
Abstract
Background
Five years after successful malaria elimination, Aneityum Island in Vanuatu experienced an outbreak of Plasmodium vivax of unknown origin in 2002. Epidemiological investigations revealed several potential sources of P. vivax. We aimed to identify the genetic origin of P. vivax responsible for the resurgence.
Methods
Five P. vivax microsatellite markers were genotyped using DNA extracted from archived blood samples. A total of 69 samples from four P. vivax populations was included: 29 from the outbreak in 2002, seven from Aneityum in 1999 and 2000, 18 from visitors to Aneityum in 2000, and 15 from nearby Tanna Island in 2002. A neighbour-joining phylogenetic tree was constructed to elucidate the relationships among P. vivax isolates. STRUCTURE and principal component analysis were used to assess patterns of genetic structure.
Results
Here we show distinct genetic origins of P. vivax during the outbreak on Aneityum. While the origin of most P. vivax lineages found during the outbreak remains unidentified, limited genetic diversity among these lineages is consistent with a rapid expansion from a recent common ancestor. Contemporaneous P. vivax from neighboring Tanna and potential relapse of P. vivax acquired from other islands in 1999 and 2000 are also identified as minor contributors to the outbreak.
Conclusions
Multiple reintroductions of P. vivax after elimination highlight the high receptivity and vulnerability to malaria resurgence in island settings of Vanuatu, despite robust surveillance and high community compliance to control measures.
Plain language summary
Plasmodium vivax is one of several parasite species that cause malaria. On Aneityum Island in Vanuatu, malaria had been eliminated in 1997, but an outbreak was reported in 2002 despite protective measures still being in place. Here, we analysed DNA of parasites from the outbreak to understand its origin, since parasites of different origins will have slight differences in their DNA. Most parasites had similar DNA suggesting they had a recent shared common ancestor whose origin remains unidentified. From this analysis we were also able to find a minority of parasites that likely came from Tanna in 2002, while another small group of parasites may have originated from parasites imported to Aneityum in 1999 or 2000. This illustrates the difficulty of maintaining a malaria-free status in resource-limited areas and the threat of imported malaria to elimination efforts.
Similar content being viewed by others

Introduction
Plasmodium vivax is the predominant cause of malaria outside sub-Saharan Africa1. In countries with co-endemic P. falciparum and P. vivax, the proportion of P. vivax infections has consistently increased as the total malaria burden decreases, highlighting the considerable adaptive potential and relative resilience of this2,3. P. vivax infections are characterised by the dormant and persistent hypnozoite form in the liver, and its activation can cause recurrent blood-stage infections known as relapses, hampering efforts to control and eventually eliminate the species4. The re-emergence of P. vivax in many regions following successful elimination programmes serves as a crucial reminder of the propensity of the parasite to resurge in areas where it has almost been eliminated5,6. After malaria elimination, preventing the re-establishment of Plasmodium parasites and their transmission is crucial for sustaining malaria remission. As most of the eliminated areas are positioned along the margins of areas of endemicity, investigating the origins of imported parasites can help design promising targeted interventions7.
Islands provide natural ecological experiments with considerable potential for malaria intervention studies8 and some have demonstrated early success towards malaria elimination9,10. Vanuatu is an archipelago of 82 islands spanning 1176 km in the South Pacific. It straddles the Buxton line (170°E, 20°S; Fig. 1), which delineates the extension of Anopheles mosquitoes11. Malaria is endemic on most islands of Vanuatu. Both P. falciparum and P. vivax historically contributed to the overall malaria burden. Malaria incidence typically peaks in March and April, with a more conspicuous increase in P. falciparum than in P. vivax12. High population coverage of long-lasting insecticidal nets and widespread deployment of malaria rapid diagnostic tests and artemisinin-based combination therapies have substantially reduced malaria incidence since 201013. In 2021, Vanuatu reported 312 indigenous P. vivax cases, corresponding to an annual parasite incidence of 0.98 cases per 1000 population1.

Islands from which P. vivax was sampled are labelled. The inset shows the location of Vanuatu in the Southwest Pacific. All of the maps were drawn with the free software DIVA-GIS version 7.5 (http://www.diva-gis.org/).
