
Abstract
The biological effects of indium-tin-oxide (ITO) are of considerable importance because workers exposed to indium compounds have been diagnosed with interstitial lung disease or pulmonary alveolar proteinosis; however, the pathophysiology of these diseases is undefined. Here, mice intraperitoneally inoculated with ITO-nanoparticles (ITO-NPs) resulted in peritonitis dependent in NLRP3 inflammasome, with neutrophils recruitment and interleukin-1β (IL-1β) production. Withal peritoneal macrophages exposed ex vivo to ITO-NPs caused IL-1β secretion and cytolysis. Further, alveolar macrophages exposed to ITO-NPs in vitro showed ITO-NP endocytosis and production of tumor necrosis factor-α (TNF-α) and IL-1β, ensued cell death by cytolysis. This cell death was RIPK1-independent but caspase1-dependent, and thus identified as pyroptosis. Endocytosis of ITO-NPs by activated THP-1 cells induced pyroptosis with IL-1β/TNF-α production and cytolysis, but not in activated THP-1 cells with knockdown of NLRP3, ASC, or caspase1. However, exposing activated THP-1 cells with NLRP3 or ASC knockdown to ITO-NPs resulted in cell death but without cytolysis, with deficiency in IL-1β/TNF-α, and revealing features of apoptosis. While, mesenchymal stem cells (MSCs) co-cultured with macrophages impaired both inflammation and cell death induced by ITO-NPs. Together, our findings provide crucial insights to the pathophysiology of respiratory diseases caused by ITO particles, and identify MSCs as a potent therapeutic.
Similar content being viewed by others


Introduction
Recently, respiratory diseases were reported in workers exposed to indium compounds such as indium oxide (InO) and indium tin oxide (ITO) particles1. These workers showed interstitial lung diseases within 4 to 13 years after the first exposure or pulmonary alveolar proteinosis just 1 to 2 years after the first exposure. As compared with the most prevalent form of silicosis, which generally results from exposure to silica (SiO2) particles for 20 years or more2, the onset of lung diseases among workers exposed to indium compounds seems to occur promptly after exposure1.
Current studies have revealed that activation of NOD-like receptor (NLR), pyrin domain-containing 3 (NLRP3) inflammasome in macrophages often results in excessive inflammation responsible for various diseases3,4. As well, the development of pulmonary fibrosis after exposure to SiO2 particles or asbestos fibers may depend on NLRP3 inflammasome activation5,6,7. Sintered-ITO was suggested to cause in vitro adverse inflammatory responses in macrophages and epithelial cells that could involve in part inflammasome activation8. However, whether ITO-NPs can activate the NLRP3 inflammasome in vivo is unknown. Moreover, the molecular mechanisms implicated in the recognition and endocytosis of ITO-NPs by alveolar macrophages and monocyte-derived macrophages leading to caspase1-dependent inflammation and cell death are unclear.
Fundamentally, NLRP3 inflammasome is an intracellular heteromeric complex expressed in immune cells that contains the NLRP3 protein, apoptosis-associated speck-like protein containing the caspase recruitment domain (ASC) and pro-caspase1. Following NLRP3 inflammasome activation and assembly, pro-caspase1 is cleaved via autocatalytic processes to result in the active form of caspase19. NLRP3 inflammasome is activated by diverse agents including adverse signals such as extracellular ATP via the purinergic P2×7 receptor (P2×7R) associated with pannexin1 (Panx1) K+ hemichannels10,11. In particular, NLRP3 inflammasome is activated by crystalline and particulate substances that are produced in excess by dysfunctional tissues such as monosodium urate (MSU) crystals in gout12, and amyloid fibrils in type-2 diabetes13, or inorganic xenogenous compounds such as SiO2 particles and asbestos fibers6. Activation of NLRP3 inflammasome leads to the maturation, via caspase1, of pro-interleukin (pro-IL-1β) and pro-IL-18 cytokines into their active and secreted forms, IL-1β and IL-1814. Eventually, activation of NLRP3 inflammasome results in the execution of a regulated cell death (RCD) known as pyroptosis through cleavage of Gasdermin D (GSDMD) by caspase1, which ultimately causes the loss of plasma membrane integrity15,16,17. In contrast to a RCD event such as apoptosis, which is non-lytic and non-immunogenic16, pyroptosis is completed by the rupture of the plasma membrane, with potent immunogenicity and with implications in the pathogenesis of various diseases15.
To initiate inflammation, particles and fibers must be detected and engulfed by macrophages and this can require surface receptors to recognize xenogenous compounds18. Scavenger receptors (SRs) such as SR class-A (SR-A) MARCO and CD204 or SR class-B (SR-B) CD36, have been suggested to facilitate the uptake of SiO2 particles and asbestos fibers by macrophages18,19,20,21,22. Further, CD36 directly coordinates NLRP3 inflammasome activation mediated by oxidized low-density lipoprotein (LDL), amyloid-β and amylin peptides, in sterile inflammation related-diseases such as atherosclerosis, Alzheimer’s disease and type-2 diabetes13. Therefore, SRs could function upstream of NLRP3 inflammasome and caspase1, which suggests significant therapeutic targets in treating respiratory diseases due to chronic inhalation of xenogenous compounds13,18,21,22.
This study is motivated to provide molecular insights to the pathophysiology of respiratory diseases caused by occupational exposure to indium compounds and to identify a therapeutic strategy for these diseases. Our hypothesis is that resident macrophages and monocyte-derived macrophages, exposed to small-sized particles of ITO induce NLRP3-ASC inflammasome activation resulting in severe inflammation and pyroptotic cell death. These conditions are particularly detrimental as they can allow a prolonged inflammation, tissue damages and fibrosis23,24. Moreover, we hypothesized that mesenchymal stem cells (MSCs), well-known for their immunosuppressive and regenerative properties25,26, could prevent both inflammation and cell death of macrophages upon exposure to ITO particles.
Therefore, this work aimed to elucidate the mechanisms of ITO-NP–mediated inflammation in vivo in mice and in vitro via extracellular recognition by SRs of ITO-NPs by alveolar macrophages to caspase1-dependent pyroptosis execution. We also aimed to examine the path of ITO-NP uptake by human monocyte-derived macrophages to intracellular NLRP3-ASC inflammasome activation and caspase1-dependent pyroptosis execution. We compared the effects of ITO-NPs in alveolar macrophages and monocyte-derived macrophages with inflammatory compounds such as silica nanoparticles (SiO2-NPs) and MSU crystals. Finally, we evaluated human bone marrow-derived mesenchymal stem cells (MSCs) as suppressors of pyroptosis in co-culture with alveolar macrophages and monocyte-derived macrophages exposed to ITO-NPs, SiO2-NPs or MSU.
Here, we report that 1) ITO-NPs induce peritonitis in mice with neutrophil recruitment and IL-1β production that depends on NLRP3 inflammasome and that, 2) endocytosis of ITO-NPs induce caspase1-dependent pyroptosis in macrophages via extracellular interaction with at least SR-A CD204 and caspase1 function, involving intracellular activation of NLRP3-ASC inflammasome and that, 3) MSCs act as a pyroptosis suppressor. The pyroptosis induced by ITO-NPs like SiO2-NPs in macrophages may be a mechanism of exacerbated inflammation in lung diseases caused by inhalation of inorganic particles such as ITO and SiO2, and MSCs treatment may be a potential therapeutic suppressor of the pyroptosis.
Results
Intraperitoneal inoculation in mice of ITO-NPs induces peritonitis dependent on NLRP3 inflammasome activation
MSU crystals cause an innate immune response via inflammasome, because mice deficient in ASC (a key component of inflammasome) show impaired neutrophil recruitment after intraperitoneal (i.p.) inoculation of MSU crystals as compared with WT mice12. Using a comparable approach, we found that mice with i.p. ITO-NP inoculation showed induced peritonitis with inflammatory cell recruitment (Fig. 1a–g and Supplemental Fig. S1a,b). Indeed, among CD45+CD11b+ peritoneal cells of mice inoculated i.p. with ITO-NPs, we found a marked increase of neutrophils phenotypically characterized as CD11b+Gr-1+Ly-6G+ and inflamed monocytes characterized as CD11b+Gr-1+ Ly-6G− (Fig. 1b,c,f). Along with the efflux of neutrophils and inflamed monocytes, the level of IL-1β was significantly increased within peritoneal fluid of mice receiving ITO-NPs (Fig. 1g). By contrast, in control mice, CD45+CD11b+ peritoneal cells were negative for the Gr-1 marker, indicating an absence of neutrophils or inflamed monocytes (Fig. 1b,f). Among these peritoneal cells, a subset expressed a high density of CD11b antigens, and were further phenotypically characterized as CD11b+F4/80+ (Fig. 1e and Supplemental Fig. S1a,b), representing resting resident peritoneal macrophages27.

(a) Schematic diagram of the experimental procedures used in vivo and ex vivo in Balb/c mice to examine the role of ITO-NPs in inducing NLRP3 inflammasome-dependent peritonitis. (b) Flow cytometer analysis of peritoneal cells of control Balb/c mice (black dots), (c) mice receiving ITO-NPs (red dots) and (d) both NLRP3 inflammasome inhibitor glybenclamide (Gly) and ITO-NPs (green dots). (b) Dot-plots show peritoneal cells of control mice stained for CD11b, Gr-1 and Ly-6G; about 20% of the CD45+ cells were peritoneal macrophages phenotypically characterized as positive for CD11b and negative for Gr-1 and Ly-6G. (c) 6-h post-inoculation i.p. of ITO-NPs in Balb/c mice, CD11b+Gr-1+ increases, to reach about 60% Ly-6G positive (neutrophils) and 13% Ly-6G negative (inflamed-monocytes). (d) Balb/c mice receiving i.p. Gly and ITO-NPs show reduced number of CD11b+Ly-6G+ neutrophils. (b–d) One representative experiment out of 4 is shown. (e) Offset histogram overlays representing cell surface density of CD11b antigens with mean fluorescence intensity (MFI x103) on CD45+ peritoneal cells from control mice (black), ITO-NP–inoculated mice (red), Gly– and ITO-NP–receiving mice (green). (f) Absolute number of cells phenotypically characterized as CD11b+Gr-1+, CD11b+Gr-1+Ly-6G+ (neutrophils) and CD11b+Gr-1+Ly-6G− (inflamed-monocytes) among CD45+ peritoneal cells from control mice and mice receiving ITO-NPs or both Gly and ITO-NPs. (g) IL-1β detected in peritoneal fluid from control mice, mice receiving ITO-NPs and both Gly and ITO-NPs. (h) Release of IL-1β, TNF-α and of LDH in mice peritoneal macrophages stimulated ex vivo with phorbol 12-myristate 13-acetate (PMA) with or without 500 μg/mL of ITO-NP exposure for 16 h. (b–h), n = 4 ± SEM, *p < 0.05. Schematic diagram abbreviations, MΦ = macrophages, Neu = neutrophils, Mo = monocytes, BMCs = bone marrow cells, BMDMs = bone marrow-derived macrophages, PMΦ = peritoneal macrophages.
