
Abstract
Study design:
Experimental animal study.
Objectives:
Quantitative analysis of secondary changes in lesion size after experimental spinal cord injury (SCI) in the rat, with special emphasis to the formation of dorsal column lesions.
Setting:
Slovakia.
Methods:
After SCI in the rat, animals survived for different periods ranging from 5 min to 7 days. Their whole spinal cords were cut transversally into 1 mm thick slabs. On each slab, the lesion profile was outlined. The overall shape of the lesion was reconstructed from a series of consecutive profiles and its length was measured.
Results:
Immediately after injury, a spindle-shaped hemorrhagic contusive lesion was observed, with the length of ~15 mm. After a quiescent phase lasting for at least 1 h, there was a dramatic secondary enlargement of the lesion and its length increased up to 40 mm between 1 and 48 h. The fully developed lesion consisted of the spindle-shaped epicenter and long cranial and caudal protrusions located in the midline between dorsal columns.
Conclusion:
We propose that secondary enlargement of the lesion can be explained by posttraumatic swelling. The expanding tissues are pushed out in longitudinal axis along the mechanically weakest parts of the spinal cord. Additional data that support this hypothesis are presented. Our findings indicate that malignant posttraumatic edema might have an important role in pathomechanisms of secondary injury after SCI.
Similar content being viewed by others


Introduction
In our previous experiments with the rat SCI model,1 we have evaluated recovery of neurological functions during long-term survival periods (4–6 weeks).1, 2, 3, 4, 5 After survival, the lesion site was histologically analyzed in order to quantitate the amount of spared tissue at the epicenter of the lesion. At the contusion site, we have observed typical spindle-shaped central cavity. At more distant segments, there were small dorsal column lesions (DCLs), occurring both cranially and caudally from the epicenter. After more detailed examination, we have found that DCLs occur surprisingly far from the site of injury. It was obvious that such a distant pathology cannot be caused directly by tissue contusion. Therefore, we have decided to study the development of DCLs at different posttraumatic periods in order to bring more details about their formation. In addition, we have measured the craniocaudal size of the lesion and documented the time course of lesion enlargement.
It has been recognized that, after contusive SCI, the injured tissues undergo a unique process in which the site of injury is gradually transformed into a large, cavity-filled lesion. Immediately after injury, overt pathology is limited to central areas of the spinal cord where shearing and stretching cause direct tissue disruption, bleeding and ischemia. Within several hours, petechial hemorrhages emerge in the more distant areas, eventually coalescing into the characteristic lesion of hemorrhagic necrosis.6 As the lesion develops, injury involves the white matter as well.7, 8, 9 This slow evolution of the lesion has been referred to as progressive hemorrhagic necrosis10, 11, 12, 13 or ‘autodestruction’.14 Progressive hemorrhagic necrosis results in secondary loss of vital spinal cord tissue and, in some species including humans, leads to formation of cystic cavity surrounded by glial scar tissue.
The processes involved in secondary injury have been extensively studied, as they represent a promising therapeutic target after SCI. However, we are not aware of a quantitative morphometric study that would clearly define the time course of the lesion enlargement. This is probably caused by technical difficulties associated with poor fixation and subsequent histological processing of the injured tissue. The rat spinal cord is approximately 11 cm long, and systematic cutting of such a big specimen is a challenging task. It must be divided into smaller pieces to make it suitable for cutting on standard cutting devices for histology. However, the lesioned tissue is not self-supporting, and the pieces of spinal cord would bend and distort during dehydration and paraffin infiltration. Therefore, with standard tissue-processing techniques, serial transversal cutting of a 11 cm long specimen is virtually impossible. In the present study, aligned spinal cords were embedded into a protein matrix that formed a semirigid block enveloping the whole spinal cord. The mass of protein matrix was firmly attached to the surface of the spinal cord tissue. It has facilitated serial transversal cutting and kept the floating sections of weakened tissue in their natural position. Therefore, protein matrix embedding was a crucial step that enabled systematic analysis and measurement of the whole lesion (several centimeters long) ‘in toto’.