Aneityum Island in Tafea Province is the southernmost inhabited island in Vanuatu and the only malaria-endemic island outside the Buxton line (Fig. 1). Tanna, which lies northwest of Aneityum, is the most populous island in the province and home to the provincial administrative capital Isangel and the main commercial town of Lenakel. Residents from outlying islands travel to Tanna to access services unavailable on their home islands13. On Aneityum, an integrated malaria elimination project with mass drug administration, sustained vector control interventions, and community-directed case surveillance initiated in 1991 led to the microscopically-confirmed elimination of P. falciparum in 1992 and P. vivax in 199714 (Fig. 2). The community-directed case surveillance programme was conducted by volunteers, who were selected by the local health committee and trained in malaria microscopy to serve as community microscopists. These community microscopists collaborated with a registered nurse on the island to examine all arrivals (active case detection) and febrile cases (passive case detection) for Plasmodium infection. Community-directed case surveillance has been maintained since 199215.

A Timeline of implementation of malaria elimination interventions, major milestones and relevant events, and cross-sectional surveys on Aneityum Island from 1991 to 2002 and B Flowchart of P. vivax sample selection for microsatellite analysis
In August 2000, a church meeting was held on Aneityum and attended by visitors from several islands in Vanuatu. Polymerase chain reaction (PCR) detected Plasmodium infections in 10.2% (28/274) of visitors who might have reintroduced P. vivax to Aneityum and triggered the outbreak in early 200216. From January to March 2002, community microscopists reported 67 P. vivax-positive cases among 240 blood samples collected from febrile residents on Aneityum, confirming an outbreak of vivax malaria of unknown origin.
Subsequent population-wide cross-sectional community- and school-based surveys on Aneityum in July 2002 revealed P. vivax prevalence of 10.1% (77/759) by PCR, with 71.4% (55/77) of the infections considered as sub-microscopic (PCR-positive but microscopy-negative)16. Analyses of sequence polymorphisms in the genes for the surface antigens P. vivax merozoite surface protein-1 and P. vivax circumsporozoite protein from microscopically positive cases revealed fewer haplotypes in parasites from Aneityum than those from other islands. Shared haplotypes among parasites from Aneityum and other islands indicated that the parasites responsible for the outbreak in 2002 observed on Aneityum were imported. However, the exact source(s) could not be determined.
Neutral microsatellites have been used extensively in studies of the genetic diversity of Plasmodium populations to assess transmission levels and patterns17,18, determine the multiplicity/complexity of infections19,20, and infer the origin and spread of drug-resistant parasites21,22. Furthermore, partly owing to their high mutation rates23, microsatellites are useful genetic markers for investigating recent malaria outbreaks24.
We analysed P. vivax microsatellite markers from the 2002 Aneityum outbreak and compared them with P. vivax populations from earlier periods and other islands to identify the origin of the parasites responsible for the 2002 outbreak. We tested the following non-mutually exclusive hypotheses: (A) the outbreak was caused by persistent P. vivax that had been circulating on Aneityum in 1999 and 2000; (B) the outbreak was caused by the reintroduction of P. vivax by church meeting attendees in 2000; and (C) the outbreak was caused by a recent P. vivax reintroduction from neighbouring Tanna Island in 2002.
Results from this study reveal distinct genetic origins of P. vivax responsible for the outbreak on Aneityum. While the origin of most P. vivax lineages cannot be identified, limited genetic diversity among these lineages suggests a common ancestor recently introduced to the island. A recent importation of P. vivax from Tanna to Aneityum (hypothesis C) is implicated by the sharing of an identical microsatellite haplotype among small subsets of parasite isolates from these two islands. Phylogenetic and population structure analyses provide support for another more distant P. vivax importation event in 1999 or 2000 (hypotheses A and B).
Methods
Ethics statement
The Ethics Committee of Tokyo Women’s Medical University and the Ministry of Health in Vanuatu approved the study. Ethical approval to analyse previously collected samples was provided by the Ethics Committee at Osaka Metropolitan University (approval number 3206). Oral informed consent was obtained from all adult participants and parents in the case of children due to the lack of formal writing systems for local 'kastom' languages in Vanuatu. The study’s purpose, procedure, and potential risks and benefits were explained to participants in Bislama (lingua franca understood by most adults in Vanuatu) and local languages by ni-Vanuatu collaborators. The consent was witnessed by a neutral third party (e.g. teacher, village chief) who recorded the name of each participant at study enrolment. All study procedures were conducted in adherence to the Declaration of Helsinki.