Peritonitis induced by ITO-NPs was diminished by impeding NLRP3 inflammasome activation by a sulfonylurea pharmacological inhibitor, glybenclamide (Gly) (Fig. 1d–g). Gly acts upstream of NLRP3 inflammasome by inhibiting K+ efflux mediated via the Panx1 ion channel28,29. Here, Gly greatly reduced the number of neutrophils without affecting that of inflamed monocytes in mice receiving ITO-NPs (Fig. 1c–f). Thus, the peritoneal neutrophil/monocyte cell ratio was about 4:1 in mice receiving ITO-NPs but <2:1 in mice receiving both Gly and ITO-NPs (Supplemental Fig. S1c). Furthermore, Gly restrained IL-1β production in ITO-NPs receiving mice (Fig. 1g). Here, our data indicate that ITO-NPs activate NLRP3 inflammasome in vivo to exacerbate inflammation.
To verify whether resident peritoneal macrophages could secrete pro-inflammatory cytokines and undergo cytolysis in response to ITO-NPs, we examined their ability ex vivo to secrete pro-inflammatory cytokines IL-1β and TNF-α and to release lactate dehydrogenase (LDH) after exposure to ITO-NPs. We found that peritoneal macrophages exposed to ITO-NPs resulted in IL-1β and TNF-α production and release of LDH (Fig. 1h), similar results were observed when peritoneal macrophages were exposed to SiO2-NPs and MSU (Supplemental Fig. S1d). As well, to exclude any endotoxin-mediated effect resulting from contamination of ITO-NPs, splenocytes and bone marrow cells (BMCs) isolated from naïve mice were exposed to ITO-NPs or lipopolysaccharide (LPS) and examined for their viability and ability to secrete TNF-α. Splenocytes and BMCs responded vigorously to LPS in terms of TNF-α production but were unable to secrete TNF-α on exposure to ITO-NPs at 50, 100 and 500 μg/mL, whilst cell viability was not disturbed for any treatments tested (Supplemental Fig. S2a,b). Thus, ITO-NPs were excluded from contamination with endotoxins and ITO-NPs could not affect splenocytes, or immature BMCs. Yet, in agreement with the findings obtained with resident peritoneal macrophages, ITO-NPs affected bone marrow-derived macrophages (BMDMs) by stimulating the release of IL-1β and TNF-α, and by decreasing cell viability through cytolysis (Supplemental Fig. S2c,d). Here, our findings indicate that ITO-NPs affect specifically macrophages. These unfavorable effects of ITO-NPs on macrophages might be related to their capability for particles uptake.
Endocytosis of ITO-NPs by alveolar macrophages provokes TNF-α and IL-1β production and caspase1-dependent cell death
Next, we compared the molecular mechanisms leading to inflammation and cell death in alveolar macrophages (AMs) exposed to ITO-NPs, SiO2-NPs and MSU crystals (Fig. 2a). Dysfunction of AMs is often associated with the pathogenesis of respiratory diseases30. Particularly, AMs prevent xenogenous compounds from accumulating into alveolar sacs, and AM endocytic potentials provide a vital defense30. We used transmission electronic microscopy (TEM) to investigate the uptake of ITO-NPs by MH-S cells, an established murine cell line of AMs. At 6-h post-exposure to 50 μg/mL ITO-NPs, MH-S cells exhibited endosomes containing ITO-NPs appearing as aggregated electronic dense spots (Fig. 2b,c). At 24-h post-exposure, endosomes were enlarged and contained an increased amount of ITO-NPs (Fig. 2c). With higher concentrations of ITO-NPs, MH-S cells showed endosomes comprising an augmented amount of ITO-NPs (Supplemental Fig. S3). Thus, the size of endosomes containing ITO-NPs was dose and time-dependent. As well, MH-S cells stimulated with LPS and exposed to ITO-NPs exhibited endosomes containing ITO-NPs (Supplemental Fig. S4a). Additionally, MH-S cells were capable of uptake of SiO2-NPs and MSU crystals (Supplemental Fig. S4b), compounds with a strong ability to trigger activation of NLRP3 inflammasome14. MH-S cells exposed to ITO-NPs dose- and time-dependently secreted TNF-α (Fig. 2d). In parallel, intracellular TNF-α level was increased (Fig. 2e), which demonstrates de novo protein production by macrophages in response to ITO-NPs. As well, intracellular and secreted TNF-α level was increased in MH-S cells exposed to SiO2-NPs or MSU crystals (Supplemental Fig. S4d–f).

(a) Schematic diagram of the experimental procedures to consider alveolar macrophage response upon exposure to ITO-NPs, SiO2-NPs or MSU. (b) TEM of non-exposed MH-S cells shows preserved cytoplasmic organelles, regular microvilli protruding, and well-delineated nucleus with heterochromatin. (c) MH-S cells exposed for 6 h to 50 μg/mL ITO-NPs exhibited early endosomes containing ITO-NPs appearing as dark spots. At 24 h post-exposure to ITO-NPs at 50 μg/mL, endosomes in MH-S contained an increased amount of ITO-NPs. (b,c) Original magnification x3000, x4000 and x8000, 1 representative TEM image is shown from at least 3. (d) MH-S cell production of TNF-α upon exposure to increasing doses, i.e. 50, 100 and 500 μg/mL, of ITO-NPs. MH-S cells exposed to ITO-NPs at 500 μg/mL and examined at 3, 6, 12 and 16 h for secretion of TNF-α. (e) Intracellular TNF-α level found in MH-S cells exposed ITO-NPs compared to unstimulated MH-S. (f) Secreted and intracellular TNF-α levels in MH-S cells treated with dexamethasone (DEX) and exposed to ITO-NPs. (g) IL-10 production in MH-S cells exposed to ITO-NPs with DEX treatment and compared to controls. (h) MH-S cells treated with or without Cyto D and exposed to ITO-NPs for 12 h were analyzed in 3 representative areas (RA) on x500 TEM images. The number of endocytic cells containing ITO-NPs was compared to the total number of nucleated cells in RA; data are given as percentage of endocytic cells. Cyto D effect was evaluated for TNF-α secretion and intracellular TNF-α expression in MH-S upon exposure to ITO-NPs. (i) Intracellular IL-1β level was evaluated in MH-S cells with ITO-NP exposure; with or without DEX and Cyto D treatment compared to controls. The concentration of ITO-NPs was 500 μg/mL and time of exposure 16 h, unless stated otherwise. (b–i) n = 3–8 ± SEM, *p < 0.05. TEM abbreviations, Cyto = cytoplasm, N = nucleus, M = mitochondria and E = endosome.
Further, we examined dexamethasone (DEX) as a potent immunosuppressive molecule that could prevent ITO-NPs–induced inflammatory signaling in macrophages. DEX diminished the intracellular and secreted TNF-α level in MH-S cells exposed to ITO-NPs (Fig. 2f), but secretion of the immunosuppressive cytokine IL-10 was increased (Fig. 2g). As well, DEX lessened TNF-α secretion in MH-S cells exposed to SiO2-NPs (Supplemental Fig. S4e). Next, we examined ITO-NP uptake blockade by using an inhibitor of actin polymerization, cytochalasin D (Cyto D). Cyto D prevented endocytosis of ITO-NPs in MH-S cells (Fig. 2h and Supplemental Fig. S4c), and significantly decreased TNF-α secretion (Fig. 2h). However, and by contrast to DEX, Cyto D did not counter the intracellular overexpression of TNF-α in MH-S cells exposed to ITO-NPs (Fig. 2h). As compared with Cyto D, DEX may regulate inflammatory proteins at the transcriptional level in preventing inflammatory signaling induced by ITO-NPs. In accordance with this data, MH-S cells exposed to ITO-NPs or SiO2-NPs showed upregulated IL-1β expression (Fig. 2i and Supplemental Fig. S4g), and DEX but not Cyto D markedly inhibited IL-1β overexpression (Fig. 2i). Here, blockade of endocytosis indicated that ITO-NPs could interact with surface receptors expressed on AMs, which was sufficient to transduce intracellular pro-inflammatory signaling. Moreover, DEX might counteract these inflammatory signaling at the transcriptional level.
