylate was produced from glucose with a yield as high as 1.76 mol/mol (Table 3). The absence of the reduced products of glucose catabolism in the medium indicated, in turn, that respiration with exogenous nitrate as the only terminal electron acceptor was able to successfully maintain the overall intracellular redox balance in the PA4FPSALD strain.
At the same time, PA4FPSALD did not consume all of the available glucose (55.5 mM), as was also the case with its precursors (Fig. 1 and Table 3), although the Nar and Nrf nitrate and nitrite respiration systems can reoxidize 250 mM NADH with 50 mM of NaNO3 added to the medium [19], which correspond to the number of reduced equivalents formed during glycolytic utilization of 125 mM glucose. Thus even upon efficient reoxidation of glycolytically formed NADH via anaerobic respiration with external electron acceptor, an excessive intracellular ATP levels remained, as in the case of the PA4FPSA strain, the key factor limiting glucose consumption under anoxic conditions by its promising, directly engineered derivative, PA4FPSALD.
Several approaches to the intensification of the glycolytic carbon flux in recombinant E. coli strains based on the decreasing of intracellular ATP levels have been described to date. The main approach is based on manipulation of components of the (F1F0) H+-ATP synthase complex in order to prevent oxidative phosphorylation and/or enhance ATP hydrolysis. Overexpression of the genes encoding the components of the cytoplasmic F1 subunit comprising the catalytic site should promote ATP hydrolysis. Conversely, inactivation of the genes encoding the components of the membrane-bound F0 subunit, which forms the proton channel, will block ATP production via oxidative phosphorylation with a concomitant increase in ATPase activity. These two strategies have both been successfully implemented to improve the biosynthetic performance of recombinant E. coli strains engineered to produce pyruvic acid from glucose under aerobic conditions [9, 28]. A rather promising, alternative approach implies enforced ATP hydrolysis in the cell due to the action of futile cycles [29].
It was previously demonstrated that the expression of the heterologous pyruvate carboxylase (EC 6.4.1.1) in E. coli strains deficient in main mixed-acid fermentation pathways provoked the emergence of an artificial futile cycle pyruvic acid–oxaloacetic acid (OAA)–malic acid–pyruvic acid during the anaerobic utilization of glucose [30]. In this cycle, OAA produced from pyruvic acid by ATP-dependent pyruvate carboxylase was converted into malic acid through the NADH-consuming initial reaction of the reductive branch of the TCA cycle, which is catalyzed by the malate dehydrogenase Mdh (EC 1.1.1.37) and then, was decarboxylated to pyruvic acid by the NAD+-dependent malic enzyme MaeA (EC 1.1.1.39), which is constitutively expressed in E. coli, resulting in the hydrolysis of a single ATP molecule in the absence of NADH generation. Inactivation of this cycle promoted the anaerobic production of four-carbon dicarboxylic acids by the corresponding recombinant strains [31], whereas its activation improved pyruvic acid production [18, 30].
The pyruvic acid–PEP–pyruvic acid futile cycle may serve as an alternative to the pyruvic acid–OAA–malic acid–pyruvic acid cycle, which functions exclusively under anaerobic conditions when the reductive branch of the TCA cycle is active. Given the presence of glucose in the medium, the pyruvic acid–PEP–pyruvic acid futile cycle may be active both under aerobic and anaerobic conditions. In this cycle, pyruvate kinase (EC 2.7.1.40) catalyzes the ADP-dependent dephosphorylation of PEP to pyruvic acid with the formation of a single ATP molecule, while phosphoenolpyruvate synthase (EC 2.7.9.2) phosphorylates pyruvic acid, using ATP as a cofactor and breaking it into AMP and inorganic phosphate. AMP regeneration to ATP by adenylate kinase (EC 2.7.4.3) requires two ATP molecules. The potential of this cycle to improve the production of the target compound from glucose under anoxic conditions by decreasing the intracellular ATP pool was recently demonstrated with a lactic acid producing E. coli strain as an example [32].