Sample collection and detection of Plasmodium infections
Cross-sectional, community- and school-based malariometric surveys were conducted on Aneityum Island in June 1999, August 2000, July 2002, and Tanna Island in July 2002. In 2000, the survey on Aneityum included 274 individuals from other islands (Santo, Malo, Ambrym, Malakula, Paama, Tongoa, Tongariki, Epi, Efate, Erromango, Tanna, Aniwa, and Futuna) who had come to Aneityum to attend a church meeting (August 23-28, 2000). P. vivax populations are henceforth denoted by the year and island origin (e.g., 1999 Aneityum). P. vivax isolates detected among church meeting attendees in 2000 are collectively referred to as 2000 non-Aneityum.
Demographic information of the survey participants, including age, sex, and village of residence, was recorded. Using microscopy and PCR, capillary blood samples were collected via finger pricking to examine Plasmodium infections. For microscopic examination, the thin blood film was fixed with methanol, and both thick and thin films were stained with 10% Giemsa solution for 10 min. An experienced microscopist examined the stained blood films. Two additional blood samples (70 μL each) were collected as dried blood spots (DBS) on Whatman ET31 Chr filter paper (Whatman International, Maidstone, UK) using a 75-mm heparinised haematocrit capillary tube (Thermo Fisher Scientific, MA, USA). After drying, DBS was stored in individual plastic bags at ambient temperature in the field and then at −20 °C upon arrival at the laboratory in Japan. DNA was extracted from a quartered DBS (corresponding to 17.5 μL of blood) using the QIAamp Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Plasmodium infections were detected using the nested PCR method that targets the mitochondrial cytochrome c oxidase-3 (cox3) gene, using 4 μL of extracted DNA (equivalent to 0.47 μL of blood) as template25.
P. vivax nuclear DNA quality verification
To ensure the presence and quality of P. vivax nuclear DNA, all mitochondrial cox3 PCR-positive samples were subject to nested PCR amplifications of three nuclear loci, msp1F1, msp1F3 and MS1626. Each 10-μL reaction consisted of 5 μL of the GoTaq Green Master Mix (Promega, WI, USA) and 1.25 μM of each primer (Supplementary Data 1). We used 3 μL of DNA (equivalent to 0.35 μL of blood) as a template for the primary PCR and 1 μL of the primary PCR product for the nested PCR. The cycling conditions for the primary PCR were as follows: 95 °C for 1 min; 40 cycles at 95 °C for 15 s, 59 °C for 30 s, and 72 °C for 30 s, and 72 °C for 30 s, and a final extension at 72 °C for 5 min. The cycling conditions for the nested PCR were identical to those of the primary PCR, except that only 30 cycles were performed. The presence of PCR amplicons after the nested round was detected using the LabChip GX Touch (PerkinElmer, MA, USA). Samples that failed to amplify two or more quality verification nuclear markers were excluded from microsatellite analysis.
Genotyping of microsatellite markers
Samples positive for two or more quality verification nuclear markers were used for microsatellite analysis. Twelve validated highly polymorphic microsatellite markers (MS1, MS2, MS5, MS6, MS7, MS8, MS9, MS10, MS12, MS15, MS20 and MS3.27) were amplified using hemi-nested PCR27. Each 10-μL reaction consisted of 5 μL of the PrimeSTAR Max Premix (Takara-Bio, Kusatsu, Japan) and 1.25 μM of each primer (Supplementary Data 1). The primary PCR used 3 μL of DNA as a template, while the nested PCR used 1 μL of the primary PCR product. The cycling conditions for the primary PCR were as follows: 95 °C for 1 min; 40 cycles at 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s, and a final extension at 72 °C for 5 min. The cycling conditions for the nested PCR were as follows: 95 °C for 1 min, 35 cycles at 95 °C for 15 s, 61 °C for 30 s, 72 °C for 45 s and a final extension at 72 °C for 5 min. The nested PCR products were resolved on an Applied Biosystems 3730xl DNA Analyzer using the GeneScan 600 LIZ size standard (Thermo Fisher Scientific). Allele sizes were determined using Peak Scanner 2 (Thermo Fisher Scientific). Alleles with a minimum of one-third of the relative fluorescence units of the primary allele were recorded as secondary alleles.