ITO-NPs induce caspase1-dependent pyroptosis in alveolar macrophages involving scavenger receptor class-A (SR-A) CD204
We next investigated ITO-NP–mediated cell death in AMs and whether caspase1, endocytosis and SRs are involved in this process. In addition to increasing pro-inflammatory cytokine production, ITO-NPs inhibited cell proliferation of MH-S cells (Supplemental Fig. S5a) and, LPS exacerbated this inhibition (Supplemental Fig. S5b). DEX did not prevent cell proliferation inhibition in MH-S exposed to ITO-NPs (Supplemental Fig. S5c). This cell proliferation inhibition was accompanied by a decrease in cell viability (Supplemental Fig. S5d,g), and consistently, LPS intensified the decreased cell viability (Supplemental Fig. S5e,h). Of note, DEX could not reverse the loss of cell viability induced by ITO-NPs in MH-S cells (Supplemental Fig. S5f,i). Furthermore, we examined the ability of 51Cr-radiolabeled MH-S cells exposed to ITO-NPs to release 51Cr into the extracellular milieu, which represents a direct measurement of AM plasma membrane rupture. 51Cr-radiolabeled MH-S exposed to ITO-NPs dose-dependently released 51Cr into the extracellular milieu (Fig. 3a), and LPS augmented the 51Cr release (Fig. 3b). Of note, DEX did not prevent MH-S cells from releasing 51Cr into the extracellular milieu with ITO-NP exposure (Fig. 3c). On the other hand, pharmacological blockade of caspase1 activity by Ac-YVAD significantly reduced 51Cr release resulting from exposure of 51Cr-radiolabeled MH-S cells to ITO-NPs (Fig. 3d). Consistently, exposed MH-S cells released LDH; Ac-YVAD but not DEX prevented the LDH release (Fig. 3e). Ac-YVAD diminished TNF-α secretion of exposed MH-S cells (Fig. 3f). Together, these data reveal that AMs exposed to ITO-NPs undergo a regulated cell death (RCD), with plasma membrane rupture, dependent on caspase1 that is evocative of pyroptosis31.

(a) MH-S cell release of 51Cr increased in dose-dependently on exposure to 50, 100, and 500 μg/mL of ITO-NPs. (b) MH-S cells treated with LPS and exposed to 50 μg/mL ITO-NPs were compared to control for release of 51Cr. (c) DEX treated MH-S were evaluated for 51Cr release on exposure to ITO-NPs. (d) Blockade of caspase1 with Ac-YVAD in MH-S cells exposed to increasing doses of ITO-NPs were evaluated for preventing 51Cr release. (e) LDH release was examined in MH-S cells exposed to 500 μg/mL ITO-NPs for 24 h with Ac-YVAD or DEX treatment. (f) TNF-α secretion from MH-S cells exposed to 500 μg/mL ITO-NPs with or without Ac-YVAD, 16 h post-exposure, were measured and compared to controls. (g) The effect of ITO-NPs endocytosis blockade with Cyto D were evaluated in MH-S cells for LDH release at 16 and 24 h post-exposure to 500 μg/mL ITO-NPs. (h) ROS scavenger N-acetyl-(L)-cysteine (NAC) and blockade of RIPK1 with necrostatin-1 (NEC-1) were tested in MH-S cells at 24 h post-exposure to 500 μg/mL ITO-NPs for LDH release. (i) MH-S cells exposed to ITO-NPs for 8 h compared to unstimulated MH-S were analyzed after Annexin V and propidium iodide (PI) staining on flow cytometer. Dot plot of Annexin V and PI staining of MH-S cells exposed to ITO-NPs (red) compared to control (black). FSC/SSC shows MH-S with ITO-NPs uptake with increasing granularity compared to control and cells noted E(+) are for cells containing endosome. Percentage of E(+) cells are given. (j) Percentage of total cells being stained with Annexin V and PI are given. An increase in the percentage of cells double positive Annexin V+PI+ among MH-S exposed to ITO-NPs, suggest a cell death with plasma membrane rupture. (a–j) n = 3–6 ± SEM, *p < 0.05.
Next, we sought to precise the molecular pathway involved in AM cell death induced by ITO-NPs. Cyto D prevented the LDH release in MH-S exposed to ITO-NPs (Fig. 3g), which demonstrates a critical role for ITO-NP endocytosis in triggering pyroptosis. Comparable protective effects of Cyto D were observed in MH-S cells exposed to SiO2-NPs (Supplemental Fig. S6a,b) or MSU crystals (Supplemental Fig. S6c,d). Furthermore, we examined whether necroptosis, a RCD that depends on receptor-interacting Ser/Thr-protein kinase 1 (RIPK1)32, or accidental cell membranes rupture due to oxygen species overproduction33, are involved in ITO-NPs–induced cell death. Necrostatin-1 (NEC-1), a potent RIPK1 inhibitor, did not neutralize LDH release in MH-S exposed to ITO-NPs (Fig. 3h), nor did neutralizing reactive oxygen species (ROS) with N-acetyl-(L)-cysteine (NAC), a ROS scavenger, diminish cell death by plasma membrane rupture in exposed MH-S cells (Fig. 3h). These data highlight a central role for caspase1 in inducing a RCD in AMs exposed to ITO-NPs, and excludes a cell death via RIPK1-mediated necroptosis or a cell death resulting from cell membranes damage due to ROS overproduction. Further, MH-S cells exposed to ITO-NPs weakly exhibited phosphatidylserine (PS) phospholipids on their outer leaflet membrane as revealed by a modest increase in the number of AnnexinV+PI− cells (Fig. 3i,j). Whereas, AnnexinV+PI+ cells were further represented in MH-S exposed to ITO-NPs (Fig. 3i,j), indicating breakage in plasma membrane integrity and featuring pyroptotic cells. These data suggest a RCD occurring in MH-S cells on ITO-NP exposure that is not necroptosis nor apoptosis, but a RCD caspase1-dependent, with plasma membrane disruption, typical of pyroptosis16.
To identify the receptors expressed by AMs that could enable ITO-NPs to induce pyroptosis, we first examined whether MH-S cells could express the collagenous scavenger receptor class-A (SR-A) MARCO, CD204 and the non-collagenous SR-B CD36 and whether ITO-NPs could interact with SRs. MH-S cells expressed SR-A CD204 and SR-B CD36, but SR-A MARCO were almost not detectable on cell surface (Fig. 4a and Supplemental Fig. S7a). Furthermore, MH-S cells exposed to ITO-NPs and SiO2-NPs showed a marked decrease in SR-A CD204 surface expression and to a lesser extent SR-B CD36 expression (Fig. 4a–d and Supplemental Fig. S7b–e). Hence, we examined whether neutralizing SR-A MARCO, CD204 or SR-B CD36 with specific monoclonal antibodies (mAbs) could reverse ITO-NP– and SiO2-NP–induced pyroptosis. Remarkably, blockade of SR-A CD204 with 2F8 mAb significantly prevented the death of MH-S cells exposed to ITO-NPs or SiO2-NPs as measured by LDH release (Fig. 4e and Supplemental Fig. S7f). However, blockade of SR-B CD36 or SR-A MARCO with JC63.1 or ED31 mAbs, respectively, did not prevent plasma membrane disruption of MH-S cells exposed to ITO-NPs and SiO2-NPs (Fig. 4f and Supplemental Fig. S7g–i). Thus, our data indicate that ITO-NPs may interact with AMs through SR-A CD204 leading ultimately to caspase1-dependent pyroptosis.

(a) Cell surface expression in resting MH-S of SR-A (collagenous) CD204 and SR-B (non-collagenous) CD36, was assessed by flow cytometer analysis (dark grey). Histograms in light grey represent isotype control. (b) Flow cytometer analysis (red) of CD204 and CD36 surface expression in MH-S cells exposed to ITO-NPs. (a,b) FSC/SSC dot-plots show MH-S with ITO-NPs uptake with increasing granularity compared to control and cells noted E(+) represented cells with endosomes. Percentage of E(+) cells are given. (c) Histograms showing the percentage of CD204+ cells and their respective MFI in unstimulated MH-S cells and MH-S cells exposed to ITO-NPs. (d) Percentage of CD36+ and their respective MFI in unstimulated MH-S cells compared to MH-S cells exposed to ITO-NPs. (e) Blockade of CD204 using purified monoclonal antibodies (mAbs) clone 2F8 (anti-CD204) or (f) blockade of SR-B CD36 with mAbs clone JC63.1 (anti-CD36) in MH-S cells exposed to ITO-NPs. (e,f) MH-S cells were examined for LDH release at 16 and 24-h post-exposure to ITO-NPs compared to controls. Concentration of ITO-NPs was 500 μg/mL, time of exposure was 16 h unless stated otherwise. n = 4–6 ± SEM, *p < 0.05.
Endocytosis of ITO-NPs by phorbol 12-myristate 13-acetate (PMA)-activated monocytes is required for caspase1-dependent pyroptosis execution
We next investigated the ITO-NP effects in human monocyte-derived macrophages (Fig. 5a). Cell proliferation of non-activated THP-1 cells was poorly affected by ITO-NPs (Supplemental Fig. S8a). However, when THP-1 cells were activated with PMA, cell proliferation was significantly inhibited (Supplemental Fig. S8b). THP-1 cells lacked ITO-NP uptake capability as assessed by TEM (Fig. 5b,c). However, when activated with PMA, THP-1 cell ability for endocytosis of ITO-NPs was greatly increased (Fig. 5b,c), which resulted in increased production of TNF-α and IL-1β (Fig. 5d), and ultimately cell death via cytolysis as assessed by LDH release (Fig. 5e). Similar results were found with PMA-activated THP-1 cells exposed to SiO2-NPs or MSU crystals (Supplemental Fig. S8c–e).

(a) Schematic diagram indicating the experimental procedures used to examine monocyte (THP-1 WT) and knockdown monocyte (THP-1 genetically deficient) for Caspase1 or NLRP3 or ASC response upon exposure to ITO-NPs, SiO2-NPs or MSU. (b) THP-1 cells (−) or (+) PMA uptake capability of ITO-NPs was evaluated by TEM. TEM analysis shows that activation of THP-1 cells with PMA potentiates endocytosis of ITO-NPs. Percentages of phagocytic cells are given for (−) or (+) PMA conditions in THP-1 cells exposed to ITO-NPs at 12 h post-exposure. c, TEM of ITO-NPs present outside (black arrow) THP-1 cells with non-activated (−) PMA compared to (+) PMA showing an endosome containing ITO-NPs; 1 representative TEM image in at least 3 is shown, original magnification ×3000. (d) Production of TNF-α and IL-1β and (e) release of LDH by PMA-activated THP-1 cells exposed to ITO-NPs compared to controls. (d,e) PMA potentiates THP-1 intracellular expression of IL-1β; further exposure of PMA-activated THP-1 cells with ITO-NPs augments IL-1β production and release of LDH. (f) DEX effects in PMA-activated THP-1 exposed to ITO-NPs in terms of TNF-α, IL-1β and LDH release. While DEX impairs TNF-α and IL-1β production, DEX does not prevent cytolysis. (g) Cyto D blockade of ITO-NP uptake (black arrow) by PMA-activated THP-1 cells at 12 h post-exposure; 1 representative TEM image in at least 3 is shown. (h) Mean percentages of endocytic cells at 12 h post-exposure to ITO-NPs are given for PMA-activated THP-1 with or without cyto D; and effects of ITO-NPs endocytosis blockade in PMA-activated THP-1 in production of IL-1β and LDH release. Overall, concentration of ITO-NPs was 500 μg/mL and time of exposure was 16 h, except if stated otherwise. (b–h) n = 3–9 ± SEM, *p < 0.05. TEM abbreviations, Cyto = cytoplasm, N = nucleus and E = endosome.
