
Abstract
Glioblastoma (GBM) remains one of the most fatal human malignancies due to its high angiogenic and infiltrative capacities. Even with optimal therapy including surgery, radiotherapy and temozolomide, it is essentially incurable. GBM is among the most neovascularised neoplasms and its malignant progression associates with striking neovascularisation, evidenced by vasoproliferation and endothelial cell hyperplasia. Targeting the pro-angiogenic pathways is therefore a promising anti-glioma strategy. Here we show that tivozanib, a pan-inhibitor of vascular endothelial growth factor (VEGF) receptors, inhibited proliferation of GBM cells through a G2/M cell cycle arrest via inhibition of polo-like kinase 1 (PLK1) signalling pathway and down-modulation of Aurora kinases A and B, cyclin B1 and CDC25C. Moreover, tivozanib decreased adhesive potential of these cells through reduction of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). Tivozanib diminished GBM cell invasion through impairing the proteolytic cascade of cathepsin B/urokinase-type plasminogen activator (uPA)/matrix metalloproteinase-2 (MMP-2). Combination of tivozanib with EGFR small molecule inhibitor gefitinib synergistically increased sensitivity to gefitinib. Altogether, these findings suggest that VEGFR blockade by tivozanib has potential anti-glioma effects in vitro. Further in vivo studies are warranted to explore the anti-tumour activity of tivozanib in combinatorial approaches in GBM.
Similar content being viewed by others

Introduction
Gliomas are the most common primary brain tumours with more than 20000 new cases each year in the United States. According to World Health Organization (WHO) prognostic grading system, glial tumours are classified into four grades (grade I–IV), with the most aggressive tumours being grade 4 astrocytomas (also known as glioblastoma; GBM)1. GBM has a poor median survival due to its rapid growth, angiogenesis, invasiveness and therapeutic resistance. Treatment of GBM includes maximal surgical resection followed by radiotherapy with concurrent and adjuvant chemotherapy. Regardless of initial response, virtually all patients experience disease relapse2. Therefore, there is a pressing need to develop improved therapeutic options for GBM patients.
Angiogenesis, a multi-step process by which tumours develop new vasculature, is a fundamental driver for tumour growth and malignant progression3,4. The vascular endothelial growth factor (VEGF) pathway is the most promising angiogenic target due to its key roles in angiogenesis and tumour growth. The VEGF family consists of seven ligands including VEGFA, VEGFB, VEGFC, VEGFD, VEGFE, placenta growth factor (PlGF) 1 and PlGF2. The tyrosine kinase receptors in this family include VEGF receptor type 1 (VEGFR1), VEGFR2 and VEGFR35.
There is evidence that enhanced expression of the VEGF family promotes malignant progression and correlates with poor prognosis in GBM6,7. Extensive endothelial proliferation and vascular permeability leading to vasogenic brain oedema, a major cause of neurologic morbidity, are hallmarks of GBM. This is mainly due to elevated expression of VEGFA and signalling through endothelial VEGFR28,9. The degree of VEGFA expression and microvascular density correlate with malignant potential and aggressive behaviour of GBM cells as reflected in disease relapse and overall survival rate9,10. Consistent with this, blockade of the VEGF pathway has been shown to normalise tumour vessels, improve radiotherapy outcome and extend survival in murine orthotopic models of GBM11.
Anti-angiogenic strategies are promising approaches for the treatment of GBM due to the highly vascular nature of these tumours and evidence has determined dependence of glioma growth on tumour-associated angiogenesis12,13,14. GBM patients treated with bevacizumab (anti-VEGFA mAb) alone or in combination with irinotecan chemotherapy have demonstrated improvement in progression-free survival15,16,17. However, lack of improvement in overall survival, frequent development of resistance and incomplete VEGF pathway blockade emphasize the need for more efficacious anti-angiogenic therapies12,18,19. In this regard, an agent with the ability to block all the three VEGF receptors is thought to have improved anti-tumour activity20.
Tivozanib (AV-951; AVEO pharmaceuticals) is a pan-VEGFR inhibitor with potential anti-angiogenic and anti-neoplastic activities21. Tivozanib has shown anti-tumour activity in xenograft models of prostate, breast, lung, pancreas, glioblastoma and renal cell carcinoma. In both phase I and II clinical trials, it has been found to be well tolerable with manageable side effects and durable clinical activity22,23. Tivozanib is currently under investigation in a phase II study in patients with recurrent GBM (NCT01846871)12. In the present study, we examined the mechanistic activity of tivozanib in human GBM cell lines.
Results
Tivozanib inhibits proliferation, clonal growth and anoikis resistance
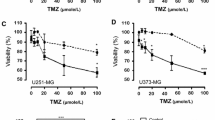
MTT assay was carried out to determine the effects of tivozanib on proliferation of the GBM cells. Treatment of these cells with tivozanib inhibited their growth (Fig. 1A,B). Moreover, the results of a colony formation assay demonstrate that tivozanib reduced their clonogenic survival (Fig. 1C,D).

(A) MTT assay was applied to estimate cell viability after 48 h of treatment with tivozanib. (B) The GBM cells were treated with tivozanib for 48 h, stained with crystal violet and imaged by an inverted microscope (images acquired at 10x magnification). (C,D) Clonogenic assay was conducted to evaluate the effects of tivozanib on clonal proliferation. (E) Anoikis resistance assay was performed with cell culture on poly-HEMA–coated culture dishes for 48 h and the proportion of viable cells was measured by MTT assay. Data are given as mean ± SD, normalized to the untreated control group. Statistically significant values of *p < 0.05, **p < 0.01, and ***p < 0.001 were determined compared with the control.