The potential of the pyruvic acid–OAA–malic acid–pyruvic acid and pyruvic acid–PEP–pyruvic acid futile cycles to improve anaerobic glucose consumption by the engeneered recombinant pyruvic acid producers was tested by enabling their functionality in both the core PA4FPSA strain and its derivative PA4FPSALD strain, which lacks the respiratory lactate dehydrogenases. It order to ensure the activity of pyruvic acid–OAA–malic acid–pyruvic acid futile cycle, the pPYC plasmid [33], expressing B. subtilis pycA gene, was introduced in the strains resulting in the corresponding derivative strains PA4FPSAPyc and PA4FPSALDPyc. The functional activity of the pyruvic acid–PEP–pyruvic acid futile cycle was ensured in the PA4FPSAPps and PA4FPSALDPps strains by the constitutive expression of the ppsA gene, which encodes phosphoenolpyruvate synthase, under the control of the phage lambda PL promoter.
The anaerobic consumption of glucose by the obtained PA4FPSAPyc, PA4FPSALDPyc, PA4FPSAPps, and PA4FPSALDPps strains increased dramatically as compared to their precursor strains (Figs. 1 and 2) with pyruvic acid yields remained at the same high level (Table 4). This indicated the positive effect of enforced ATP hydrolysis resulting from the action of each of the futile cycles on the intensity of carbon flux through the cascade of glycolytic reactions, leading to the target production of pyruvic acid as the main product of carbon substrate utilization.

Concentrations (mM) of pyruvic (1), lactic (2), and malic (3) acids secreted by the PA4FPSAPyc, PA4FPSAPps, PA4FPSALDPyc, and PA4FPSALDPps strains during the anaerobic glucose utilization (4) upon respiration with sodium nitrate as an external electron acceptor and enforced ATP hydrolysis resulted from the action of the futile cycles pyruvic acid–OAA–malic acid–pyruvic acid (PA4FPSAPyc and PA4FPSALDPyc strains) and pyruvic acid–PEP–pyruvic acid (PA4FPSAPps and PA4FPSALDPps strains).
Nevertheless, the effect of the introduction of the respective futile cycles was somewhat different for the different strains. In particular, the PA4FPSAPyc and PA4FPSALDPyc strains with the active futile cycle of pyruvic acid–OAA–malic acid–pyruvic acid secreted malic acid as one of the products of glucose utilization (Fig. 2). On the one hand, this indicated the functionality of this particular futile cycle in the recombinant strains, but, on the other hand, it pointed to the insufficient activity of the malic enzymes in the cells. At the same time, the carbon recovery, which characterize the completeness of substrate conversion into the detected products, increased in the case of the PA4FPSAPyc and PA4FPSALDPyc strains as compared to the parental strains, while remained almost unchanged for the PA4FPSAPps and PA4FPSALDPps strains (Tables 3 and 4).
Since the functioning of the pyruvic acid—PEP–pyruvic acid futile cycle did not result in the formation of the new glucose-utilization products, it could indicate that some intermediates of glycolysis were involved in the side reactions of substrate consumption, which resulted in unproductive carbon loss in the corresponding recombinant strains. With the absence of pyruvate dehydrogenase in the studied strains and, consequently, the inactivity of the oxidative branch of the TCA cycle even under the respirative conditions, the partially activated oxidative branch of the pentose phosphate pathway (PPP) could be responsible for the carbon loss due to the release of CO2 in the reaction catalyzed by 6-phosphogluconate dehydrogenase (EC 1.1.1.44), and then for the decrease in carbon recovery values. The activity of the pyruvic acid–OAA–malic acid–pyruvic acid futile cycle decreased the contribution of the oxidative branch of PPP to the nonproductive carbon loss by the recombinant strains, whereas the enforced pyruvic acid–PEP interconversion had almost no effect on the intensity of the corresponding process. At the same time, the PA4FPSALDPps strain, which hydrolyzed excessive ATP via the pyruvic acid–PEP–pyruvic acid futile cycle, produced pyruvic acid with an increased yield (1.77 mol/mol), secreted no detectable amounts of any byproducts, and consumed the available glucose almost completely, in contrast to the PA4FPSALDPyc strain, which reduced the intracellular pool of the corresponding cofactor as a result of the activity of the pyruvic acid–OAA–malic acid–pyruvic acid futile cycle. Conversely, the high carbon recovery (96%, Table 4) demonstrated by the PA4FPSALDPyc strain upon the anaerobic glucose utilization under nitrate-respiration conditions suggested that the yield of pyruvic acid achieved by the strain (1.78 mol/mol, Table 4) could be further elevated by an increase in the activity of the malic enzymes in the cell. However, the pyruvic acid–OAA–malic acid–pyruvic acid futile cycle, which ensures enforced ATP hydrolysis in the PA4FPSALDPyc strain, is inactive in the absence of the available CO2 source in the medium, while the addition of bicarbonate to the medium will increase the total cost of the target compound synthesis.