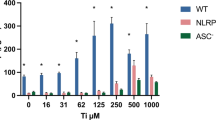
Then, we considered caspase1-dependent pyroptosis in activated monocytes, to elucidate the molecular pathway of pro-inflammatory cytokines production and LDH release following ITO-NPs endocytosis. As described for AMs, DEX inhibited production of both TNF-α and IL-1β in PMA-activated THP-1 cells exposed to ITO-NPs but did not prevent cell death (Fig. 5f). Importantly, Cyto D obstructed endocytosis of ITO-NPs in PMA-activated THP-1 cells, restrained IL-1β secretion and prevented LDH release (Fig. 5g,h). By contrast, NAC and NEC-1 did not impede cell death in PMA-activated THP-1 cells exposed to ITO-NPs (Fig. 6a). Consistent with our findings regarding AMs, pharmacological blockade of caspase1 activity using Ac-YVAD, significantly restricted cell death and diminished IL-1β and TNF-α secretion in PMA-activated THP-1 cells exposed to ITO-NPs (Fig. 6a,b). Similar beneficial effect of Ac-YVAD was found in PMA-activated THP-1 cells with SiO2-NP or MSU crystal exposure (Supplemental Fig. S9a–b). To reveal whether caspase1 is specifically indispensable to pyroptosis in activated-monocytes ensuing ITO-NPs endocytosis, and to exclude a role of caspase4 and caspase5 in participating in this event17, we examined the response of THP-1 genetically knockdown for caspase1 (i.e. deficient in caspase1, abridged def-Casp1) exposed to ITO-NPs. We found that while PMA-activated wild-type (WT) THP-1 exposed to increasing doses of ITO-NPs produced IL-1β in a dose-dependent manner, PMA-activated THP-1 knockdown for caspase1 were unable to secrete IL-1β (Fig. 6c). As well, PMA-activated THP-1 caspase1 knockdown were prevented from plasma membrane rupture resulting of ITO-NPs exposure, as assessed by an absence of LDH release compared to WT control (Fig. 6d).

(a) LDH release as measured in PMA-activated THP-1 cells exposed to ITO-NPs with caspase1 activity blockade using Ac-YVAD, compared with neutralizing ROS with NAC or RIPK1 with NEC-1 (b) IL-1β and TNF-α secretion in PMA-activated THP-1 cells exposed to ITO-NPs with caspase1 activity blockade using Ac-YVAD. (c) Effect of increasing doses of ITO-NPs in PMA-activated THP-1 with a stable knockdown in caspase1 (def-Casp1) compared to wild-type (WT) PMA-activated THP-1 in terms of IL-1β secretion and (d) LDH release. (e) Intracellular expression and release of IL-1α alarmin in PMA-activated THP-1 exposed to ITO-NPs compared to controls. (f) Caspase1 activity blockade with Ac-YVAD or (g) endocytosis of ITO-NPs blockade with Cyto D, reduced IL-1α release by PMA-activated THP-1 cells exposed to ITO-NPs. Concentration of ITO-NPs was 500 μg/mL, and time of exposure was 16 h, unless stated otherwise. (a–g), n = 3–4 ± SEM, *p < 0.05.
Thereby the pyroptosis is achieved through plasma-membrane rupture, and thus of the release of intracellular contents that can include molecules such as alarmin IL-1α34,35. IL-1α is considered to have a potent role in inducing tissue damage and fibrosis34,36. Therefore, we examined the ability of activated monocytes exposed to ITO-NPs to produce and release IL-1α. IL-1α production and release was significantly upregulated in PMA-activated THP-1 cells exposed to ITO-NPs, and similar results were found with SiO2-NP or MSU crystal exposure (Fig. 6E and Supplemental Fig. S9c,d). Impeding caspase1 activity or endocytosis with Ac-YVAD or Cyto D prevented IL-1α release of PMA-activated THP-1 cells exposed to ITO-NPs (Fig. 6f,g). These findings indicate that both the release of IL-1α and the cell death through cytolysis, induced by ITO-NPs in macrophages, is dependent on activity of caspase1. As well, endocytosis of ITO-NPs by PMA-activated THP-1 cells was required for IL-1α release, which is consistent with our data showing that blockade of endocytosis, prevented the rupture of plasma membrane in macrophages exposed to ITO-NPs, thus impeding intracellular content release of IL-1α.
PMA-activated monocytes with NLRP3 or ASC knockdown show ITO-NP uptake with apoptotic cell death rather than pyroptosis
Because NOD-like receptor NLRP3 and its adaptor ASC are critical components of the NLRP3 inflammasome permitting caspase1 activation4,14,37, we next investigated whether ITO-NPs could specifically require NLRP3-ASC-Caspase1 axis for inducing pyroptosis in macrophages. We compared the effects of ITO-NPs in THP-1 cells with stable knockdown of NLRP3 (def-NLRP3) or ASC (def-ASC) and WT THP-1 cells. On exposure to ITO-NPs, PMA-activated THP-1 cells with NLRP3 or ASC deficiency were unable to release LDH, thus showing for unaltered plasma membranes (Fig. 7a). Furthermore, PMA-activated THP-1 WT cells secreted IL-1β and TNF-α with increasing doses of ITO-NPs (Fig. 7b,c); whereas PMA-activated THP-1 cells with NLRP3 or ASC deficiency were unable to secrete IL-1β or TNF-α (Fig. 7b,c).

(a) LDH release capabilities as measured in PMA-activated THP-1 either wild-type (WT) or NLRP3 knockdown (def-NLRP3), or ASC knockdown (def-ASC) on exposure to increasing doses of ITO-NPs. (b) PMA-activated THP-1 WT cells exposed to increasing doses of ITO-NPs but not cells with NLRP3 knockdown or ASC knockdown released IL-1β dose-dependently. (c) Similarly, PMA-activated THP-1 WT cells exposed to increasing doses of ITO-NPs but not cells with NLRP3 knockdown or ASC knockdown released TNF-α dose-dependently. (d) PMA-activated THP-1 WT cells or with NLRP3 knockdown or ASC knockdown exposed to ITO-NPs were assessed for cell viability by trypan blue dye exclusion cell count on light microscope. Results are given as number of cells alive and dead cells (×106) with or without exposure to ITO-NPs. (e) PMA-activated THP-1 WT cells or cells with NLRP3 knockdown or ASC knockdown exposed to ITO-NPs were stained with Annexin V and PI to determine the relative number of single positive cells Annexin V+PI− (apoptotic cells) compared to the number of cells double positive Annexin V+PI+ (pyroptotic cells). Results are given as a percentage of total cells with or without exposure to ITO-NPs. (f) TEM analysis of PMA-activated THP-1 WT cells exposed to ITO-NPs exhibited almost exclusively endocytic cells containing ITO-NPs with plasma membrane being ruptured; black arrow shows ITO-NPs being released after plasma membrane has dislocated (red arrow). By contrast, PMA-activated THP-1 cells with NLRP3 knockdown or ASC knockdown exhibited cells with capabilities of ITO-NPs uptake (black arrow), showing morphological features of apoptosis, such as membrane blebs and apoptotic bodies (blue arrow). TEM original magnification x3000 and x8000; 1 representative image is shown in at least 3. Concentration of ITO-NPs was 500 μg/mL, and time of exposure was 16 h, unless stated otherwise. (a–f) n = 3–8 ± SEM, *p < 0.05. TEM abbreviations, Cyto = cytoplasm, N = nucleus and E = endosome.
Nonetheless, ITO-NPs increased cell death among both THP-1 WT and NLRP3 or ASC knockdown THP-1 cells with PMA activation, as assessed by optical microscope cell count on trypan blue dye exclusion (Fig. 7d). As cell viability decline was not associated with LDH release in NLRP3 or ASC knockdown THP-1 cells with PMA activation and exposed to ITO-NPs (Fig. 7a), we examined whether these cells were subjected to apoptosis. We found that WT PMA-activated THP-1 cells, upon exposure to ITO-NPs, exhibited a modest increase in AnnexinV+PI− apoptotic cells as compared to Annexin V+PI+ pyroptotic cells (Fig. 7e). On the contrary, NLRP3 or ASC knockdown THP-1 cells with PMA activation, ITO-NP exposure resulted in a marked increase of Annexin V+PI− apoptotic cells, while Annexin V+PI+ pyroptotic cells were not augmented compared to WT control (Fig. 7e). Consistently, TEM of PMA-activated THP-1 cells with NLRP3 or ASC deficiency exposed to ITO-NPs were capable of ITO-NPs uptake, and often exhibited apoptotic morphological features among macrophages with endosomes containing ITO-NPs, such as membrane blebs and apoptotic bodies (Fig. 7f). Conversely, among PMA-activated THP-1 WT cells exposed to ITO-NPs, unfit endocytic cells often appeared with disrupted plasma membranes (Fig. 7f). Our data suggest that circumvention of NLRP3 or ASC inflammasome components in macrophages exposed to ITO-NPs could prevent pyroptosis but facilitate alternatively apoptosis. Moreover, ITO-NP endocytosis by macrophages did not directly or, indirectly through ROS overproduction, causes damage to cell membranes, but rather demonstrated that NLRP3 inflammasome activation and caspase1 function are specifically required for pyroptosis. Blockade of inflammation using DEX or even specific blockade of NLRP3 inflammasome components might not be optimal in preventing cell death in macrophages exposed to ITO-NPs.