Detachment of adherent cells from the extracellular matrix induces anoikis, a special type of apoptosis. Acquisition of resistance to anoikis is a prerequisite for tumour cell survival and spread24. Tivozanib diminished anoikis resistance in the GBM cells (Fig. 1E). Taken together, these data illustrate that tivozanib decreased proliferation, clonal growth and anoikis resistance in the GBM cells.
Tivozanib induces G2/M cell cycle arrest
Due to the anti-proliferative effects of tivozanib, we sought if it inhibits cell cycle progression or affects apoptosis. In A172 and T98G cells, tivozanib increased the percentage of cells in G2/M phase while decreasing the G1/S fraction. Moreover, a small number of cells underwent cell death, as indicated by appearance of a sub-G0/G1 population. Tivozanib-treated U87MG cells displayed an increase in the G2/M population (Fig. 2A).

(A) Following treatment with tivozanib for 48 h, the cell pellets were fixed and incubated with PI to analyse the cell cycle distribution on a flow cytometer. The graphs are representative of three independent experiments with similar results. (B) The cells were treated with tivozanib for 48 h then total RNA was harvested for qRT-PCR analysis. (C) Protein lysates from tivozanib-treated cells were subjected to Western blotting and probed with the indicated antibodies. β-actin was used as the loading control. The blots are representative of three independent experiments with similar outcomes. Data are given as mean ± SD. Statistically significant values of *p < 0.05, **p < 0.01, and ***p < 0.001 were determined compared with the control. AURK, aurora kinase; FOXM1, forkhead box M1; CCNB, cyclin B; NEK2, NIMA related kinase 2; CENPF, centromere protein F; IL, interleukin; CDKN1A, cyclin-dependent kinase inhibitor 1 A; CDK, cyclin-dependent kinase; CCNA2, cyclin A2; GADD45A, growth arrest and DNA damage inducible alpha; CHEK, checkpoint kinase; CDC25, cell division cycle 25; MYT1, myelin transcription factor 1; SFN, stratifin.
We next determined the effects of tivozanib on expression of genes and proteins that regulate the G2/M transition. Activation of CDK1/cyclin B complex is a pivotal step in mitotic initiation. WEE1 and myelin transcription factor 1 (MYT1) block mitotic entry via phosphorylation of CDK1, a process reversed by CDC25C25,26. The phosphatase activity of CDC25C is pre-empted by checkpoint kinase 1 (CHEK1)27. Moreover, polo-like kinase 1 (PLK1) drives entry into mitosis via phosphorylation of CDC25C as well as inactivation of WEE1 and MYT128. Both serine/threonine kinases Aurora A and B activate PLK129. Forkhead box protein M1 (encoded by FOXM1) is a member of the FOX family of transcription factors that regulates expression of a large array of G2/M-specific genes such as PLK1, cyclin B2 (encoded by CCNB2), NIMA related kinase 2 (encoded by NEK2) and centromere protein F (encoded by CENPF)30.
Tivozanib inhibited p-PLK1 and decreased expression of Aurora A, Aurora B, CDC25C and cyclin B1 (Fig. 2C, Supplementary Fig. 1). Both mRNA and protein levels of the G2/M checkpoint regulator p21 (encoded by CDKN1A) were increased by tivozanib treatment (Fig. 2B,C, Supplementary Fig. 1). In addition, tivozanib reduced the mRNA levels of FOXM1, CCNB2, NEK2, CENPF, CDK1 and CDK2 (Fig. 2B). These data suggest that tivozanib inhibited proliferation of the GBM cells through a G2/M cell cycle arrest.
Tivozanib reduces adhesive and invasive abilities of the GBM cells
Adhesion of GBM cells to extracellular matrix (ECM) followed by migration along the ECM and invasion into the adjacent brain tissue is a hallmark of GBM and represents the major obstacle to successful chemo- and surgical therapy31. Cell adhesion molecules such as ICAM-1 (intercellular adhesion molecule-1) and VCAM-1 (vascular cell adhesion molecule-1) play key roles in adhesion of GBM cells to the ECM32. To investigate whether tivozanib impairs adhesive properties of the GBM cells, we assayed adhesion of tivozanib-treated cells to collagen I which is a substrate for a range of cell adhesion molecules. Tivozanib decreased cell adhesion to collagen I, concomitant with significant reduction of ICAM-1 and VCAM-1 (Fig. 3A–C, Supplementary Fig. 2).

(A) Tivozanib-treated cells were seeded into collagen I-coated culture dishes then the adhesive cells were stained, lysed and the optical densitometry was read. (B,C) The effects of tivozanib on mRNA and protein levels of ICAM-1 and VCAM-1 were measured by qRT-PCR and Western blot analysis. β-actin was used as the loading control. The blots are representative of three independent experiments with similar results. (D) Equal amounts of secreted protein from treated cells were incubated with a synthetic substrate labelled with amino-4-trifluoromethyl coumarin (AFC). The substrate is cleaved by cathepsin B to release AFC, which is fluorometrically detected. (E) The conditioned media from each sample was subjected to a chromogenic substrate, which is cleaved by active uPA and produces a colorimetrically detectable product. (F) The conditioned media was collected and separated on a non-reducing polyacrylamide gel containing gelatin A. Gelatinolytic activities are visualized as clear bands against the blue background of stained gelatin. The zymograms are representative of three independent experiments with similar results. The gels were cropped and the full-length gels are presented in Supplementary Fig. 3. (G) The cells were placed into 8-μm porous culture inserts, treated with tivozanib and allowed to migrate for 48 h. The migrated cells on the lower surface of the inserts were fixed with methanol, stained with crystal violet, lysed with 30% acetic acid and the optical densitometry was measured at 590 nm. (H) For invasion assay, the cells were placed into matrigel-coated inserts and allowed to invade through the matrigel layer for 48 h. Data are given as mean ± SD. Statistically significant values of *p < 0.05, **p < 0.01, and ***p < 0.001 were determined compared with the control.