Materials and methods
Spinal cord compression injury model
We have used the rat SCI model described in detail previously.1 Briefly, after induction of anesthesia with 4% halothane in room air, animals were maintained in halothane anesthesia (1.5–2% halothane in room air delivered via face mask) during the operation. Skin on the spine was cut over the L1–T10 spinous processes, the soft tissue was removed and a 2F Fogarty catheter was inserted into the epidural space cranially via a small hole drilled in the vertebral arch of T10. The catheter was advanced for 1 cm, so that the center of the balloon was resting at T8–T9 level of the spinal cord. After stabilization of the body temperature at 37 °C, the balloon was rapidly inflated with 12.5 μl of saline for 5 min, which corresponds to intermediate degree of injury (partial recovery of locomotion with permanent neurological deficit). After that, the balloon was deflated and the catheter removed, soft tissues and skin sutured in anatomical layers and the animal was disconnected from anesthesia. All experiments were performed in accordance with European Community legislation and were approved by Ethics Committee at the Institute of Neurobiology and the State Veterinary and Alimentary Administration of the Slovak Republic. We certify that all applicable institutional and governmental regulations concerning the ethical use of animals were followed during the course of this research.
Experimental groups
After SCI, animals were allowed to survive for 5 min, 1 h, 1day, 2, 3, 4, 5 and 7 days (n=5 in each group). In addition, two animals were subjected to more severe injury (contusion with 20 μl balloon) and allowed to survive for 2 days.
At the end of their survival period, all animals were deeply anesthetized with ketamine/xylazine and transcardially perfused with saline followed by 4% paraformaldehyde in 0.1 M phosphate buffer. The perfused tissues were stored at 4 °C overnight. The spinal cord was then removed from the bone and postfixed in the same fixative for at least 24 h.
Spinal cord processing
To facilitate systematic analysis, whole spinal cords were embedded in a protein matrix as described by Herzog and Brosamle.15 Before embedding, the dura mater was carefully removed with the aid of ophtalmic scissors and sharp tweezers under operating microscope. Care was taken to cut (rather than tear off) all the rootlets attached to the dura mater in order to avoid postmortem mechanical damage to the lesioned spinal cord parenchyma. The whole spinal cord was then blotted dry and suspended on two ligatures in a custom-made mold with a dimension 10 × 10 mm. The whole mold was then filled with glutaraldehyde-polymerized protein matrix prepared as follows: stock solution was prepared by dissolving 0.75 g of gelatine (150 bloom, BDH Ltd, Atherstone, UK) and 30 g of sucrose in 50 ml of warm 0.1 M phosphate buffer. Separately, 57 g of chicken egg albumin (BDH Ltd) were dissolved in 100 ml of 0.1 M phosphate buffer. Both these solutions were mixed and filtered through muslin. For embedding, 9 parts of the gelatine–albumin solution were mixed with 1 part of ice-cold 25% glutaraldehyde. After stirring quickly, the solution was poured over the specimen and allowed to polymerise at room temperature. After approximately 10 min, the hardened matrix with the embedded spinal cord was removed from the mold and stored in 4% paraformaldehyde in 0.1 M phosphate buffer. The whole block was placed into a metallic Tissue matrix (TM-1000 10 × 10 mm2 chamber with 1 mm slices, ASI Instruments, USA). The block was then cut with a microtome blade into 1 mm thick slabs. Individual slabs were collected and stored in the fixative in standard 24-well microplates. Serial macrophotographic pictures were prepared from these slabs on a black glass plate. The lesion size was evaluated directly from the high-resolution macrophotographs.
Some selected slabs were further subjected to histological processing. The whole slabs were flat-embedded in Durcupan and cut into 1 μm thick semithin sections that were stained with toluidine blue.