Mesenchymal stem cells prevent pyroptosis in alveolar macrophages and activated monocytes with ITO-NP, SiO2-NP or MSU crystal exposure
Next, we examined whether mesenchymal stem cells (MSCs) could prevent both the inflammatory response and cell death in macrophages undergoing pyroptosis (Fig. 8a). MSCs possess immunosuppressive and regenerative properties capable of controlling inflammatory disorders such as graft versus host disease, Crohn’s disease and chronic obstructive pulmonary disease25,26,38. Here, primary MSCs were derived from adult human bone marrow mononuclear cells39. MSCs were expanded in vitro and established as CD14-CD34-CD45- non-hematopoietic cells and phenotypically characterized as CD73+CD90+CD105+ cells (Fig. 8b). On TEM, MSCs were large with a clear cytoplasm containing an eccentric nucleus and were morphologically discernable from macrophages (Fig. 8c,e). MSCs co-cultured with MH-S or PMA-activated THP-1 cells exposed to ITO-NPs were analyzed by TEM. MH-S and PMA-activated THP-1 cells but not MSCs were capable of ITO-NP endocytosis (Fig. 8d,e). Furthermore, TEM of co-cultured MSCs and macrophages showed tight cell-to-cell interaction between MSCs and macrophages exhibiting endosomes containing ITO-NPs, without displaying plasma membrane rupture or apoptosis morphological features (Fig. 8e). Furthermore, MH-S cells exposed to ITO-NPs released a reduced amount of LDH, which was directly proportional to the number of MSCs present (Fig. 8f). As well, TNF-α secretion decreased significantly when MSCs were co-cultured with ITO-NP-exposed MH-S cells (Fig. 8g). Similar results were found for MH-S exposed to SiO2-NPs and MSU crystals on co-culture with MSCs (Supplemental Fig. S10a,b). Consistently, MSCs alleviated LDH release and TNF-α secretion resulting from exposure to ITO-NPs in PMA-activated THP-1 cells (Fig. 8h,i). Similar protective effect of MSCs was found in PMA-activated THP-1 cells upon exposure to SiO2-NPs or MSU crystals (Supplemental Fig. S10c,d). Concomitantly, we found a significant increase in secretion of the immunosuppressive cytokine IL-10 in PMA-activated THP-1 cells co-cultured with MSCs and exposed to ITO-NPs, SiO2-NPs or MSU compared to controls (Fig. 8j and Supplemental Fig. S10e). Therefore, MSCs possess potent ability to protect macrophages against both arms of pyroptosis – inflammation and cell death – induced by ITO-NPs, SiO2-NPs or MSU crystals and might favor tissue regeneration by increasing IL-10 secretion to further promote tissue healing40.

(a) Schematic diagram indicating the experimental procedures used to examine the effects of MSCs in suppressing pyroptosis, upon exposure of macrophages to ITO-NPs, SiO2-NPs or MSU. (b) Cytometer analysis of MSCs shows cells negative for hematopoietic markers CD14, CD34 and CD45, but positive for MSC markers CD73, CD90, and CD105; 1 representative histogram in 4 for each marker is shown. (c) TEM of MSCs shows large cells with a clear cytoplasm and eccentric nucleus; 1 representative image in 3 is shown; original magnification ×1500. (d) MSCs appear on TEM as poorly capable of ITO-NP uptake (black arrow) at 12 h post-exposure; 1 representative image in 3 is shown; original magnification ×500. (e) TEM of MSCs co-cultured with MH-S or PMA-activated THP-1 cells exposed to ITO-NPs. TEM reveals tight cell-to-cell contacts (orange arrow) between MSCs and macrophages containing ITO-NP endosomes (black arrow); original magnification ×3000 and ×8000, 1 representative TEM image in 3 is shown. (f) MSCs co-cultured with MH-S cells exposed to ITO-NPs exhibited LDH release that was inversely proportional to the number of MSCs present. (g) MSCs effect on TNF-α release by MH-S cells exposed to ITO-NPs. (h) MSCs co-cultured with PMA-activated THP-1 cells exposed to ITO-NPs were tested for LDH release, and the release of LDH was inversely proportional to the number of MSCs present. (i) TNF-α release by PMA-activated THP-1 cells exposed to ITO-NPs in co-culture with MSCs. (j) MSCs co-cultured with PMA-activated THP-1 cells exposed to ITO-NPs were examined for IL-10 release. MSCs alone are represented as an empty green circle on the graph. Concentration of ITO-NPs was 500 μg/mL, and exposure time 16 h, unless stated otherwise. (b–j) n = 3–6 ± SEM, *p < 0.05. TEM abbreviations, Cyto = cytoplasm, N = nucleus and E = endosome.
Discussion
Cummings K. and colleagues revealed that occupational inhalation of InO and ITO particles cause respiratory diseases1. These respiratory diseases are characterized by a rapid evolution: the median age at diagnosis is 35 years old, with cases of premature death1,41,42. To provide further insights into the pathophysiology for respiratory diseases; we examined the inflammatory ability of ITO-NPs in vivo in mice and in vitro in AMs and monocyte-derived macrophages.
We first show that ITO-NP–caused peritonitis in mice depended on NLRP3 inflammasome. Resident macrophages first in contact with ITO-NPs may undergo cell death via cytolysis, which can have a considerable impact by inducing prolonged inflammation with tissue damage and fibrosis. Consistently, a mouse model of bleomycin-induced pulmonary fibrosis showed that ATP released from injured cells amplified NLRP3 inflammasome43. Previous studies have shown that extracellular ATP availability and subsequent activation of the P2×7R/Panx1 pathway predominantly enhanced neutrophil accumulation in inflammatory diseases10,12,44,45. This agrees with our results showing that blockade of NLRP3 inflammasome with glybenclamide in the ITO-NP–mediated peritonitis mouse model principally reduced the influx of neutrophils and IL-1β production.
Then, we examined the ITO-NP effects in AMs and monocyte-derived macrophages. Particularly, AMs exposed to ITO-NPs, SiO2-NPs, or MSU crystals provoked a regulated cell death (RCD) distinguished by cytolysis that depended on caspase1 activity; this RCD was identified as pyroptosis16. Necroptosis, which is a RIPK1–dependent and caspase1–independent RCD that resembles to pyroptosis, was not involved in mediating cell death of AMs exposed to ITO-NPs. Indeed, blockade of RIPK1 with NEC-1 failed to rescue AMs from cytolysis. As well, blockade of ROS with NAC in AMs exposed to ITO-NPs did not impede cytolysis, which demonstrates the major role of caspase1 in inducing pyroptosis. Furthermore, ITO-NPs and SiO2-NPs induced pyroptosis in AMs via scavenger receptor class-A (SR-A) CD204, and endocytosis of these particles was required for execution of pyroptosis. The blockade of CD204, but not of SR-A MARCO or SR-B CD36, reduced the cytolysis induced by ITO-NPs or SiO2-NPs in AMs. Thus, SR-A CD204 could have a role in AMs as an extracellular pattern-recognition receptor46,47,48, acting upstream of NLRP3 inflammasome, to detect and facilitate endocytosis of ITO-NPs like SiO2-NPs.
Further, endocytosis of ITO-NPs by macrophages results in NLRP3 inflammasome activation, resulting in the production of IL-1β, IL-1α and TNF-α, and cytolysis, both events – inflammation and cell death – being caspase1-dependent. Moreover, ITO-NP–induced pyroptosis was abrogated in PMA-activated THP-1 cells with knockdown of NLRP3, ASC or caspase1. Thus, NLRP3 inflammasome was indispensable in mediating caspase1-dependent pyroptosis in macrophages exposed to ITO-NPs. Concurrently, we have excluded other inflammasome pathways such as NLRP1, NLRC4, NLRP6 or AIM214,37,49 participating in caspase1-dependent pyroptosis induced by ITO-NPs. In addition, PMA-activated THP-1 cells with caspase1 knockdown and ITO-NP exposure were unable to undergo pyroptosis. This finding excludes the implication of caspase4 or caspase5, caspases known for inducing pyroptosis independently to NLRP3 inflammasome and caspase150,51, in the cell death of PMA-activated THP-1 cells with ITO-NP exposure. Altogether, it appears that pyroptosis induced in macrophages upon exposure to ITO-NPs is strictly dependent on endocytosis of the particles, NLRP3 inflammasome activation and caspase1 function. However, both NLRP3 and ASC knockdown THP-1 cells, with PMA activation and with ITO-NP exposure underwent a cell death with features of apoptosis. While PMA-activated THP-1 cells with NLRP3 or ASC deficiency and ITO-NP exposure did not secrete IL-1β/TNF-α or undergo cytolysis, cell viability has declined with an increase in AnnexinV+PI− apoptotic cells. Moreover, TEM of PMA-activated THP-1 cells knockdown with NLRP3 or ASC and ITO-NP exposure exhibited morphological features of apoptosis, with endocytic cells showing membrane blebs and apoptotic bodies. This finding agrees with the hypothesis that NLRP3 inflammasome might play a role as a molecular switch in guiding inflammatory (i.e., pyroptosis) and non-inflammatory (i.e., apoptosis) RCD52,53.