GBM cells secrete proteinases such as cysteine proteinase cathepsin B, serine proteinase urokinase-type plasminogen activator (uPA) and matrix metalloproteinases (MMPs) to remodel the ECM and drive tumour invasion. Cathepsin B stimulates pro-uPA, which leads to proteolytic activation of pro-MMPs and dissolution of the ECM. In this regard, the proteolytic cascade of cathepsin B/uPA/MMP is a potential therapeutic target to restrain GBM invasion33. Tivozanib reduced the enzymatic levels of cathepsin B, uPA and MMP-2 (Fig. 3D–F). Moreover, our data demonstrate that tivozanib-mediated inhibition of the proteases was associated with attenuation of cell migration and invasion (Fig. 3G,H).
Tivozanib enhances sensitivity to gefitinib
Emerging evidence indicates that the VEGF ligands act as survival factors for tumour cells that express the receptors34,35,36. In leukaemia cells, the VEGF family opposes apoptosis via induction of the anti-apoptotic protein Bcl-2 and blockade of VEGFR3 induces chemosensitisation in ovarian cancer cells37,38. These studies suggest that the VEGF family plays important roles in therapy resistance.
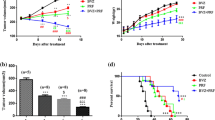
We thus determined the effects of tivozanib on proliferative response of T98G (temozolomide-resistant) and U87MG (radio- and temozolomide-resistant) cells to gefitinib, irinotecan and temozolomide. The tivozanib-gefitinib combination therapy had synergistic effects on growth inhibition and activation of caspase-3, an indicator of apoptosis (Fig. 4A–C, Table 1). Moreover, tivozanib synergistically increased irinotecan anti-tumour activity in T98G and sensitivity to temozolomide in U87MG cells (Fig. 4D–G, Supplementary Table 1,2). These data suggest that VEGFR blockade by tivozanib enhances chemosensitivity in the GBM cells.

(A) The effect of tivozanib-gefitinib combination therapy on cell proliferation was investigated by MTT assay and shown by EC50 shift analysis. (B) Normalised isobolograms of combination of tivozanib (10 μM) and gefitinib (1, 2, 5, 10, 20 and 50 μM). The data were analysed using the CalcuSyn software. The connecting line represents additivity. Data points located below the line indicate a synergistic drug-drug interaction and data points above the line indicate an antagonistic drug-drug interaction. (C) The effect of combined tivozanib-gefitinib treatment on activation of caspase 3 was measured by a colorimetric caspase 3 activity assay. (D) The effect of tivozanib-irinotecan combination therapy on cell proliferation was investigated by MTT assay and shown by EC50 shift analysis. (E) Normalised isobolograms of combination of tivozanib (10 μM) and irinotecan (0.1, 0.5, 1, 2.5, 5, 10 and 25 μg/mL). (F) The effect of tivozanib-temozolomide combination therapy on cell proliferation was investigated by MTT assay and shown by EC50 shift analysis. (G) Normalised isobolograms of combination of tivozanib (10 μM) and temozolomide (50, 100, 200, 500, 1000 and 2500 μM). Data are given as mean ± SD. Statistically significant values of *p < 0.05 and **p < 0.01 were determined compared with the control.
Discussion
Angiogenesis blockade is considered the most effective targeted therapy for GBM39. The anti-glioma effects of anti-angiogenics include inhibition of tumour-associated neoangiogenesis, disruption of tumour stem cell microvascular niche and tumour vascular normalisation40,41. VEGF pathway inhibitors may also sensitise glioma-associated endothelial cells to cytotoxic therapies42,43. Moreover, these agents reduce VEGFA-induced brain oedema and thereby, improve chemotherapy delivery to tumour cells44,45,46. Despite this, the direct anti-glial effects of angiogenic inhibitors on VEGFR-expressing GBM cells are largely unknown47. In the present study, we investigated the effects of the pan-VEGFR inhibitor tivozanib on the GBM cells.
There is evidence that aberration in cell cycle regulatory network is a frequent event in the pathogenesis of GBM48. Alteration of mitotic regulatory proteins such as Aurora kinases A and B, survivin and PLK1 correlates with a dismal survival in GBM49,50,51. Depletion of AURKA and AURKB, blocking PLK1 and shRNA suppression of CCNB1 retards tumour growth and increases chemosensitivity52,53,54,55. For instance, inhibition of PLK1 in GBM cells inhibits cell proliferation and enhances radiosensitisation56. Altogether, these findings imply that the cell cycle regulatory machinery is a potential therapeutic target in GBM in order to interrupt tumour growth57,58. In agreement, our findings reveal that tivozanib diminished proliferation of the GBM cells through a G2/M cell cycle arrest via up-regulation of p21 and down-modulation of Aurora kinases A and B, cyclin B1 and CDC25C as well as inhibition of p-PLK1.
The initiation, progression and therapeutic resistance of GBM are associated with a subset of tumour cells called glioma-initiating cells (GICs) with an extensive self-renewal capacity59,60. These glioma stem-like cells (GSCs) have the ability to transdifferentiate into tumour endothelium and contribute to neoangiogenesis and resistance to anti-angiogenics61,62. Targeted inhibition of signalling networks that drive the self-renewal of GICs is a novel therapeutic strategy in GBM63. An autocrine VEGF/VEGFR2 axis is thought to promote the self-renewal capacity and viability of GSCs and its abrogation reduces their survival and tumorigenicity64. In addition, the survival of GSCs is strongly dependent on the mitotic regulatory proteins and these cells have been shown to be sensitive to Aurora kinase A and PLK1 inhibitors65. Aurora kinase A regulates tumorigenicity of GICs by activating Wnt signalling pathway and its knockdown inhibits their “stemness”, the self-renewal capacity and tumorigenicity66. Our present results reveal that tivozanib inhibits p-PLK1 and decreases Aurora kinase A in the GBM cells, suggesting that tivozanib might have anti-tumour activity in GICs cells.