Evaluation of the lesion size
The longitudinal size of the lesion was determined as the number of sections (that is, millimeters) where the lesion was present. The intergroup differences between consecutive survival times were compared by the unpaired Student’s t-test. On the digital image of each slab, the boundaries of hemorrhagic necrosis were delineated manually, and the medio-lateral size of the lesion was measured. These measurements were used to prepare a schematic drawing from each animal and the lesion shape was projected on the spinal cord silhouette. For documentation, the drawings from all animals in one group were digitally superposed.
Intraparenchymal injection of gelatine
In this pilot experiment, we wanted to document the distribution of a fluid injected locally into the central gray matter. Five control animals received intraspinal injection of colored gelatine. The anesthetized animals were fixed in a sterotaxic frame, and a small laminotomy was made in T9 vertebra. A glass micropipette with a bevelled tip (50 μm diameter) was connected to a microsyringe filled with 2% gelatine solution mixed with Indian ink. Under operating microscope, the dura mater was incised with a tip of hypodermic needle (400 μm lateral from the midline), and the micripipette was inserted at 15° angle (1.1 mm deep) into the central part of the gray matter. In order to prevent leaking of the injected fluid, the micropipette was glued to the dura with a droplet of cyanoacrylate glue, and 10 μl of gelatine was injected slowly (within 10 min) into the spinal gray matter. The micropipette was then removed. After injection, the tissues were perfusion-fixed and the spinal cords were processed as described above.
Results
Development of the lesion
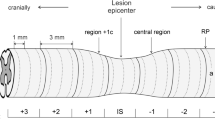
After 5 min and 1 h, the picture of the lesion was almost identical. At the epicenter, the whole gray matter area was filled with hemorrhage, and the contused parenchyma was apparently pushed away from the site of compression. The white matter remained mostly preserved. Both cranially and caudally from the epicenter, there were scattered small hemorrhages, distributed mostly in the central and dorsal parts of the spinal cord (Figure 1). The craniocaudal size of the whole lesion was 15.1±2.2 mm after 5 min and 14.8±1.7 mm after 1 h.

SCI+1 h survival. Consecutive sections (1 mm thick) are aligned in columns (cranial end is left and up). Assembly of 40 sections represents a 4 cm long segment of the spinal cord. Note an ‘empty’ hemorrhagic epicenter of injury and numerous smaller hemorrhages. Structure of the spinal cord in regions more distant from the central injury is intact. Scale bar, 3 mm.
After 24 h, the extent of the lesion changed dramatically. The epicenter was edematous and the hemorrhages increased in number and size. A large part of the white matter encircling the epicenter of injury remained relatively well preserved. The most striking change was the development of cranial and caudal protrusions of the lesion. These newly formed lesions were invariably located in the central part of the dorsal white matter. On transversal sections, the DCL looked like a symmetrical round-shaped necrosis occuppying a relatively small cross-sectional area. Closer inspection revealed that the center of this formation contains a mass of poorly defined tissue. This mass oppressed the surounding white matter, causing its mechanical injury and small local bleedings from the ruptured vessels. These DCLs added considerably to the length of the whole injury that was now 28.3±9.8 mm (Figure 2).

SCI+1 day survival. Consecutive sections (1 mm thick) are aligned in columns (cranial end is left and up). Assembly of 60 sections represents a 6 cm long segment of the spinal cord. Note edematous epicenter, increased number of hemorrhages and prominent cranial and caudal protrusions in dorsal columns (sections 6–23 and 37–47, respectively). Scale bar, 3 mm.
After 2 days, the lesion size further increased. The epicenter was edematous and disintegrated. In more distant sections, some hemorrhagic areas turned into cavities. During manipulation with the tissue slabs, loose detritic material sometimes came out from the DCL like a toothpaste. The average length of the lesion was 40.2±12.1 mm (Figure 3).