Thus, controlling prolonged inflammation induced by xenogenous compounds implies targeting both inflammation and cell death54. Although inflammation can be impeded by directly targeting inflammatory pathways, cell death might still occur among immune cells, thus disorganizing the innate immunity16,54. Indeed, DEX moderates lung fibrosis in rodents55,56, and glucocorticoids are used as treatment for certain respiratory diseases57; however, the actual benefit of DEX is still poorly defined58. In our study, we examined DEX properties in the context of pyroptosis induced by ITO-NPs, SiO2-NPs and MSU crystals in macrophages. We ascertained that DEX inhibits the production of IL-1β and TNF-α in macrophages exposed to ITO-NPs, SiO2-NPs or MSU crystals. Yet, DEX did not neutralize caspase1-dependent cell death induced by ITO-NPs in macrophages. Consistently, in a model of Mycobacterium tuberculosis (MTb) infection of microglial cells, which vigorously activate caspase1 and result in IL-1β and TNF-α upregulation, pre-treating microglial cells with DEX and infection with MTb downregulated IL-1β and TNF-α transcription59. In contrast, DEX can increase NLRP3 transcriptional expression in macrophages60, rendering macrophages even more responsive to caspase1-dependent cell death. Agreeing with this, our findings indicate that blockade of inflammatory pathways by DEX or by circumventing NLRP3 inflammasome somehow did not protect macrophages exposed to ITO-NPs against cell death. Thus, we sought an approach with MSCs to control both arms of pyroptosis –inflammation and cytolysis while preventing apoptosis.
Here, we show that MSCs can prevent pyroptosis in alveolar macrophages and monocyte-derived macrophages induced by ITO-NPs, SiO2-NPs and MSU crystals. MSCs cooperate with macrophages containing endosomes with ITO-NPs likely via cell-to-cell contact as assessed by TEM, ultimately resulting in decreased pro-inflammatory cytokines release, cell death and enhanced IL-10 immunosuppressive cytokine production. Hence, IL-10 is critical for both reducing inflammation and favoring tissue healing61,62. Most importantly, MSCs reduced cytolysis of macrophages exposed to ITO-NPs without increasing apoptosis. The anti-apoptotic properties of MSCs towards immune and non-immune cells are well established63,64,65. Here, we show that MSCs possess potent anti-pyroptotic effect in macrophages.
Altogether, our findings indicate that respiratory diseases induced by ITO-NPs can be mediated NLRP3-ASC assembly and caspase1-dependent pyroptosis in macrophages, and identifies MSCs as promising therapeutic to suppress pyroptosis (Supplemental Fig. S11). These conclusions may be extended to other inflammatory disorders involving pyroptosis such as in silicosis and gout. Further investigations on the molecular pathways involved in MSCs for preventing pyroptosis in cells are critical to improve MSC-based therapeutic settings in inflammatory diseases and in regenerative medicine.
Methods
ITO-NPs, SiO2-NPs and MSU
Indium (III) oxide: Tin (IV) oxide (ITO) nanoparticles (ITO-NPs) are particles with size <50 nm (Sigma-Aldrich). Silicon (IV) oxide SiO2-NPs are particles with diameter <100 nm (Invivogen). Monosodium urate (MSU) is a solid crystal (Adipogen). ITO-NPs and SiO2-NPs apparent size were assessed on transmission electron microscopy. ITO-NPs and SiO2-NPs were used as particles in suspension, while MSU was soluble in complete medium. Complete medium consisted of RPMI 1640 (Sigma-Aldrich), 10% heat-inactivated FBS (Gibco), L-glutamin, streptomycin and penicillin (Sigma-Aldrich). Solution containing ITO-NPs were UV light-treated and absence of endotoxin-content were assessed on mouse splenocytes compared to stimulation with positive controls containing lipopolysaccharide endotoxins.
Mouse peritonitis model
All animal experiments, including procedures, sampling and animal cares, were approved by the ethical committee of Kochi Medical School (Japan), in accordance with guidelines and regulations. Mice used in this study were purchased from SLC, Inc (Japan), and were allowed one week for acclimation to the animal facility (Kochi Medical School) before studies were initiated. Peritonitis was induced by injection of 2 mg of ITO-NPs in 1 mL PBS into the peritoneum cavity of male Balb/c mice (7–12 weeks). 6 h post-inoculation, mice were sacrificed and peritoneal cavities were washed with 1 mL of PBS. Peritoneal lavage fluids were centrifuged and supernatants were kept at −20 °C for ELISA. Repeated peritoneal lavage (5 times) with 1 mL PBS was performed to obtain peritoneal cells. Total peritoneal cells were used for flow cytometer analysis. Blockade of NLRP3 inflammasome was achieved by injecting i.p. 200 μL/mouse of a glybenclamide (Sigma-Aldrich) solution in PBS (Gly) representing a dose of 50 mg/kg, at least 45 min before ITO-NP injection.
Cells
Peritoneal cells were from naïve Balb/c mice and seeded at 2 × 106 cells/mL complete medium, allowed to adhere in culture dishes for 1 h in an incubator. Adherent cells were peritoneal macrophages assessed as CD11b+F4/80+cells by flow cytometer. Splenocytes were from spleen and bone marrow cells were obtained by flushing tibiae and femora of naïve Balb/c mice. Bone marrow–derived macrophages (BMDMs) were obtained by culture of bone marrow cells in a BMDM medium consisting of RPMI 1640, 10% FBS, 1% penicillin/streptomycin, 0.01% 2-β mercaptoethanol (Sigma-Aldrich), and 20 ng/mL of recombinant murine GM-CSF (Biolegend). After 4 days, BMDMs medium was replaced, and adherent cells were harvested after 8 days as BMDM. MH-S and THP-1 wild-type (WT), both from ATCC, were used as alveolar (mouse) and monocyte-like (human) macrophages. THP-1 cells knockdown in NLRP3, ASC and caspase1 were from Invivogen. Cells were cultured in RPMI 1640 complete medium. Hygromycin B selective antibiotic (Sigma-Aldrich) was used at 100 μg/mL for knockdown THP-1 cells. Lipopolysacharride (LPS) from Escherichia coli 0111:B4 (Sigma-Aldrich) was used at 100 ng/mL in some experiments. Phorbol 12-myristate 13-acetate (PMA) from Sigma-Aldrich, was used at 20 ng/mL to activate THP-1 cells; about 1 h before experiments.
Mesenchymal stem cells (MSCs) and co-culture with macrophages
Approval for collecting procedures and use of human biological samples were obtained from the medical ethics committee at the University Hospital of Toulouse (France), with written informed consents obtained from all donors. Human marrow cells were from femoral heads adults undergoing orthopedic surgery. Mononuclear cells contained in marrow cells were isolated by Ficoll-Histopaque 1077 (Sigma-Aldrich), plated in culture flasks at 105 cells/cm2, in α-MEM (Gibco), 10% heat-inactivated FBS, and 1 ng/mL human recombinant basic fibroblast growth factor (b-FGF, RD Systems). The culture was depleted of hematopoietic cells by successive medium removal made every other day. A confluent spindle/fibroblastic cell monolayer was obtained about 15 day after initial plating and were considered as MSCs passage 0 (P0). MSCs were used at P4 to P8 for phenotyping, characterization and co-culture experiments. MSC were allowed to adhere before co-culture with macrophages in RPMI 1640 complete medium. For TEM, M:MSCs co-culture were carried out with M:MSC of 2:1, M = 106 cells in 6-well plate. For ELISA, M:MSCs co-culture experiments were carried out in 24-well plates at M:MSC of 2:1, M = 106 cells. For LDH release, MSCs were co-cultured with macrophages at M:MSC 5:1, 2:1 and 1:1, M = 105 cells in round-bottom 96-well plates.
Pharmacological inhibitors and blockade with antibodies
Inhibitors (all from Sigma-Aldrich) and antibodies were added to cells at least 30 min before stimulation. Dexamethasone (DEX) was used at 100 ng/mL. Ac-Tyr-Val-Ala-Asp-chloromethylketone (Ac-YVAD), inhibitor of caspase1, was used at 10 μg/mL. Cytochalasin D (Cyto D) was used at 20 μM. Necrostatin-1 (NEC-1), an inhibitor of RIPK1 was used at 50 μM. N-Acetyl-(L)-cysteine (NAC) was used at 20 μg/mL. Purified monoclonal antibodies (mAbs) for blockade of MARCO (Clone ED31) and CD204 (Clone 2F8) were from AbD Serotec, and purified mAbs for CD36 (Clone JC63.1) were from Cayman Chemical. All mAbs were used at 2 μg/mL.
Cell viability
Cells were plated at 105 cells per well in a final volume of 200 μL. CCK-8 solution (Sigma-Aldrich) was added then plates were incubated for 4 h and read on a microplate reader (Multiskan, Thermo Scientific) at 450 nm. Data were obtained as OD and given as % of control, with control = cell in complete medium and, % of control = (OD sample/OD mean of control) ×100.
[3H]-Thymidine incorporation
Cells were inoculated into round-bottom 96-well plates at 2.5 × 104 cells per well in of 200 μL, stimulated under various conditions and allowed to incubate at 37 °C for 6 h, then pulsed with 0.1 μCi [3H]-Thymidine (Perkin-Elmer) for 18 h at 37 °C. Cells were harvested onto FilterMat A filters (Wallac) by using an automated cell harvester (Harvester 96, Tomtec). The FilterMat A was then dried and a solid scintillator (Meltlex, Perkin-Elmer) was used according to the manufacturer’s instructions. β-scintillation counts, counts per minute (c.p.m), of [3H]-Thymidine incorporated into DNA were determined with a MicroBeta reader (Wallac, Perkin-Elmer).
[51Cr] Chromium release
Cells were labeled with [51Cr] (Perkin-Elmer) by incubating 106 cells in RPMI 1640, 1% heat-inactivated FBS and 50 μCi of [51Cr] for 1 h at 37 °C. [51Cr]-labeled cells were washed 3 times with PBS and re-suspended in complete medium. Cells were inoculated in 96-well round-bottom plates, 2.5 × 104 cells/well were stimulated in a volume of 200 μL and incubated at 37 °C for 6 h. Then, 30 μL of supernatants from each well were transferred into a scintillator-coated plate (Lumaplate, Perkin-Elmer) and incubated overnight at room temperature. [51Cr] was counted by β-scintillation, counts per minute (c.p.m) with use of a Microbeta reader (Wallac, Perkin-Elmer). Cells treated with 2% Tween 20 (Sigma-Aldrich) in distilled water represented a measure for maximal [51Cr] release, and cells in complete medium were a measure of spontaneous [51Cr] release. Results were given as raw c.p.m or as [51Cr]-release % of maximum release = (sample [51Cr]-release)-(spontaneous [51Cr]-release)/(maximal [51Cr]-release)-(spontaneous [51Cr]-release) ×100.