Diffuse infiltration into the normal brain tissue and metastasis throughout the cerebrospinal fluid and leptomeninges are major reasons for tumour recurrence and treatment failure in GBM67. The extracellular proteases cathepsin B, uPA and MMP-2 allow the aggressive infiltration by degradation of the ECM components and promoting GBM cell invasion68. Elevated expression of cathepsin B correlates with a poor patient survival and increased invasiveness of GBM cells and its depletion interrupts GBM growth, invasion and angiogenesis69,70,71. uPA transcript, protein and enzymatic levels correlate with tumour grade and its inhibition decreases GBM growth and invasion in vitro and tumorigenicity in vivo72,73,74. In addition, MMP-2 activity is augmented in GBM tumours compared to normal brain and its expression levels is associated with malignant progression75. Inhibition of MMP-2 activity or its knockdown reduces GBM invasion76,77. In ovarian carcinoma cells, the VEGF/VEGFR loop promotes tumour invasion through induction of uPA and MMP-2, suggesting that blockade of the VEGF family might hinder tumour invasiveness78. In concert with this, the results of the current study demonstrate that tivozanib decreased invasive potential of the GBM cells via inhibition of the enzymatic levels of cathepsin B, uPA and MMP-2. These finding further suggest that tivozanib could be applied in combination with radio- and chemotherapies in order to halt intrinsic and post-treatment invasion of GBM.
Amplification and overexpression of EGFR are observed in 50% of gliomas and associate with a worse prognosis79. Despite this, clinical trials with EGFR-targeted therapies have not shown marked activity in GBM patients80,81. Evidence indicates that increased tumour angiogenesis drives resistance to anti-EGFR therapies82,83. Elevated expression of VEGFA contributes to development of gefitinib-resistant colon cancer cells, which was abrogated by treatment with a VEGFR2 inhibitor84. Our findings suggest that tivozanib potentiates anti-tumour activity of gefitinib in the GBM cells.
Members of the Bcl-2 family of proteins play central roles in treatment response in GBM patients85. High levels of BAX correlate with better clinical outcome86. ABT737, a Bcl-2 family inhibitor, sensitises GBM cells to both anti-cancer drugs and the death ligand TRAIL87. In addition, the inhibitor of apoptosis protein survivin (encoded by BIRC5) has been shown to mediate radioresistance in GBM cells88. These findings suggest that targeting the apoptotic machinery enhances GBM responsiveness to pro-apoptotic agents89. Consistent with this, our data demonstrate that tivozanib-induced sensitisation to gefitinib might be through down-regulation of the anti-apoptotic proteins survivin and Bcl-2.
Taken together, our data demonstrate that inhibition of VEGF receptors by tivozanib reduced proliferative and invasive characteristics of the GBM cells and provide new insight into the mechanistic activity of tivozanib (Fig. 5). Combination of tivozanib with gefitinib synergistically increased the anti-tumour activity of gefitinib on cell growth and induction of apoptosis. These findings suggest that tivozanib has potential anti-glioma effects. Further in vivo studies are warranted to elucidate the anti-tumour activity of tivozanib in combinatorial approaches in GBM.

Tivozanib induced a G2/M cell cycle arrest through up-regulation of p21 and suppression of Aurora kinase A, Aurora kinase B, p-PLK1, cyclin B1 and CDC25C. Tivozanib decreased adhesive and invasive potential of the GBM cells via inhibition of ICAM-1, VCAM-1 and enzymatic levels of MMP-2. Furthermore, tivozanib increased anti-tumour activity of gefitinib on cell growth and induction of apoptosis through down-regulation of the anti-apoptotic proteins survivin and Bcl-2.
Materials and Methods
Antibodies and chemicals
Antibodies were obtained as follows: p-PLK1 (Thr210) (Cell Signalling Technology); Aurora A (Millipore); Aurora B (Abcam); PLK1 (clone F-8), VCAM-1 (clone H-276), ICAM-1 (clone H-108), Bcl-2 (clone N-19), Bax (clone N-20), survivin (clone FL-142), p21 (clone C-19), cyclin B1 (clone GNS1), Wee1 (clone C-20), CDC25 (clone C-20), c-Myc (clone 9E10) and β-actin (Santa Cruz Biotechnology).
Tivozanib was purchased from AdooQ BioScience (Irvine, CA, USA). Gefitinib (EGFR small molecule inhibitor) and temozolomide (a DNA alkylating agent) were obtained from ChemieTek (Indianapolis, IN, USA). Irinotecan (a topoisomerase I inhibitor) was purchased from pharmacy of Shariati hospital (Tehran, Iran). Poly-hydroxyethylmethacrylate polymer (poly-HEMA) was obtained from Santa Cruz Biotechnology.
Human glioblastoma cell lines
Human glioblastoma cell lines A172, T98G and U87MG were obtained from National Cell Bank of Iran (NCBI, Tehran, Iran). All the cell lines were authenticated by STR profiling using Cell IDTM system (Promega) and were routinely checked for mycoplasma infection. Cell cultures were maintained at 37 °C in 5% CO2 in a humidified incubator and cultured according to NCBI recommendations.