SCI+2 days survival. Consecutive sections (1 mm thick) are aligned in columns (cranial end is left and up). Assembly of 60 sections represents a 6 cm long segment of the spinal cord. The lesion has reached its maximal size. Note swollen tissues at the epicenter, a starting liquefaction of some necrotic parts and extremely long protrusions in dorsal columns. Scale bar, 3 mm.
After 3 days, the length of the lesion remained unchanged. In some necrotic areas, hemorrhagic foci changed their color from red to yellowish and/or turned into cavities. The length of the lesion was 40.0±10.5 mm.
After 4 and 5 days, the necrotic tissues were gradually removed. The edema has receded, and decoloration was more pronounced. In the most severely affected areas, cavities were formed. Obviously, the necrotic tissues were undergoing the processes of liquefaction and resorption.
After 7 days, there was further progress in cavitation and debris removal.
In a plot summarizing the changes in the lesion size within the first 3 days (Figure 4), it is obvious that the lesion size is expanding dramatically within the first 2 days, when its size reached maximum. After that, only regressive changes were observed, and the necrosis underwent a process of clearance and formation of demarcated cavities. The pattern of the expanding lesion is illustrated on superimposed drawings (Figure 5).

Plot showing the changes of the lesion size within the first 3 days after injury. Data are means±s.e.m. *P<0.05; **P<0.01.

Variability of the lesion sizes. In each group, five drawings from individual animals are superposed.
Severe injury experiment
After severe injury (20 μl balloon volume and 2 days survival), the pattern of the lesion was identical, but the overall volume of the hemorrhagic necrosis was apparently larger. The cranial and caudal protrusions extended to the cervical and lumbar intumescences, respectively (Figure 6). At their endings, the DCLs formed a bulb-like enlargement. The cross-sectional area of the DCL occuppied the whole perimeter of the dorsal funiculi, touching the dorsal surface of the spinal cord. Consequently, the DCLs were clearly visible from the dorsal aspect after removal of the dura mater, providing a good overall picture of the fully developed lesion (Figure 7).

Experiment with severe SCI+2 days survival. Consecutive sections (1 mm thick) are aligned in columns (cranial end is left and up). Assembly of 60 sections represents a 6 cm long segment of the spinal cord. The length of the lesion is almost 60 mm. In comparison to moderate SCI, the DCL is bigger, filling up the whole height of the dorsal columns. Scale bar, 3 mm.

Spinal cord of an animal with severe SCI+2 days survival before embedding and sectioning. The dura has been removed. After severe injury, both epicenter and protrusions are clearly visible from dorsal aspect.
Histological observations
In semithin sections across the developing DCL (24 h after injury), we have evaluated subcellular details of the lesion. It was obvious that the core contained a mass of tissue inserted between the dorsal funiculi. This mass was composed of poorly identifiable cellular debris mixed with red blood cells. There was prominent vacuolization in the whole DCL (Figure 8).

Durcupan-embedded tissue cut into semithin section and stained with toluidine blue. DCL, 2 days after SCI. The mass of the lesion consists of tissue debris containing a large number of vacuoles. GM, gray matter; WM, white matter; scale bar, 100 μm.
Pattern of gelatine distribution
In all animals, the injected gelatine was distributed longitudinally (Figure 9). The fluid has found its way into the space between dorsal columns and here it advanced in cranial and caudal directions from the site of injection. Thus 10 μl of gelatine has formed an elongated structure with the length of approximately 30 mm.

Gelatine distribution after unilateral injection into the central gray matter. The fluid fills up the space between dorsal columns and here it advances longitudinally; (a) dorsal aspect; (b) consecutive sections (1 mm thick) are aligned in columns (cranial end is left and up); (c) site of injection. Scale bar, 3 mm.