Population genetic analysis
Because most samples (68.2%; 75/110) included in the microsatellite analysis were sub-microscopic and likely contained limited P. vivax DNA, PCR amplification success rates varied among markers despite multiple attempts (Supplementary Data 2). To include as many samples as possible in subsequent analyses, five markers (MS2, MS5, MS6, MS7 and MS8) with the most genotyped samples were included. The microsatellite haplotype was constructed for each sample using only the primary allele at each marker.
The genetic diversity of each microsatellite locus was assessed by calculating the number of alleles and expected heterozygosity (HE) in each population. In addition, haplotype diversity (h) was used to assess P. vivax population diversity. HE and h were calculated using the following equations:
Where n represents the number of analysed samples, and Pi represents the frequency of the ith allele or ith haplotype in the population28. The mean number of allele differences between pairs of haplotypes in each population was calculated using Arlequin 3.5.2.229.
Multilocus linkage disequilibrium (LD) among microsatellites was assessed using LIAN 3.730. The standardised index of association (ISA) was calculated as follows:
Where n is the number of loci analysed, VD is the observed variance in the number of loci differing between each pair of haplotypes, and Ve is the expected variance under the linkage equilibrium. To investigate whether the observed data differed from random expectations, we compared VD values with the distribution of Ve values using Monte Carlo simulations with 10,000 random resamplings of simulated datasets, in which alleles at each locus were randomly reshuffled among genotypes.
Genetic differentiation among populations was assessed using pairwise FST genetic distances, as described in Arlequin 3.5.2.229. The statistical significance of the FST genetic distances was evaluated by randomly permuting haplotypes between sites approximately 10,000 times to generate a null distribution against which the observed values were compared.
To determine relationships among P. vivax microsatellite haplotypes, Populations 1.2.3231 was used to calculate Nei’s DA genetic distance28,32 and MEGA1133 was used to construct an unrooted neighbour-joining tree. To detect the P. vivax population genetic structure, we used the admixture model34 with island as prior information (LOCPRIOR) in STRUCTURE version 2.3.435. The number of genetic clusters (K) was set from 1 to 10 and estimated using a Markov Chain Monte Carlo algorithm with 50,000 burn-in and 50,000 iterations per replication over 20 replications for each K. Using STRUCTURE HARVESTER Web v0.6.9436, we calculated ∆K37, the rate of change in the log probability between successive Ks, to estimate the optimal K. Principal component analysis (PCA) was used as a non-model-based alternative to STRUCTURE to infer the P. vivax population genetic structure. PCA was performed using the Adegenet package38 in R version 4.2.1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Detection of Plasmodium infections using microscopy and PCR
Of 2608 samples available from the 1999–2002 period in Vanuatu (Fig. 2), PCR detected 219 (8.4%) Plasmodium infections, of which 57 (2.8%) were also detected by microscopy. P. vivax accounted for most microscopic (75.4%; 43/57) and sub-microscopic (85.8%; 139/162) infections. Throughout the study period, neither microscopy nor PCR detected any species other than P. vivax on Aneityum Island, and the prevalence of P. vivax infection by PCR increased from 0.8% (6/709) in 1999 to 10.1% (77/759) in 2002 (Table 1).
Selection of P. vivax samples for microsatellite analysis
PCR amplification of at least two nuclear loci used for DNA quality verification failed in 72 of the 182 mitochondrial cox3 PCR-positive P. vivax samples. Of the remaining 110 samples subject to PCR amplification of 12 microsatellites, complete genotyping at five markers (MS2, MS5, MS6, MS7 and MS8) was successful in 69 samples (24 microscopic and 45 sub-microscopic): five from 1999 Aneityum, two from 2000 Aneityum, 18 from 2000 non-Aneityum, 15 from 2002 Tanna and 29 from 2002 Aneityum. Due to small sample sizes, the 1999 and 2000 Aneityum samples were combined as 1999–2000 Aneityum and analysed as a single 'pre-outbreak' population (Fig. 2).