Cell proliferation
The cells in logarithmic growth phase were plated in 96-well plates. After incubation at 37 °C for 24 h, the cultures were exposed to desired concentrations of the drugs for 48 h and the proportion of viable cells was determined by MTT assay. Synergism was determined by calculation of combination index (CI) according to Chou and Talalay90 using CalcuSyn software (Biosoft, Cambridge, UK). CI < 1, CI = 1, and CI > 1 represent synergism, additive effects and antagonism of two drugs, respectively.
Crystal violet staining
The cells were plated at a density of 6 × 104 cells in 6-well plates and treated with tivozanib for 48 h. The cultures were fixed with ice-cold methanol and stained with crystal violet (0.5% w/v). The images were acquired with an inverted microscope.
Colony formation assay
Cells were seeded onto 6-well plates with a density of 1000–2000 cell/well. After 12 h, the cells were treated with tivozanib for 48 h. The media was then changed to drug-free media and the cells were incubated at 37 °C in 5% CO2 for 10 d. The cultures were fixed in ice-cold methanol and stained with crystal violet solution (1% w/v). The colonies were counted and surviving fraction (SF) was estimated as: (mean colony counts)/(cells plated) × (plating efficiency), where plating efficiency (PE) was determined as (mean colony counts)/(cells plated for untreated controls)91.
Anoikis resistance assay
To mimic anchorage-independent growth conditions, culture dishes were coated with poly-HEMA, as described by Sher et al.92. Approximately 5 × 103 cells were seeded under anchorage-independent conditions with tivozanib treatment for 48 h. Cell viability was determined by MTT assay.
Analysis of gene expression by quantitative reverse transcription-PCR
The quantitative reverse transcription-PCR (qRT-PCR) analysis was performed on a LightCycler® 96 instrument (Roche Molecular Diagnostics) using RealQ SYBR Green PCR reagents (Ampliqon, Copenhagen, Denmark). The primers used are listed in Table 2. Expression of target mRNA was normalized to the expression of beta-2-microglobulin (B2M) for generation of ΔCT values and relative mRNA expression was quantified using the ΔΔCT method, where CT is cycle threshold93.
Western blot analysis
The cells were lysed for 30 min in ice-cold RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate and 0.1% SDS) containing protease and phosphatase inhibitors (Roche Molecular Biochemicals). Equal amounts of protein (50 μg) were separated on SDS-PAGE, transferred to PVDF membrane (Membrane Solutions, TX, USA) then probed with primary and horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma). β-actin was used as the loading control and proteins were detected using a BM chemiluminescence detection kit (Roche Molecular Biochemicals).
Cell cycle analysis
Cell cycle analysis was performed using propidium iodide (PI) staining. Harvested cells were washed in ice-cold PBS, fixed in 70% ethanol and stored at −20 °C overnight. The cell pellets were then incubated with RNase A (100 μg/mL) (Sigma), PI (50 μg/mL) (Sigma) and 0.05% Triton X-100. Cellular DNA content was analysed on a FACSCalibur (BD Bioscience) flow cytometer equipped with CellQuest Pro software.
Zymography
Equal amounts of protein from the supernatants were applied to polyacrylamide gels copolymerized with gelatin A (Sigma). After electrophoresis, the gels were rinsed in re-activation buffer overnight and then stained with Coomassie Brilliant Blue. Areas of enzymatic activity appeared as clear bands over the dark background94.
Urokinase-type plasminogen activator activity assay
Urokinase-type plasminogen activator (uPA) activity was assayed with a uPA-specific chromogenic substrate according to the manufacturer’s instructions (Millipore). Equal amounts of protein from the conditioned media were added to the chromogenic substrate and incubated at 37 °C for 1 h. The samples were read at 405 nm.
Cathepsin B activity assay
To monitor the effect of tivozanib on activity of secreted cathepsin B, a cathepsin B activity assay (Abcam) was applied following the manufacturer’s instructions. Briefly, equal amounts of protein from the supernatants were incubated with a specific substrate of cathepsin B at 37 °C for 2 h. The substrate contains amino-4-trifluoromethyl coumarin, which is released because of cathepsin B activity. The samples were then read in a Synergy HT fluorescent microplate reader (BioTek Instruments) with a 400 nm excitation filter and a 505 nm emission filter. The cathepsin B activity was obtained by comparing the relative fluorescence units of the samples with the control group.
Cell adhesion
Cell adhesion assay was carried out as described by Ueno et al.95. The cells were treated with tivozanib for 48 h and then seeded in collagen I-coated 60 mm dishes (Biocoat Cell Environments; Becton Dickinson). After incubation for 15 min at 37 °C, cells were washed twice with cold PBS, stained with 1% crystal violet, lysed with 30% acetic acid and the optical densitometry was measured at 590 nm.
Cell migration and invasion
Transwell cell migration and invasion assays were carried out as described earlier96.
Caspase 3 activity assay
A colorimetric caspase 3 activity assay (Sigma) was conducted to quantitatively determine apoptotic cell death. Cell lysates from both adherent and floating cells were centrifuged at 20000 × g for 10 min. Twenty μg of the supernatant was incubated with 85 μL of assay buffer plus 10 μL of caspase 3 substrate acetyl-Asp- Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) in a 96-well plate at 37 °C for 12 h. The samples were then read at 405 nm in an ELISA reader.
Statistical analysis
All data were evaluated in triplicate against untreated control cells and collected from three independent experiments. Data were graphed and analysed by GraphPad Prism Software 7.0a using one-way ANOVA and the unpaired two-tailed Student’s t test. All data are presented as mean ± standard deviation (SD).