Discussion
The phenomenon of secondary injury has been first postulated in 1911 by Allen when he found that early myelotomy and removal of the posttraumatic hematomyelia resulted in improvement of neurological function in dogs subjected to experimental acute SCI.16 Secondary pathology at the site of injury has been qualitatively described in detail,6, 16, 17, 18, 19 but systematic quantitative studies documenting the details of progressive enlargement of the lesion are missing. The most detailed study known to us has been published by Liu et al.20 Their schematic drawings of the growing lesion indicate that there is rapid expansion within the first 24 h and that the lesion size further increases but more slowly.
Our data show that secondary enlargement of the lesion is not a linear process. Immediately after SCI, there is a quiescent period when the primary lesion remains virtually unchanged for at least 1 h. Its enlargement starts after this delay and the lesion size grows up to 48 h. In our experiments, the secondary enlargement of the central lesion was always associated with the formation of DCLs that could extend as far as to the levels of cervical and lumbar intumescences. Detailed observation revealed that DCLs are longitudinal protrusions growing out of the central lesion and that the process of their formation is characterized by some typical features: (1) DCLs are spreading both cranially and caudally, (2) develop concurrently with secondary enlargement of the epicenter; (3) form symmetrical round-shaped ‘tunnels’ interconnected with the central necrosis; (4) contain a mass of tissue inserted between distorted but relatively intact dorsal funiculi; and (5) their distant endings form a hemorrhagic bulb-like enlargement.
In the present study, we describe for the first time that DCL is not a necrosis of dorsal funiculi per se, but that it contains a mass of inserted tissue. This important detail could be recognized because we have examined unprocessed spinal cords. Routine histological processing is detrimental for edematous tissues. In histological sections prepared early after injury, this important detail has not been detected, as DCL appeared as a pale-staining region where the cellular details were blurred. In later stages, there was complete loss of cellularity and cavitation.7, 21
These findings led us to formulate a hypothesis that secondary enlargement of the lesion is driven by increased intramedullary pressure. Owing to posttraumatic edema, the volume of the injured tissue gradually increases. In the spinal cord, lateral swelling is confined by the tough dura mater. The expanding tissue therefore ‘herniates’ cranially and caudally along the midline between the posterior funiculi. This hypothesis would explain both the timing and pattern of the fully developed lesion. In this way, relatively big volumes of tissue debris can be distributed in longitudinal directions. In experimental studies, this pattern of injury could be easily overlooked, because the swelling is delayed, and the tissue debris is transported far from the contused area. After longer survival periods, the DCLs can collapse and become hardly detectable.
In the literature, there are several observations that indirectly support our hypothesis. The round-shaped DCLs have been described in various SCI experiments, in histopathological studies7, 8, 9, 21 and in MRI visualizations,22, 23 indicating that this phenomenon is not restricted to our model. Previously, it has been speculated that DCLs are Wallerian degenerations that somehow turn into necrosis,21 but to the best of our knowledge, the timing and localization of DCLs do not correspond to Wallerian degeneration. The timing of secondary enlargement corresponds well with water content changes described in comparable models of SCI. After injury, the water content gradually increases, peaks at 24 h and remains elevated at 3–7 days.24, 25, 26
Longitudinal spreading of the lesion after SCI has been observed in human patients, as well.27 In a neuropathological study, Ito et al.28 have described DCLs in the spinal cords of patients who died early after SCI. The character of these lesions was identical to our findings. The DCLs were named ‘pencil-shaped necrosis’, and the authors proposed for the first time the mechanism driven by increased intramedullary pressure. They argued that the posterior funiculus is the mechanically weakest part of the spinal cord, because similar pattern of syrinx formation has been described in various pathological situations where blood or neoplasms expanded within the spinal cord parenchyma.29, 30, 31 Our experiment with gelatine injection clearly supports this notion.