Microsatellite diversity in P. vivax populations
Unbiased HE of the five microsatellite markers in each population is summarised in Table 2. Except for MS8, HE was consistently lower in the 2002 Tanna and 2002 Aneityum populations. Limited polymorphisms were observed in MS5 (HE ± SE: 0.229 ± 0.003) and MS6 (0.254 ± 0.004) in 2002 Aneityum, and MS7 (0.342 ± 0.028) in 2002 Tanna.
Fifty-three unique microsatellite haplotypes were identified in 69 samples (Supplementary Data 3). In the 1999–2000 Aneityum and 2000 non-Aneityum samples, each P. vivax isolate harboured a unique haplotype (Table 2). In 2002 Tanna, 12 unique haplotypes were identified among 15 samples (h ± SE: 0.971 ± 0.011), and three haplotypes (26, 28 and 29) were each shared between two samples. In 2002 Aneityum, 17 unique haplotypes were identified in 29 samples (0.836 ± 0.027). Haplotype 39 was found in 41.4% (12/29) of the samples, whereas another closely related haplotype 47 was shared by two samples. Haplotype 26 was shared between two 2002 Tanna and one 2002 Aneityum samples. Haplotype 42, which differed from haplotype 26 at MS8, was found in another 2002 Aneityum sample (Supplementary Data 3).
The mean number of allele differences between the pairs of samples in each population was calculated as another measure of within-population genetic variation (Table 3). The mean was lower in the 2002 Tanna (mean ± SD: 2.857 ± 1.267) and 2002 Aneityum (2.091 ± 1.562) populations than in the 1999–2000 Aneityum (4.238 ± 0.865) and the 2000 non-Aneityum (4.562 ± 0.538) populations. Consistent with this finding, significant LD among loci was observed only in 2002 Tanna (p = 2.6 × 10−3) and 2002 Aneityum (p < 2.0 × 10−5) (Table 4).
Genetic relatedness and structuring of P. vivax populations
FST analyses of pairwise genetic distances indicated that the 2002 Aneityum and the 2002 Tanna populations differed significantly from each other and other populations (p < 0.05) (Table 3). However, no statistically significant difference was observed between the 1999–2000 Aneityum population and the 2000 non-Aneityum population and between microscopic (n = 13) and sub-microscopic (n = 16) infections within the 2002 Aneityum population (FST = 0.001; p = 0.42).
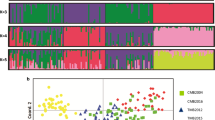
Two major clades were identified in the unrooted neighbour-joining tree. The first comprised all 15 isolates from 2002 Tanna, two from 2002 Aneityum (haplotypes 26 and 42), and one from 2000 non-Aneityum. The second consisted of six of seven isolates from 1999–2000 Aneityum and 16 of 18 isolates from 2000 non-Aneityum (Fig. 3A). Both STRUCTURE and PCA revealed population structure among the four P. vivax populations. Bayesian clustering assignments revealed that the most likely number of clusters (K) was three (Fig. 3B, C). Two lineages (haplotypes 26 and 42) in the 2002 Aneityum population exhibited patterns of inferred ancestry similar to those in the 2002 Tanna population. PCA also revealed a similar pattern, with three distinct clusters; the first consisted exclusively of lineages from 2002 Aneityum, the second consisted of lineages from 2002 Tanna, and two lineages from 2002 Aneityum (haplotypes 26 and 42), and the third consisted of haplotypes from 1999 to 2000 Aneityum and 2000 non-Aneityum (Fig. 3C).

A An unrooted neighbour-joining tree based on Nei’s DA genetic distances. B Inference of population structure using the admixture model in STRUCTURE. Each bar represents an individual P. vivax isolate. The isolates are grouped by populations identified by year and geographical origin. The most likely number of ancestral populations (K) is three, each represented by a colour. C Principal component analysis (PCA) plot of P. vivax microsatellite haplotypes. Arrows indicate P. vivax isolates of likely Tanna origin in the 2002 Aneityum outbreak.
Discussion
In this study, we analysed the genetic variation at five microsatellite markers from microscopic and sub-microscopic P. vivax infections using samples collected from Aneityum and other islands before and during the 2002 Aneityum outbreak. Our results indicated that most P. vivax lineages identified during the outbreak were genetically distinct from those on Aneityum in 1999 and 2000 (hypothesis A) and among church meeting attendees in 2000 (hypothesis B). However, a small number of P. vivax isolates during the outbreak appeared to have originated from Tanna Island (hypothesis C).