Additional Information
How to cite this article: Momeny, M. et al. Blockade of vascular endothelial growth factor receptors by tivozanib has potential anti-tumour effects on human glioblastoma cells. Sci. Rep. 7, 44075; doi: 10.1038/srep44075 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Wen, P. Y. & Kesari, S. Malignant gliomas in adults. New England Journal of Medicine 359, 492–507 (2008).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New England Journal of Medicine 352, 987–996 (2005).
Wang, Z. et al. Broad targeting of angiogenesis for cancer prevention and therapy. Seminars in cancer biology. 35, S224–S243 (2015).
Folkman, J. Angiogenesis. Annu. Rev. Med. 57, 1–18 (2006).
Lange, C., Storkebaum, E., de Almodóvar, C. R., Dewerchin, M. & Carmeliet, P. Vascular endothelial growth factor: a neurovascular target in neurological diseases. Nature Reviews Neurology (2016).
Grau, S. et al. Expression of VEGFR3 in glioma endothelium correlates with tumor grade. Journal of neuro-oncology 82, 141–150 (2007).
Godard, S. et al. Classification of human astrocytic gliomas on the basis of gene expression A correlated group of genes with angiogenic activity emerges as a strong predictor of subtypes. Cancer research 63, 6613–6625 (2003).
Dvorak, H. F. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. Journal of clinical oncology 20, 4368–4380 (2002).
Jain, R. K. et al. Angiogenesis in brain tumours. Nature Reviews Neuroscience 8, 610–622 (2007).
Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 438, 932–936 (2005).
Winkler, F. et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer cell 6, 553–563 (2004).
Lu-Emerson, C. et al. Lessons From Anti–Vascular Endothelial Growth Factor and Anti–Vascular Endothelial Growth Factor Receptor Trials in Patients With Glioblastoma. Journal of Clinical Oncology 33, 1197–1213 (2015).
Stefanik, D. F. et al. Monoclonal antibodies to vascular endothelial growth factor (VEGF) and the VEGF receptor, FLT-1, inhibit the growth of C6 glioma in a mouse xenograft. Journal of neuro-oncology 55, 91–100 (2001).
Szabo, E. et al. Autocrine VEGFR1 and VEGFR2 signaling promotes survival in human glioblastoma models in vitro and in vivo . Neuro-oncology now043 (2016).
Friedman, H. S. et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. Journal of Clinical Oncology 27, 4733–4740 (2009).
Kreisl, T. N. et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. Journal of Clinical Oncology 27, 740–745 (2009).
Vredenburgh, J. J. et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. Journal of Clinical Oncology 25, 4722–4729 (2007).
Chinot, O. L. et al. Bevacizumab plus radiotherapy–temozolomide for newly diagnosed glioblastoma. New England Journal of Medicine 370, 709–722 (2014).
Gilbert, M. R. et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. New England Journal of Medicine 370, 699–708 (2014).
Takahashi, H. & Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clinical science 109, 227–241 (2005).
Jamil, M. O., Hathaway, A. & Mehta, A. Tivozanib: Status of Development. Current oncology reports 17, 1–7 (2015).
Eskens, F. A. et al. Biologic and clinical activity of tivozanib (AV-951, KRN-951), a selective inhibitor of VEGF receptor-1,-2, and-3 tyrosine kinases, in a 4-week-on, 2-week-off schedule in patients with advanced solid tumors. Clinical Cancer Research 17, 7156–7163 (2011).
Nosov, D. A. et al. Antitumor activity and safety of tivozanib (AV-951) in a phase II randomized discontinuation trial in patients with renal cell carcinoma. Journal of clinical oncology 30, 1678–1685 (2012).
Paoli, P., Giannoni, E. & Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1833, 3481–3498 (2013).
Chow, J. P. & Poon, R. Y. The CDK1 inhibitory kinase MYT1 in DNA damage checkpoint recovery. Oncogene 32, 4778–4788, doi: 10.1038/onc.2012.504 (2013).
Boutros, R., Lobjois, V. & Ducommun, B. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer 7, 495–507, doi: 10.1038/nrc2169 (2007).
Sanchez, Y. et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277, 1497–1501 (1997).
Zitouni, S., Nabais, C., Jana, S. C., Guerrero, A. & Bettencourt-Dias, M. Polo-like kinases: structural variations lead to multiple functions. Nature Reviews Molecular Cell Biology 15, 433–452 (2014).
Archambault, V. & Carmena, M. Polo-like kinase-activating kinases: Aurora A, Aurora B and what else? Cell Cycle 11, 1490–1495 (2012).
Laoukili, J. et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nature cell biology 7, 126–136 (2005).
Cuddapah, V. A., Robel, S., Watkins, S. & Sontheimer, H. A neurocentric perspective on glioma invasion. Nature Reviews Neuroscience 15, 455–465 (2014).
Nakada, M. et al. Molecular targets of glioma invasion. Cellular and molecular life sciences 64, 458–478 (2007).
Rao, J. S. Molecular mechanisms of glioma invasiveness: the role of proteases. Nature Reviews Cancer 3, 489–501 (2003).
Calvani, M., Trisciuoglio, D., Bergamaschi, C., Shoemaker, R. H. & Melillo, G. Differential involvement of vascular endothelial growth factor in the survival of hypoxic colon cancer cells. Cancer Research 68, 285–291 (2008).
Pidgeon, G. P., Barr, M. P., Harmey, J. H., Foley, D. A. & Bouchier-Hayes, D. J. Vascular endothelial growth factor (VEGF) upregulates BCL-2 and inhibits apoptosis in human and murine mammary adenocarcinoma cells. British Journal of Cancer 85, 273 (2001).
Hua, K.-T. et al. Vascular endothelial growth factor-C modulates proliferation and chemoresistance in acute myeloid leukemic cells through an endothelin-1-dependent induction of cyclooxygenase-2. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1843, 387–397 (2014).