Increased intraspinal pressure was mentioned in early reports by Allen, who described that, after median longitudinal incision was made early after traumatic injury in dogs, there was ‘great outpouring of blood from the injured area posteriorly through incision’. In the same paper, he mentions a patient with SCI who was operated 4 h after injury, and ‘when the cord was incised, the blood spurted out as if under great pressure’.32 In recent animal studies, optimal time for myelotomy has been determined as 24 h after injury,33 and in controlled conditions, beneficial effect of myelotomy on edema reduction and functional recovery after SCI has been demonstrated.34
In this context, it is of interest that the only approved pharmacological treatment for spinal cord injury was application of methylprednisolone, reviewed by Hall and Springer.35 High doses of methylprednisolone reduced disability when applied within the first 8 h after injury, but the precise mechanisms of protection is not clear. It might act by reducing inflammation, glutamate release and free-radical accumulation.36 In addition to this, methylprednisolone is known to ameliorate edema associated with brain tumors,37 and methylprednisolone pretreatment is a standard of care to prevent intraoperative and postoperative brain swelling. It is thus tempting to assume that edema reduction contributes to the protective effect of methylprednisolone therapy.
Recently, hypertonic saline therapy has become a means for reducing intracranial pressure to treat cerebral edema after traumatic brain injury.38, 39, 40 Hypertonic saline lowers intracranial pressure by establishing an osmotic gradient between the intracellular and intravascular space. Experimental studies indicate that hypertonic saline therapy after SCI can improve behavioral and histopathological outcomes,41, 42, 43, 44 indicating that posttraumatic edema indeed contributes significantly to secondary injury after SCI.
In conclusion, we have measured the secondary expansion of the lesion after spinal cord trauma. With aid of a new embedding technique, we could perform a systematic morphometric analysis of the whole spinal cord that confirmed a dramatic secondary enlargement of the lesion within the first 48 h after injury. After 48 h, only regressive changes were observed. The specific pattern and timing of the progressive necrosis indicate that the enlargement of the lesion is mainly driven by posttraumatic swelling that transports the tissue debris for long distances along the craniocaudal axis. The DCL is inserted into the newly formed space between dorsal funiculi and seems to cause a relatively small local damage to the dorsal white matter. However, one can envisage that, at the epicenter of injury, delayed swelling can be particularly dangerous to the peripherally located white matter. When the central gray matter expands, it would push the intact white matter against the meninges. This would cause secondary hypoperfusion/ischemia in the white matter that survived the mechanical insult. In this way, posttraumatic edema can cause additional ischemic injury of long axonal projections and contribute significantly to the final neurological loss. After contusive SCI, malignant posttraumatic edema thus can be an important component in the pathomechanisms of secondary injury.
Data archiving
There were no data to deposit.
References
Vanicky I, Urdzikova L, Saganova K, Cizkova D, Galik J . A simple and reproducible model of spinal cord injury induced by epidural balloon inflation in the rat. J Neurotrauma 2001; 18: 1399–1407.
Vanicky I, Ondrejcak T, Ondrejcakova M, Sulla I, Galik J . Long-term changes in spinal cord evoked potentials after compression spinal cord injury in the rat. Cell Mol Neurobiol 2006; 26: 1521–1539.
Vanický I, Urdzíková L, Saganová K, Maršala M . Intrathecal methylprednisolone does not improve outcome after severe spinal cord injury in the rat. Neurosci Res Commun 2002; 31: 183–191.
Saganova K, Orendacova J, Cizkova D, Vanicky I . Limited minocycline neuroprotection after balloon-compression spinal cord injury in the rat. Neurosci Lett 2008; 433: 246–249.
Saganova K, Orendacova J, Sulla I Jr, Filipcik P, Cizkova D, Vanicky I . Effects of long-term FK506 administration on functional and histopathological outcome after spinal cord injury in adult rat. Cell Mol Neurobiol 2009; 29: 1045–1051.
Balentine JD . Pathology of experimental spinal cord trauma. I. The necrotic lesion as a function of vascular injury. Lab Invest 1978; 39: 236–253.
Guth L, Zhang Z, Steward O . The unique histopathological responses of the injured spinal cord. Implications for neuroprotective therapy. Ann NY Acad Sci 1999; 890: 366–384.