CONCLUSIONS
Thus, the potential for the industrial use of each of the approaches based on enforced ATP hydrolysis achieved due to the action of artificially created futile cycles to obtain efficient anaerobic production of pyruvic acid from glucose by recombinant E. coli strains should be evaluated considering both the potential benefits and the possible increase in the operating costs.
The results of the present study clearly demonstrate the potential of enforced ATP hydrolysis, due to the use of ATP-consuming futile cycles, to improve the biosynthetic characteristics of recombinant E. coli strains directly engineered for the anaerobic production of pyruvic acid from glucose during anaerobic respiration with an external electron acceptor.
REFERENCES
Ingram, L.O., Conway, T., Clark, D.P., Sewell, G.W., and Preston, J.F., Appl. Environ. Microbiol., 1987, vol. 53, no. 10, pp. 2420–2425.
Eram, M.S. and Ma, K., Biomolecules, 2013, vol. 3, no. 3, pp. 578–596.
Reiße, S., Haack, M., Garbe, D., Sommer, B., Steffler, F., Carsten, J., Bohnen, F., Sieber, V., and Brück, T., Front. Bioeng. Biotechnol., 2016, vol. 4, no. 74. https://doi.org/10.3389/fbioe.2016.00074
Li, Y., Chen, J., and Lun, S.Y., Appl. Microbiol. Biotechnol., 2001, vol. 57, no. 4, pp. 451–459.
Park, H.S., Lee, J.Y., and Kim, H.S., Biotechnol. Bioeng., 1998, vol. 58, nos. 2–3, pp. 339–343.
Zhang, Y., Tao, F., Du, M., Ma, C., Qiu, J., Gu, L., He, X., and Xu, P., Appl. Microbiol. Biotechnol., 2010, vol. 86, no. 2, pp. 481–489.
Saper, R.B., Eisenberg, D.M., and Phillips, R.S., Am. Fam. Physician, 2004, vol. 70, no. 9, pp. 1731–1738.
Maleki, N. and Eiteman, M.A., Fermentation, 2017, vol. 3, no. 8. https://doi.org/10.3390/fermentation3010008
Causey, T.B., Shanmugam, K.T., Yomano, L.P., and Ingram, L.O., Proc. Natl. Acad. Sci. U S A, 2004, vol. 101, no. 8, pp. 2235–2240.
Koebmann, B.J., Westerhoff, H.V., Snoep, J.L., Nilsson, D., and Jensen, P.R., J. Bacteriol., 2002, vol. 184, no. 14, pp. 3909–3916.
Vemuri, G.N., Altman, E., Sangurdekar, D.P., Khodursky, A.B., and Eiteman, M.A., Appl. Environ. Microbiol., 2006, vol. 72, no. 5, pp. 3653–3661.
Fischer, C.R., Tseng, H.C., Tai, M., Prather, K.L., and Stephanopoulos, G., Appl. Microbiol. Biotechnol., 2010, vol. 88, no. 1, pp. 265–275. https://doi.org/10.1007/s00253-010-2749-2
Skorokhodova, A.Yu., Gulevich, A.Yu., and Debabov, V.G., Appl. Biochem. Microbiol., 2017, vol. 53, no. 3, pp. 304–309.
Chen, G.Q., Microb. Cell. Fact., 2012, vol. 11, no. 111. https://doi.org/10.1186/1475-2859-11-111
Carvalho, M., Matos, M., Roca, C., and Reis, M.A., N. Biotechnol., 2014, vol. 31, no. 1, pp. 133–139.