Infrastructure was (and remains) limited on Aneityum, prompting many residents to travel to Tanna to access services including secondary education and hospital care. The observation that P. vivax isolates from Aneityum and Tanna shared an identical microsatellite haplotype 26 recapitulated the importance of human-mediated parasite movement among islands in Vanuatu39,40 and highlighted the ease with which an outbreak could be seeded. Although we could not definitively ascertain the direction of P. vivax gene flow, we noted that both haplotype 26 and the closely related haplotype 42 were nested within the 2002 Tanna clade in the unrooted neighbour-joining tree (Fig. 3A) and clustered with the Tanna lineages instead of the remaining Aneityum lineages in PCA (Fig. 3C). Phylogenetic analysis (Fig. 3A) and PCA (Fig. 3C) also indicated genetic affinity among one 2002 Aneityum sample and one 1999–2000 Aneityum and one 2000 non-Aneityum samples. The 1999–2000 Aneityum sample was derived from a male child who had moved to Aneityum from Pentecost. The child was diagnosed with P. vivax in May 1999 and again in July 1999 during our survey. The 2000 non-Aneityum sample was derived from a male adult church meeting attendee from Tanna. Not only did these infections exemplify the vulnerability of Aneityum to P. vivax importation, but they also suggested the possibility that one of the seeds for the 2002 outbreak may have been introduced in 1999 or 2000. Notably, we previously indicated the potential of P. vivax relapses occurring up to five years after the initial infection16, raising the possibility that the 2002 outbreak was partially seeded from long-latency relapses from infections initially acquired years before.
The 2002 Tanna samples included in this study were collected through cross-sectional community surveys in early July, approximately a week before the cross-sectional surveys on Aneityum that supplied the P. vivax outbreak samples. These observations suggested that the importation of P. vivax from Tanna likely had a direct, albeit minor, contribution to the outbreak in Aneityum.
Notably, the 2002 Tanna population shared many characteristics with the 2002 Aneityum population, including lower expected heterozygosity and haplotype diversity (Table 2), lower mean pairwise differences (Table 3), and significant LD among loci (Table 4). Nationally, the increase in malaria incidence during the early 2000s was attributed to the combined effect of an earthquake followed by a tsunami and a general reduction in funding for the malaria programme13,41. Lower P. vivax genetic diversity in the 2002 Tanna population was consistent with heightened transmission and rapid expansion of a limited number of parasite clones.
Although the geographic origin of most P. vivax samples on Aneityum in 2002 remains elusive, our findings suggested that this population is semi-clonal, with 41.4% (12/29) of the isolates represented by a single microsatellite haplotype (Supplementary Data 3). The samples included in this study were collected in July 2002, a few months after the onset of the outbreak reported by community microscopists in January16 and the typical peak transmission season between February and March12. While the mutation rates of microsatellites and recombination rates in P. vivax are unknown42,43, the limited microsatellite diversity observed in this population suggested that most P. vivax lineages on Aneityum in 2002 likely descended from a small number of recent common ancestors.
In our previous study, we speculated that visitors who attended the church meeting in 2000 were a potential source of the parasites responsible for the 2002 outbreak in Aneityum16. However, our findings from the current study did not support a direct causal link between P. vivax lineages among visitors and those among Aneityum residents 2 years later, as no shared haplotypes were observed between the 2000 non-Aneityum and 2002 Aneityum P. vivax populations. FST-based analysis of genetic differentiation (Table 3) and population structure using STRUCTURE and PCA (Fig. 3B, C) indicated that the two P. vivax populations were distinct.
We previously identified microscopic P. vivax infections only in individuals aged ≤20 (born after 1981) and concluded that persisting anti-P. vivax IgG antibodies among adults routinely exposed to P. vivax before elimination may have limited the infections to sub-microscopic levels in this cohort16. In this study, we did not observe significant differentiation (FST = 0.001; p = 0.42) between the lineages of microscopic and sub-microscopic infections in Aneityum in 2002. Therefore, additional microsatellite markers and the genotyping or sequencing of other host immunity-targeted P. vivax antigen genes are required to assess whether distinct parasitic clones cause microscopic and sub-microscopic infections.