James ND, Bartus K, Grist J, Bennett DL, McMahon SB, Bradbury EJ . Conduction failure following spinal cord injury: functional and anatomical changes from acute to chronic stages. J Neurosci 2011; 31: 18543–18555.
Zhang Z, Krebs CJ, Guth L . Experimental analysis of progressive necrosis after spinal cord trauma in the rat: etiological role of the inflammatory response. Exp Neurol 1997; 143: 141–152.
Fitch MT, Doller C, Combs CK, Landreth GE, Silver J . Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci 1999; 19: 8182–8198.
Nelson E, Gertz SD, Rennels ML, Ducker TB, Blaumanis OR . Spinal cord injury. The role of vascular damage in the pathogenesis of central hemorrhagic necrosis. Arch Neurol 1977; 34: 332–333.
Tator CH, Fehlings MG . Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg 1991; 75: 15–26.
Tator CH, Koyanagi I . Vascular mechanisms in the pathophysiology of human spinal cord injury. J Neurosurg 1997; 86: 483–492.
Nemecek S . Morphological evidence of microcirculatory disturbances in experimental spinal cord trauma. Adv Neurol 1978; 20: 395–405.
Herzog A, Brosamle C . 'Semifree-floating' treatment: a simple and fast method to process consecutive sections for immunohistochemistry and neuronal tracing. J Neurosci Methods 1997; 72: 57–63.
Bresnahan JC . An electron-microscopic analysis of axonal alterations following blunt contusion of the spinal cord of the rhesus monkey (Macaca mulatta. J Neurol Sci 1978; 37: 59–82.
Casella GT, Bunge MB, Wood PM . Endothelial cell loss is not a major cause of neuronal and glial cell death following contusion injury of the spinal cord. Exp Neurol 2006; 202: 8–20.
Griffiths IR, McCulloch MC . Nerve fibres in spinal cord impact injuries. Part 1. Changes in the myelin sheath during the initial 5 weeks. J Neurol Sci 1983; 58: 335–349.
Tator CH, Rowed DW . Current concepts in the immediate management of acute spinal cord injuries. Can Med Assoc J 1979; 121: 1453–1464.
Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW et al. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci 1997; 17: 5395–5406.
Zhang Z, Guth L . Experimental spinal cord injury: Wallerian degeneration in the dorsal column is followed by revascularization, glial proliferation, and nerve regeneration. Exp Neurol 1997; 147: 159–171.
Narayana P, Abbe R, Liu SJ, Johnston D . Does loss of gray- and white-matter contrast in injured spinal cord signify secondary injury? In vivo longitudinal MRI studies. Magn Reson Med 1999; 41: 315–320.
Weber T, Vroemen M, Behr V, Neuberger T, Jakob P, Haase A et al. In vivo high-resolution MR imaging of neuropathologic changes in the injured rat spinal cord. Am J Neuroradiol 2006; 27: 598–604.
Demediuk P, Lemke M, Faden AI . Spinal cord edema and changes in tissue content of Na+, K+, and Mg2+ after impact trauma in rats. Adv Neurol 1990; 52: 225–232.
Lemke M, Demediuk P, McIntosh TK, Vink R, Faden AI . Alterations in tissue Mg++, Na+ and spinal cord edema following impact trauma in rats. Biochem Biophys Res Commun 1987; 147: 1170–1175.
Lemke M, Faden AI . Edema development and ion changes in rat spinal cord after impact trauma: injury dose-response studies. J Neurotrauma 1990; 7: 41–54.
Sudo H, Taneichi H, Kaneda K . Secondary medulla oblongata involvement following middle cervical spinal cord injury associated with latent traumatic instability in a patient with ossification of the posterior longitudinal ligament. Spinal Cord 2006; 44: 126–129.
Ito T, Oyanagi K, Wakabayashi K, Ikuta F . Traumatic spinal cord injury: a neuropathological study on the longitudinal spreading of the lesions. Acta Neuropathol (Berl) 1997; 93: 13–18.