Jung, H.M., Kim, Y.H., and Oh, M.K., Biotechnol. J., 2017, vol. 12, no. 11. https://doi.org/10.1002/biot.201700121
Förster, A.H., Beblawy, S., Golitsch, F., and Gescher, J., Biotechnol. Biofuels, 2017, vol. 10, no. 65. https://doi.org/10.1186/s13068-017-0745-9
Skorokhodova, A.Y., Gulevich, A.Y., and Debabov, V.G., J. Biotechnol., 2019, vol. 293, pp. 47–55.
Unden, G. and Bongaerts, J., Biochim. Biophys. Acta, 1997, vol. 1320, no. 3, pp. 217–234.
Skorokhodova, A.Yu., Sukhozhenko, A.V., Gulevich, A.Yu., and Debabov, V.G., Biotekhnologiiya, 2019, vol. 35, no. 2, pp. 16–24.
Morzhakova, A.A., Skorokhodova, A.Yu., Gulevich, A.Yu., and Debabov, V.G., Appl. Biochem. Microbiol., 2013, vol. 49, no. 2, pp. 113–119.
Sambrook, J., Fritsch, E., and Maniatis, T., Molecular Cloning: A Laboratory Manual, 2nd ed., New York: Cold Spring Harbor Lab. Press, 1989.
Datsenko, K.A. and Wanner, B.L., Proc. Natl. Acad. Sci. U. S. A., 2000, vol. 97, no. 12, pp. 6640–6645.
Katashkina, Zh.I., Skorokhodova, A.Yu., Zimenkov, D.V., Gulevich, A.Yu., Minaeva, N.I., Doroshenko, V.G., Biryukova, I.V., and Mashko, S.V., Mol. Biol. (Moscow), 2005, vol. 39, no. 5, pp. 823–831.
Gulevich, A.Yu., Skorokhodova, A.Yu., Ermishev, V.Yu., Krylov, A.A., Minaeva, N.I., Polonskaya, Z.M., Zimenkov, D.V., Biryukova, I.V., and Mashko, S.V., Mol. Biol. (Moscow), 2009, vol. 43, no. 3, pp. 547–557.
Maklashina, E., Berthold, D.A., and Cecchini, G., J. Bacteriol., 1998, vol. 180, no. 22, pp. 5989–5996.
Nishimura, Y., Tan, I.K.P., Ohgami, Y., Kohgami, K., and Kamihara, T., FEMS Microbiol. Lett., 1983, vol. 17, nos. 1–3, pp. 283–286.
Zhu, Y., Eiteman, M.A., Altman, R., and Altman, E., Appl. Environ. Microbiol., 2008, vol. 74, no. 21, pp. 6649–6655.
Hädicke, O. and Klamt, S., Biochem. Soc. Trans., 2015, vol. 43, no. 6, pp. 1140–1145.
Skorokhodova, A.Yu., Stasenko, A.A., Gulevich, A.Yu., and Debabov, V.G., Appl. Biochem. Microbiol., 2018, vol. 54, no. 2, pp. 141–148.
Skorokhodova, A.Yu., Gulevich, A.Yu., and Debabov, V.G., Biotekhnologiya, 2018, vol. 34, no. 2, pp. 18–25.
Hadicke, O., Bettenbrock, K., and Klamt, S., Biotechnol. Bioeng., 2015, vol. 112, no. 10, pp. 2195–2199.
Skorokhodova, A.Yu., Gulevich, A.Yu., Morzhakova, A.A., Shakulov, R.S., and Debabov, V.G., Appl. Biochem. Microbiol., 2011, vol. 47, no. 4, pp. 373–380.
Funding
The work was supported by a grant from the Russian Foundation for Basic Research (project no. 18-04-01222).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
No experimentation involving animals or human was performed by any of the authors.
Conflict of Interest. The authors have no conflicts of interest.
Additional information
Translated by E. Martynova
Rights and permissions
About this article

Cite this article
Skorokhodova, A.Y., Gulevich, A.Y. & Debabov, V.G. Optimization of the Anaerobic Production of Pyruvic Acid from Glucose by Recombinant Escherichia coli strains with Impaired Fermentation Ability via Enforced ATP Hydrolysis. Appl Biochem Microbiol 57, 434–442 (2021). https://doi.org/10.1134/S0003683821040153
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0003683821040153