Our study had several limitations. First, the sample quality and availability limited our ability to genetically characterise all available P. vivax samples. Most samples in this study were obtained from asymptomatic and sub-microscopic P. vivax infections with low parasitaemia levels, which limited the number of samples and microsatellites included in our analyses. Of the 182 P. vivax-positive samples by the mitochondrial cox3 PCR, 49 (26.9%) failed to amplify any nuclear loci, while 23 (12.6%) amplified only one nuclear locus. These 72 samples were deemed to have low DNA concentrations and were excluded from microsatellite marker analysis (Fig. 2). Molecular diagnostic yields are mainly determined by blood volume analysed and the gene copy number of the amplified molecular marker44. The discrepancy in PCR amplification efficiencies in our study likely reflected such a difference in gene copy numbers between the mitochondrial cox3 and the nuclear microsatellite markers. Within each parasite, there are 20 to 150 copies of the mitochondrial cox3 gene45, but only one copy of nuclear microsatellite markers, making the former an ideal target for PCR detection of Plasmodium infections. Low P. vivax nuclear DNA concentrations in most samples, and a lack of availability of advanced methods and analytical approaches, limited our ability to gain a more comprehensive understanding of the population genomic diversity of P. vivax parasites. Recent developments in selective whole genome amplification (sWGA) of Plasmodium species from samples stored as DBS and in amplicon deep sequencing have provided opportunities to include low-parasite density infections in malaria genomic epidemiology46,47,48,49,50. Nevertheless, there is some discussion that advanced malaria genomics may have several gaps for an immediate “real-world malaria control and elimination strategy”51 and may not be necessary for malaria control or even its elimination52. Second, P. vivax samples from other islands in Vanuatu in 2000 were collected from church meeting attendees and thus were not representative of the genetic diversity of the parasites on those islands. Third, 2002 Aneityum samples were collected six months after the onset of the outbreak. Additional but undetected parasite reintroduction events between the onset of the outbreak in early 2002 and our survey in July 2002 could have obscured or even replaced the original P. vivax clone that triggered the outbreak. Lastly, the primary objective of the cross-sectional survey on Aneityum in July 2002 was to determine P. vivax prevalence in the communities. As it was not an outbreak investigation, we did not enquire about participants’ travel histories or distinguish whether an active infection resulted from a recent inoculation by an infected Anopheles vector or a relapse from activation of latent hypnozoites. In endemic countries, a substantial proportion of P. vivax cases is reportedly caused by the activation of hypnozoites with genotypes distinct from those that caused the initial infections53. Detecting potential relapses from heterologous hypnozoites among the P. vivax infections in the 2002 Aneityum samples could have complicated the utilisation of genotyping or DNA sequencing to determine the source of the parasites responsible for the outbreak. Since P. vivax infections in adults were sub-microscopic due to persisting immunity acquired before malaria elimination and undetectable by diagnostics available locally, coupled with the potential of long-latency relapse, the 2002 outbreak’s seeding event could have occurred years ago, and the outbreak itself could have been triggered by a recent relapse. Determining the proportion of infections resulting from relapse during P. vivax outbreaks can inform the most optimal response strategy, however distinguishing between primary infections and relapses in settings such as Aneityum or Vanuatu, where conditions conducive to transmission remain, is difficult.
Although we lack entomological data, the observation that most P. vivax isolates during the 2002 outbreak were genetically very similar suggests that transmission by Anopheles mosquitoes played a major role in sustaining the outbreak. Therefore, it can be inferred that the level of receptivity was probably very high in 2002. In response, a second round of mass drug administration was implemented, targeting individuals under 20 years of age. At the same time, provisions of insecticide-treated nets (ITNs) were strengthened, coupled with a robust malaria health promotion programme to raise awareness of the continuous risk of malaria resurgence among local communities15. Subsequent annual cross-sectional surveys revealed low prevalence (1.9 to 3.9%) of P. vivax infections by PCR between 2003 and 2007. Since 2010, no Plasmodium infections have been detected by microscopy and PCR54. Nonetheless, 74.1 and 67.4% of the island’s residents reported ITN use in 2016 and 2019, respectively (unpublished data). We believe that high ITN usage likely suppressed malaria receptivity on Aneityum Island.