Hashizume Y, Iljima S, Kishimoto H, Hirano A . Pencil-shaped softening of the spinal cord. Pathologic study in 12 autopsy cases. Acta Neuropathol 1983; 61: 219–224.
Hinokuma K, Ohama E, Oyanagi K, Kakita A, Kawai K, Ikuta F . Syringomyelia. A neuropathological study of 18 autopsy cases. Acta Pathol Jpn 1992; 42: 25–34.
Oyanagi K, Yamazaki K, Hinokuma K, Ito F, Ikuta S . An autopsy case of intramedullary venous malformation of the spinal cord with spreading hematomyelia. Clin Neuropathol 1990; 9: 148–151.
Allen AR . Surgery of experimental lesions of spinal cord equivalent to crush injury of fracture dislocation of spinal column. A preliminary report. J Am Med Assoc 1911; 57: 878–880.
Yang DG, Li JJ, Gu R, Yang ML, Zhang X, Du LJ et al. Optimal time window of myelotomy in rats with acute traumatic spinal cord injury: a preliminary study. Spinal Cord 2013; 51: 673–678.
Hu AM, Li JJ, Sun W, Yang DG, Yang ML, Du LJ et al. Myelotomy reduces spinal cord edema and inhibits aquaporin-4 and aquaporin-9 expression in rats with spinal cord injury. Spinal Cord 2015; 53: 98–102.
Hall ED, Springer JE . Neuroprotection and acute spinal cord injury: a reappraisal. NeuroRx 2004; 1: 80–100.
Short DJ, El Masry WS, Jones PW . High dose methylprednisolone in the management of acute spinal cord injury - a systematic review from a clinical perspective. Spinal Cord 2000; 38: 273–286.
Gomes JA, Stevens RD, JJ3 Lewin, Mirski MA, Bhardwaj A . Glucocorticoid therapy in neurologic critical care. Crit Care Med 2005; 33: 1214–1224.
De Vivo P, Del Gaudio A, Ciritella P, Puopolo M, Chiarotti F, Mastronardi E . Hypertonic saline solution: a safe alternative to mannitol 18% in neurosurgery. Minerva Anestesiol 2001; 67: 603–611.
Forsyth LL, Liu-DeRyke X, Parker D Jr, Rhoney DH . Role of hypertonic saline for the management of intracranial hypertension after stroke and traumatic brain injury. Pharmacotherapy 2008; 28: 469–484.
Ziai WC, Toung TJ, Bhardwaj A . Hypertonic saline: first-line therapy for cerebral edema? J Neurol Sci 2007; 261: 157–166.
Legos JJ, Gritman KR, Tuma RF, Young WF . Coadministration of methylprednisolone with hypertonic saline solution improves overall neurological function and survival rates in a chronic model of spinal cord injury. Neurosurgery 2001; 49: 1427–1433.
Young WF, Rosenwasser RH, Vasthare US, Tuma RF . Preservation of post-compression spinal cord function by infusion of hypertonic saline. J Neurosurg Anesthesiol 1994; 6: 122–127.
Tuma RF, Vasthare US, Arfors KE, Young WF . Hypertonic saline administration attenuates spinal cord injury. J Trauma 1997; 42: S54–S60.
Spera PA, Vasthare US, Tuma RF, Young WF . The effects of hypertonic saline on spinal cord blood flow following compression injury. Acta Neurochir (Wien) 2000; 142: 811–817.
Acknowledgements
This work has been supported by project APVV 14-0847 and project NEUREG, ITMS: 26220120063.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Tomko, P., Farkaš, D., Čížková, D. et al. Longitudinal enlargement of the lesion after spinal cord injury in the rat: a consequence of malignant edema?. Spinal Cord 55, 255–263 (2017). https://doi.org/10.1038/sc.2016.133
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sc.2016.133
- Springer Nature Limited