Dias, S., Shmelkov, S. V., Lam, G. & Rafii, S. VEGF165 promotes survival of leukemic cells by Hsp90-mediated induction of Bcl-2 expression and apoptosis inhibition. Blood 99, 2532–2540 (2002).
Lim, J., Yang, K., Taylor-Harding, B., Wiedemeyer, W. R. & Buckanovich, R. J. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia 16, 343–353. e342 (2014).
Chowdhary, S. & Chamberlain, M. Bevacizumab for the treatment of glioblastoma. Expert review of neurotherapeutics 13, 937–949 (2013).
Tong, R. T. et al. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer research 64, 3731–3736 (2004).
Jain, R. K. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307, 58–62 (2005).
Duda, D. G., Jain, R. K. & Willett, C. G. Antiangiogenics: the potential role of integrating this novel treatment modality with chemoradiation for solid cancers. Journal of Clinical Oncology 25, 4033–4042 (2007).
Kerbel, R. S. Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science 312, 1171–1175 (2006).
Jain, R. K., Tong, R. T. & Munn, L. L. Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: insights from a mathematical model. Cancer research 67, 2729–2735 (2007).
Tate, M. C. & Aghi, M. K. Biology of angiogenesis and invasion in glioma. Neurotherapeutics 6, 447–457 (2009).
Batchelor, T. T. et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer cell 11, 83–95 (2007).
Chi, A. S., Sorensen, A. G., Jain, R. K. & Batchelor, T. T. Angiogenesis as a therapeutic target in malignant gliomas. The oncologist 14, 621–636 (2009).
McLendon, R. et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008).
Tsai, H.-P. et al. Association of Aurora A and gamma-tubulin expression in astrocytomas and patient survival. Neurological research 36, 746–751 (2014).
Zeng, W. F., Navaratne, K., Prayson, R. A. & Weil, R. J. Aurora B expression correlates with aggressive behaviour in glioblastoma multiforme. Journal of clinical pathology 60, 218–221 (2007).
Cheng, M.-W., Wang, B.-C., Weng, Z.-Q. & Zhu, X.-W. Clinicopathological significance of Polo-like kinase 1 (PLK1) expression in human malignant glioma. Acta histochemica 114, 503–509 (2012).
Hata, T. et al. RNA interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer research 65, 2899–2905 (2005).
He, S.-J. et al. Inhibition of Aurora kinases induces apoptosis and autophagy via AURKB/p70S6K/RPL15 axis in human leukemia cells. Cancer Letters 382, 215–230 (2016).
Renner, A. G. et al. Polo-like kinase 1 is overexpressed in acute myeloid leukemia and its inhibition preferentially targets the proliferation of leukemic cells. Blood 114, 659–662 (2009).
Yuan, J. et al. Stable gene silencing of cyclin B1 in tumor cells increases susceptibility to taxol and leads to growth arrest in vivo . Oncogene 25, 1753–1762 (2006).
Tandle, A. T. et al. Inhibition of polo-like kinase 1 in glioblastoma multiforme induces mitotic catastrophe and enhances radiosensitisation. European Journal of Cancer 49, 3020–3028 (2013).
Thomas, A. A., Brennan, C. W., DeAngelis, L. M. & Omuro, A. M. Emerging therapies for glioblastoma. JAMA neurology 71, 1437–1444 (2014).
Alexander, B. M., Pinnell, N., Wen, P. Y. & D’Andrea, A. Targeting DNA repair and the cell cycle in glioblastoma. Journal of neuro-oncology 107, 463–477 (2012).
Stiles, C. D. & Rowitch, D. H. Glioma stem cells: a midterm exam. Neuron 58, 832–846 (2008).
Bao, S. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. nature 444, 756–760 (2006).
Ricci-Vitiani, L. et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 468, 824–828 (2010).
Wang, R. et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468, 829–833 (2010).
Vescovi, A. L., Galli, R. & Reynolds, B. A. Brain tumour stem cells. Nature Reviews Cancer 6, 425–436 (2006).
Hamerlik, P. et al. Autocrine VEGF–VEGFR2–Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. The Journal of experimental medicine 209, 507–520 (2012).
Mannino, M., Gomez-Roman, N., Hochegger, H. & Chalmers, A. J. Differential sensitivity of Glioma stem cells to Aurora kinase A inhibitors: implications for stem cell mitosis and centrosome dynamics. Stem cell research 13, 135–143 (2014).
Xia, Z. et al. AURKA governs self-renewal capacity in glioma-initiating cells via stabilization/activation of β-catenin/Wnt signaling. Molecular Cancer Research 11, 1101–1111 (2013).
Paw, I., Carpenter, R. C., Watabe, K., Debinski, W. & Lo, H.-W. Mechanisms regulating glioma invasion. Cancer letters 362, 1–7 (2015).
Vehlow, A. & Cordes, N. Invasion as target for therapy of glioblastoma multiforme. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer 1836, 236–244 (2013).
Colin, C. et al. High expression of cathepsin B and plasminogen activator inhibitor type-1 are strong predictors of survival in glioblastomas. Acta neuropathologica 118, 745–754 (2009).
Demchik, L. L., Sameni, M., Nelson, K., Mikkelsen, T. & Sloane, B. F. Cathepsin B and glioma invasion. International journal of developmental neuroscience 17, 483–494 (1999).
Lakka, S. S. et al. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene 23, 4681–4689 (2004).
Zhang, X. et al. Expression and localisation of urokinase-type plasminogen activator gene in gliomas. Journal of clinical neuroscience 7, 116–119 (2000).