LDH release
105 cells/well were stimulated in round bottom 96 well-plates in a volume of 200 μL. 100-μL of supernatant from each well was transferred into flat-bottom 96-well plates, then catalyst and dye solutions were added (LDH Detection Kit, Takara). Then 100 μL HCl 1N/well were added, and plates were read on a microplate reader (Multiskan, Thermo Scientific) at 450 nm. Results are given as % of maximum release, with medium = no cells and TX-100 = cells + 1% Triton X-100, finally % of maximum release = (OD sample − OD medium)/(OD TX-100 − OD medium) ×100.
Flow cytometry
Cells were stained in PBS with 1% FBS and Trustain FcX (Biolegend) and antibodies conjugated with FITC, PE or APC. Stained cells were fixed in PBS 1% FBS and 0.5% formaldehyde (Sigma-Aldrich). Mouse antibodies for CD11b (Clone M1/70), CD45 (Clone 30-F11), F4/80 (Clone BM8), Gr-1 (Clone RB6-8C5), or Ly-6G (Clone 1A8) were from Biolegend. Mouse antibodies for MARCO (Clone ED31) and CD204 (Clone 2F8) were from AbD Serotec. Mouse antibody for CD36 (Clone HM36) was from Biolegend. Human antibodies for CD90 (Clone DG3), CD105 (Clone 43A4E1), CD73 (Clone AD2), CD34 (Clone 581), CD45 (Clone 5B1) and CD14 (Clone M5E2) were from Becton Dickinson. Detection of phosphatidylserine (PS) was achieved with FITC-conjugated Annexin V, cells were concomitantly stained with a propidium iodide (PI), and reagents were from Sigma-Aldrich. Appropriate staining controls were performed, data were acquired on a flow cytometer (FACS Calibur, or LSR Fortessa BD Biosciences) and analyzed by FlowJo (Tree star Inc.) or Flowing (Turku Center).
Transmission electronic microscopy (TEM)
Cellular samples were fixed in 0.9% NaCl, 1% glutaraldehyde (Wako) for 1 h at 4 °C. Cells were pelleted and re-suspended in 1 mL of a 37 °C pre-warmed 1.1% agarose solution (Bio-Rad). After centrifugation (3000 rpm at 37 °C), pellets were placed at 4 °C and cut into small specimens, washed in 5% sucrose PBS (Wako) and post-fixed with 5% sucrose, 1.5% osmic acid (Nisshin EM) in 100 mM phosphate buffer (Wako) pH 7.3, for 1 h at 4 °C. Post-fixed specimens were washed in 5% sucrose PBS and dehydrated in a graded series of ethanol (Wako) and propylene oxide (Wako), then embedded in epoxy resin EPON 812 (TAAB Laboratories). Ultrathin sections were obtained with an ultra-microtome (Leica EM UC7) and subjected to double staining with a uranyl-acetate (Wako) and lead-citrate (Nacalai Tesque). Samples were observed under a JEM-1400Plus Electronic Microscope (JEOL). TEM were analyzed by collecting images of at least 3 representative areas (RA) at magnification of ×500. One RA contained 10–20 nucleated cells. Then representative images were taken at higher magnifications.
Total protein extraction and titration
Dry cell pellets were obtained after PBS washing and centrifugation of tested cells (1500 rpm, 8 min at 4 °C). Tris-HCl lysis buffer consisting of 50 mM Tris-HCl, 300 mM NaCl and 0.5% Triton X-100, pH 7.6, was added, 250 μL of lysis buffer/pellet. Cells were vortexed and kept for 1.5 h on ice. Cell lysates were sonicated with 3 pulses of 5 s, minimum power (Ultrasonic, UTH-80), and centrifuged at 16,000 g for 10 min at 4 °C. The cell extracts were placed into new test tubes. 10 μl of cell extracts was diluted into 990 μl PBS and used for determination of total protein by Bradford assay (Micro BCA, Thermo Scientific). Cell extracts were diluted with PBS from 1/4 to 1/8 and tested in ELISA for a given cytokine. ELISA data are given as picogram (intracellular cytokine) per milligram (pg/mg) total protein.
ELISA
Samples for ELISA were supernatants or cell lysates of cells stimulated under conditions with cells plated at 106 cells in 1 mL final volume per well, in 24-well plates. Cell culture supernatants or cell lysates or biological fluids from mice were assayed for mouse IL-1β, TNF-α and IL-10 (Mouse ELISA MAX Deluxe, Biolegend). Cell culture supernatants or cell lysates were assayed for human IL-1α , IL-1β, TNF-α and IL-10 (Human ELISA MAX Deluxe, Biolegend).
Statistical analysis
Data are given as mean ± SEM, unpaired t test or one-way ANOVA with a multiple comparison test were performed using GraphPad Prism 5.0 (GraphPad Software, Inc). P < 0.05 was considered statistically significant.
Ethics statement
Approval for collecting procedures and use for research purposes of bone-marrow samples were obtained from the medical ethics committee (CPP# DC-2012-1612) at the University Hospital of Toulouse (France), and written informed consents were obtained from all donors. The institutional animal care and use committee at Kochi Medical School (Japan) reviewed and approved all animal experiments in this study. Animal care, use, and treatment were strictly in compliance with guidelines and regulations.
Additional Information
How to cite this article: Naji, A. et al. Endocytosis of indium-tin-oxide nanoparticles by macrophages provokes pyroptosis requiring NLRP3-ASC-Caspase1 axis that can be prevented by mesenchymal stem cells. Sci. Rep. 6, 26162; doi: 10.1038/srep26162 (2016).
Change history
04 August 2016
The HTML version of this paper was updated after publication, following a technical error that resulted in the publication of an incorrect version of the Supplementary Information file. This has now been corrected in the HTML; the PDF version of the paper was correct from the time of publication.
References
Cummings, K. J. et al. Indium lung disease. Chest 141, 1512–1521, doi: 10.1378/chest.11-1880 (2012).
Steenland, K. & Ward, E. Silica: a lung carcinogen. CA: a cancer journal for clinicians 64, 63–69, doi: 10.3322/caac.21214 (2014).
Lamkanfi, M. & Dixit, V. M. Mechanisms and functions of inflammasomes. Cell 157, 1013–1022, doi: 10.1016/j.cell.2014.04.007 (2014).
Davis, B. K., Wen, H. & Ting, J. P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annual review of immunology 29, 707–735, doi: 10.1146/annurev-immunol-031210-101405 (2011).
Yazdi, A. S. et al. Nanoparticles activate the NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause pulmonary inflammation through release of IL-1alpha and IL-1beta. Proceedings of the National Academy of Sciences of the United States of America 107, 19449–19454, doi: 10.1073/pnas.1008155107 (2010).
Dostert, C. et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677, doi: 10.1126/science.1156995 (2008).
Baron, L. et al. The NLRP3 inflammasome is activated by nanoparticles through ATP, ADP and adenosine. Cell death & disease 6, e1629, doi: 10.1038/cddis.2014.576 (2015).
Badding, M. A. et al. Sintered indium-tin oxide particles induce pro-inflammatory responses in vitro, in part through inflammasome activation. PloS one 10, e0124368, doi: 10.1371/journal.pone.0124368 (2015).
Srinivasula, S. M. et al. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. The Journal of biological chemistry 277, 21119–21122, doi: 10.1074/jbc.C200179200 (2002).
Franceschini, A. et al. The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 29, 2450–2461, doi: 10.1096/fj.14-268714 (2015).
Katsnelson, M. A., Rucker, L. G., Russo, H. M. & Dubyak, G. R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. Journal of immunology 194, 3937–3952, doi: 10.4049/jimmunol.1402658 (2015).
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A. & Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241, doi: 10.1038/nature04516 (2006).
Sheedy, F. J. et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nature immunology 14, 812–820, doi: 10.1038/ni.2639 (2013).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832, doi: 10.1016/j.cell.2010.01.040 (2010).
Miao, E. A., Rajan, J. V. & Aderem, A. Caspase-1-induced pyroptotic cell death. Immunological reviews 243, 206–214, doi: 10.1111/j.1600-065X.2011.01044.x (2011).
Linkermann, A., Stockwell, B. R., Krautwald, S. & Anders, H. J. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nature reviews. Immunology 14, 759–767, doi: 10.1038/nri3743 (2014).
Shi, J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665, doi: 10.1038/nature15514 (2015).
Murthy, S. et al. Alternative activation of macrophages and pulmonary fibrosis are modulated by scavenger receptor, macrophage receptor with collagenous structure. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 29, 3527–3536, doi: 10.1096/fj.15-271304 (2015).
Iyer, R., Hamilton, R. F., Li, L. & Holian, A. Silica-induced apoptosis mediated via scavenger receptor in human alveolar macrophages. Toxicology and applied pharmacology 141, 84–92, doi: 10.1006/taap.1996.0263 (1996).
Beamer, C. A. & Holian, A. Scavenger receptor class A type I/II (CD204) null mice fail to develop fibrosis following silica exposure. American journal of physiology. Lung cellular and molecular physiology 289, L186–195, doi: 10.1152/ajplung.00474.2004 (2005).
Hamilton, R. F. Jr., Thakur, S. A., Mayfair, J. K. & Holian, A. MARCO mediates silica uptake and toxicity in alveolar macrophages from C57BL/6 mice. The Journal of biological chemistry 281, 34218–34226, doi: 10.1074/jbc.M605229200 (2006).
Wang, X., Chen, Y., Lv, L. & Chen, J. Silencing CD36 gene expression results in the inhibition of latent-TGF-beta1 activation and suppression of silica-induced lung fibrosis in the rat. Respiratory research 10, 36, doi: 10.1186/1465-9921-10-36 (2009).
De Nardo, D., De Nardo, C. M. & Latz, E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. The American journal of pathology 184, 42–54, doi: 10.1016/j.ajpath.2013.09.007 (2014).
Rastrick, J. & Birrell, M. The role of the inflammasome in fibrotic respiratory diseases. Minerva medica 105, 9–23 (2014).
Naji, A. et al. Concise review: combining human leukocyte antigen G and mesenchymal stem cells for immunosuppressant biotherapy. Stem cells 31, 2296–2303, doi: 10.1002/stem.1494 (2013).
Weiss, D. J. Concise review: current status of stem cells and regenerative medicine in lung biology and diseases. Stem cells 32, 16–25, doi: 10.1002/stem.1506 (2014).
Zhang, X., Goncalves, R. & Mosser, D. M. The isolation and characterization of murine macrophages. Current protocols in immunology/edited by John E. Coligan … [et al.] Chapter 14, Unit 14 11, doi: 10.1002/0471142735.im1401s83 (2008).
Laliberte, R. E., Eggler, J. & Gabel, C. A. ATP treatment of human monocytes promotes caspase-1 maturation and externalization. The Journal of biological chemistry 274, 36944–36951 (1999).
Lamkanfi, M. et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. The Journal of cell biology 187, 61–70, doi: 10.1083/jcb.200903124 (2009).
Morales-Nebreda, L., Misharin, A. V., Perlman, H. & Budinger, G. R. The heterogeneity of lung macrophages in the susceptibility to disease. European respiratory review: an official journal of the European Respiratory Society 24, 505–509, doi: 10.1183/16000617.0031-2015 (2015).
Accarias, S. et al. Pyroptosis of resident macrophages differentially orchestrates inflammatory responses to Staphylococcus aureus in resistant and susceptible mice. European journal of immunology 45, 794–806, doi: 10.1002/eji.201445098 (2015).
Newton, K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends in cell biology 25, 347–353, doi: 10.1016/j.tcb.2015.01.001 (2015).
Golstein, P. & Kroemer, G. Cell death by necrosis: towards a molecular definition. Trends in biochemical sciences 32, 37–43, doi: 10.1016/j.tibs.2006.11.001 (2007).
Rabolli, V. et al. The alarmin IL-1alpha is a master cytokine in acute lung inflammation induced by silica micro- and nanoparticles. Particle and fibre toxicology 11, 69, doi: 10.1186/s12989-014-0069-x (2014).
Rider, P. et al. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. Journal of immunology 187, 4835–4843, doi: 10.4049/jimmunol.1102048 (2011).
Lamacchia, C., Rodriguez, E., Palmer, G. & Gabay, C. Endogenous IL-1alpha is a chromatin-associated protein in mouse macrophages. Cytokine 63, 135–144, doi: 10.1016/j.cyto.2013.04.010 (2013).
Franchi, L., Eigenbrod, T., Munoz-Planillo, R. & Nunez, G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nature immunology 10, 241–247, doi: 10.1038/ni.1703 (2009).
Uccelli, A., Moretta, L. & Pistoia, V. Mesenchymal stem cells in health and disease. Nature reviews. Immunology 8, 726–736, doi: 10.1038/nri2395 (2008).
Selmani, Z. et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+CD25highFOXP3+ regulatory T cells. Stem cells 26, 212–222, doi: 10.1634/stemcells.2007-0554 (2008).
Peranteau, W. H. et al. IL-10 overexpression decreases inflammatory mediators and promotes regenerative healing in an adult model of scar formation. The Journal of investigative dermatology 128, 1852–1860, doi: 10.1038/sj.jid.5701232 (2008).
Badding, M. A., Stefaniak, A. B., Fix, N. R., Cummings, K. J. & Leonard, S. S. Cytotoxicity and characterization of particles collected from an indium-tin oxide production facility. Journal of toxicology and environmental health. Part A 77, 1193–1209, doi: 10.1080/15287394.2014.920757 (2014).
Cummings, K. J. et al. Early changes in clinical, functional, and laboratory biomarkers in workers at risk of indium lung disease. Annals of the American Thoracic Society 11, 1395–1403, doi: 10.1513/AnnalsATS.201407-346OC (2014).
Riteau, N. et al. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. American journal of respiratory and critical care medicine 182, 774–783, doi: 10.1164/rccm.201003-0359OC (2010).
da Silva, G. L. et al. P2X7 receptor is required for neutrophil accumulation in a mouse model of irritant contact dermatitis. Experimental dermatology 22, 184–188, doi: 10.1111/exd.12094 (2013).
McDonald, B. et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330, 362–366, doi: 10.1126/science.1195491 (2010).
Mukhopadhyay, S. et al. SR-A/MARCO-mediated ligand delivery enhances intracellular TLR and NLR function, but ligand scavenging from cell surface limits TLR4 response to pathogens. Blood 117, 1319–1328, doi: 10.1182/blood-2010-03-276733 (2011).
Kelley, J. L., Ozment, T. R., Li, C., Schweitzer, J. B. & Williams, D. L. Scavenger receptor-A (CD204): a two-edged sword in health and disease. Critical reviews in immunology 34, 241–261 (2014).
Todt, J. C., Hu, B. & Curtis, J. L. The scavenger receptor SR-A I/II (CD204) signals via the receptor tyrosine kinase Mertk during apoptotic cell uptake by murine macrophages. Journal of leukocyte biology 84, 510–518, doi: 10.1189/jlb.0307135 (2008).
Latz, E. The inflammasomes: mechanisms of activation and function. Current opinion in immunology 22, 28–33, doi: 10.1016/j.coi.2009.12.004 (2010).
Shi, J. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192, doi: 10.1038/nature13683 (2014).
Baker, P. J. et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. European journal of immunology 45, 2918–2926, doi: 10.1002/eji.201545655 (2015).
Aachoui, Y., Sagulenko, V., Miao, E. A. & Stacey, K. J. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Current opinion in microbiology 16, 319–326, doi: 10.1016/j.mib.2013.04.004 (2013).
He, W. T. et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell research 25, 1285–1298, doi: 10.1038/cr.2015.139 (2015).
Wang, L. et al. Potential role of apoptotic macrophages in pulmonary inflammation and fibrosis. Journal of cellular physiology 194, 215–224, doi: 10.1002/jcp.10220 (2003).
Khalil, N., Whitman, C., Zuo, L., Danielpour, D. & Greenberg, A. Regulation of alveolar macrophage transforming growth factor-beta secretion by corticosteroids in bleomycin-induced pulmonary inflammation in the rat. The Journal of clinical investigation 92, 1812–1818, doi: 10.1172/JCI116771 (1993).
Shi, K. et al. Dexamethasone attenuates bleomycin-induced lung fibrosis in mice through TGF-beta, Smad3 and JAK-STAT pathway. International journal of clinical and experimental medicine 7, 2645–2650 (2014).
Bousquet, J. Inhaled corticosteroids in severe COPD. The Lancet. Respiratory medicine 1, 177–178, doi: 10.1016/S2213-2600(13)70042-0 (2013).
Rhen, T. & Cidlowski, J. A. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. The New England journal of medicine 353, 1711–1723, doi: 10.1056/NEJMra050541 (2005).
Lee, H. M., Kang, J., Lee, S. J. & Jo, E. K. Microglial activation of the NLRP3 inflammasome by the priming signals derived from macrophages infected with mycobacteria. Glia 61, 441–452, doi: 10.1002/glia.22448 (2013).
Busillo, J. M., Azzam, K. M. & Cidlowski, J. A. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. The Journal of biological chemistry 286, 38703–38713, doi: 10.1074/jbc.M111.275370 (2011).
Deng, B., Wehling-Henricks, M., Villalta, S. A., Wang, Y. & Tidball, J. G. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. Journal of immunology 189, 3669–3680, doi: 10.4049/jimmunol.1103180 (2012).
Nakagome, K. et al. In vivo IL-10 gene delivery attenuates bleomycin induced pulmonary fibrosis by inhibiting the production and activation of TGF-beta in the lung. Thorax 61, 886–894, doi: 10.1136/thx.2005.056317 (2006).
He, A. et al. The antiapoptotic effect of mesenchymal stem cell transplantation on ischemic myocardium is enhanced by anoxic preconditioning. The Canadian journal of cardiology 25, 353–358 (2009).
Raffaghello, L. et al. Human mesenchymal stem cells inhibit neutrophil apoptosis: a model for neutrophil preservation in the bone marrow niche. Stem cells 26, 151–162, doi: 10.1634/stemcells.2007-0416 (2008).
Ramasamy, R. et al. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: impact on in vivo tumor growth. Leukemia 21, 304–310, doi: 10.1038/sj.leu.2404489 (2007).
Acknowledgements
This study was supported by the Program to Disseminate Tenure Tracking System of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. This work was supported by intramural funding to A.N. from Kochi Medical School (KMS), Kochi University, Japan.
Author information
Authors and Affiliations
Contributions
A.N., N.S. and K.-i.Y. conceived the experiments. A.N., B.A.M., K.-i.Y., F.D. and N.B. conducted the experiments. A.N., B.A.M. and F.D. analyzed the results. A.N., F.D., L.S. and N.S. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Naji, A., Muzembo, B., Yagyu, Ki. et al. Endocytosis of indium-tin-oxide nanoparticles by macrophages provokes pyroptosis requiring NLRP3-ASC-Caspase1 axis that can be prevented by mesenchymal stem cells. Sci Rep 6, 26162 (2016). https://doi.org/10.1038/srep26162
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26162
- Springer Nature Limited
This article is cited by
-
Dose–response relationship of pulmonary disorders by inhalation exposure to cross-linked water-soluble acrylic acid polymers in F344 rats
Particle and Fibre Toxicology (2022)
-
Nanomaterial-mediated autophagy: coexisting hazard and health benefits in biomedicine
Particle and Fibre Toxicology (2020)
-
Mitochondrial donation in translational medicine; from imagination to reality
Journal of Translational Medicine (2020)
-
Biological functions of mesenchymal stem cells and clinical implications
Cellular and Molecular Life Sciences (2019)
-
Stem cell-derived mitochondria transplantation: a novel strategy and the challenges for the treatment of tissue injury
Stem Cell Research & Therapy (2018)