Raghu, H., Gondi, C. S., Dinh, D. H., Gujrati, M. & Rao, J. S. Specific knockdown of uPA/uPAR attenuates invasion in glioblastoma cells and xenografts by inhibition of cleavage and trafficking of Notch-1 receptor. Molecular cancer 10, 1 (2011).
Gondi, C. S. et al. Expression of antisense uPAR and antisense uPA from a bicistronic adenoviral construct inhibits glioma cell invasion, tumor growth, and angiogenesis. Oncogene 22, 5967–5975 (2003).
Forsyth, P. et al. Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. British journal of cancer 79, 1828 (1999).
Kesanakurti, D., Chetty, C., Maddirela, D. R., Gujrati, M. & Rao, J. Functional cooperativity by direct interaction between PAK4 and MMP-2 in the regulation of anoikis resistance, migration and invasion in glioma. Cell death & disease 3, e445 (2012).
Badiga, A. V. et al. MMP-2 siRNA inhibits radiation-enhanced invasiveness in glioma cells. PloS one 6, e20614 (2011).
So, J., Wang, F.-q., Navari, J., Schreher, J. & Fishman, D. A. LPA-induced epithelial ovarian cancer (EOC) in vitro invasion and migration are mediated by VEGF receptor-2 (VEGF-R2). Gynecologic oncology 97, 870–878 (2005).
Shinojima, N. et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer research 63, 6962–6970 (2003).
Raizer, J. J. et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro-oncology 12, 95–103 (2010).
Lassman, A. B. et al. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01. Clinical Cancer Research 11, 7841–7850 (2005).
Viloria-Petit, A. et al. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo A role for altered tumor angiogenesis. Cancer research 61, 5090–5101 (2001).
Vallböhmer, D. et al. Molecular determinants of cetuximab efficacy. Journal of Clinical Oncology 23, 3536–3544 (2005).
Ciardiello, F. et al. Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clinical Cancer Research 10, 784–793 (2004).
Strik, H. et al. BCL-2 family protein expression in initial and recurrent glioblastomas: modulation by radiochemotherapy. Journal of Neurology, Neurosurgery & Psychiatry 67, 763–768 (1999).
Ruano, Y. et al. Identification of survival‐related genes of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma multiforme. Cancer 112, 1575–1584 (2008).
Tagscherer, K. E. et al. Apoptosis-based treatment of glioblastomas with ABT-737, a novel small molecule inhibitor of Bcl-2 family proteins. Oncogene 27, 6646–6656 (2008).
Chakravarti, A. et al. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene 23, 7494–7506 (2004).
Krakstad, C. & Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: opportunities for targeted therapeutics. Molecular cancer 9, 1 (2010).
Chou, T.-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer research 70, 440–446 (2010).
Franken, N. A., Rodermond, H. M., Stap, J., Haveman, J. & Van Bree, C. Clonogenic assay of cells in vitro . Nature protocols 1, 2315–2319 (2006).
Sher, I., Adham, S. A., Petrik, J. & Coomber, B. L. Autocrine VEGF‐A/KDR loop protects epithelial ovarian carcinoma cells from anoikis. International journal of cancer 124, 553–561 (2009).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative CT method. Nature protocols 3, 1101–1108 (2008).
Momeny, M. et al. Silibinin inhibits invasive properties of human glioblastoma U87MG cells through suppression of cathepsin B and nuclear factor kappa B-mediated induction of matrix metalloproteinase 9. Anti-cancer drugs 21, 252–260 (2010).
Ueno, Y. et al. Heregulin‐induced activation of ErbB3 by EGFR tyrosine kinase activity promotes tumor growth and metastasis in melanoma cells. International journal of cancer 123, 340–347 (2008).
Momeny, M. et al. Heregulin-HER3-HER2 signaling promotes matrix metalloproteinase-dependent blood-brain-barrier transendothelial migration of human breast cancer cell lines. Oncotarget 6, 3932–3946 (2015).
Acknowledgements
This study was supported by a grant from Haematology/oncology and Stem Cell Transplantation Research Centre, Shariati hospital, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran. Technical assistance of Ms. Razieh Khoshsohbat, Ms. Roghieh Koohi Ortakand and Ms. Malihe Zahiri is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
M.M. designed the research; F.M., N.K.G., H.Y., Z.S., G.Z., S.M., H.E., F.B. and L.A. conducted the research; M.G., A.R.D., S.A., S.M.T., L.D., A.H.E., K.A. and A.G. analysed the data; M.M. wrote the paper; S.H.G. had primary responsibility for the final content. All authors have reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Momeny, M., Moghaddaskho, F., Gortany, N. et al. Blockade of vascular endothelial growth factor receptors by tivozanib has potential anti-tumour effects on human glioblastoma cells. Sci Rep 7, 44075 (2017). https://doi.org/10.1038/srep44075
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep44075
- Springer Nature Limited
This article is cited by
-
ID1high/activin Ahigh glioblastoma cells contribute to resistance to anti-angiogenesis therapy through malformed vasculature
Cell Death & Disease (2024)
-
Relevance of Thymic Stromal Lymphopoietin on the Pathogenesis of Glioblastoma: Role of the Neutrophil
Cellular and Molecular Neurobiology (2024)
-
CTSB is a negative prognostic biomarker and therapeutic target associated with immune cells infiltration and immunosuppression in gliomas
Scientific Reports (2022)
-
Anti-tumor activity of cediranib, a pan-vascular endothelial growth factor receptor inhibitor, in pancreatic ductal adenocarcinoma cells
Cellular Oncology (2020)
-
Dacomitinib, a pan-inhibitor of ErbB receptors, suppresses growth and invasive capacity of chemoresistant ovarian carcinoma cells
Scientific Reports (2017)