Vanuatu aims to interrupt malaria transmission and achieve zero indigenous malaria cases nationally by the end of 2023 through a multi-stage process in which elimination is targeted first in the most peripheral provinces with low incidences (Tafea, Torba and Penama), followed by central provinces with intermediate (Shefa) and finally high incidences (Malampa and Sanma)13. Key to this strategy is preventing the reintroduction of Plasmodium parasites and re-establishing their transmission in provinces where malaria has been successfully eliminated. Case-based surveillance and rapid response are imperative to sustaining malaria elimination at the subnational level. Despite surveillance among arrivals and suspected febrile cases in communities, high coverage of ITNs, and persisting antimalarial immunity among residents, an outbreak of P. vivax was reported on Aneityum only five years after the parasite had been eliminated, highlighting both the high receptivity and high vulnerability of islands in Vanuatu to resurgence15,16. Genetic epidemiological methods have provided fruitful insights in many epidemiological investigations. However, our findings suggest that pinpointing the origins of parasites among many potential sources during a reintroduction event based on genetic or genomic methods alone can be challenging.
Data availability
Source data used for results and figures are provided in the Supplementary Data 4. The authors declare that all data supporting the findings of this study are available within the article and its Supplementary Information files are available from the authors upon request.
References
WHO. World Malaria Report 2022 (WHO, 2022).
Cotter, C. et al. The changing epidemiology of malaria elimination: new strategies for new challenges. Lancet 382, 900–911 (2013).
Price, R. N., Commons, R. J., Battle, K. E., Thriemer, K. & Mendis, K. Plasmodium vivax in the era of the shrinking P. falciparum map. Trends Parasitol. 36, 560–570 (2020).
White, N. J. The rise and fall of long-latency Plasmodium vivax. Trans. R. Soc. Trop. Med. Hyg. 113, 163–168 (2019).
Battle, K. E. & Kevin Baird, J. The global burden of Plasmodium vivax malaria is obscure and insidious. PLoS Med. 18, 1–18 (2021).
Sattabongkot, J., Tsuboi, T., Zollner, G. E., Sirichaisinthop, J. & Cui, L. Plasmodium vivax transmission: chances for control? Trends Parasitol. 20, 192–198 (2004).
Nasir, S. M. I., Amarasekara, S., Wickremasinghe, R., Fernando, D. & Udagama, P. Prevention of re-establishment of malaria: historical perspective and future prospects. Malar. J. 19, 1–16 (2020).
Bhattarai, A. et al. Impact of artemisinin-based combination therapy and insecticide-treated nets on malaria burden in Zanzibar. PLoS Med. 4, 1784–1790 (2007).
Bruce-Chwatt, L. J., Draper, C. C. & Konfortion, P. Seroepidemiological evidence of eradication of malaria from Mauritius. Lancet 302, 547–551 (1973).
Tian, H. et al. Malaria elimination on Hainan Island despite climate change. Commun. Med. 2, 1–9 (2022).
Buxton, P. A. Researches in Polynesia and Melanesia. Parts I–VII (London School of Hygiene and Tropical Medicine, 1927).
Kaneko, A. et al. Malaria epidemiology, glucose 6-phosphate dehydrogenase deficiency and human settlement in the Vanuatu Archipelago. Acta Trop. 70, 285–302 (1998).
Vanuatu Government, M. of H. National Strategic Plan for Malaria Elimination 2021-2026. Malaria and other Vector Borne Diseases Control Program (2021).
Kaneko, A. et al. Malaria eradication on islands. Lancet 356, 1560–1564 (2000).
Kaneko, A. A community-directed strategy for sustainable malaria elimination on islands: short-term MDA integrated with ITNs and robust surveillance. Acta Trop. 114, 177–183 (2010).
Kaneko, A. et al. Characteristic age distribution of Plasmodium vivax infections after malaria elimination on Aneityum Island, Vanuatu. Infect. Immun. 82, 243–252 (2014).
Kattenberg, J. H. et al. Monitoring Plasmodium falciparum and Plasmodium vivax using microsatellite markers indicates limited changes in population structure after substantial transmission decline in Papua New Guinea. Mol. Ecol. 29, 4525–4541 (2020).
Tessema, S. K. et al. Spatial and genetic clustering of Plasmodium falciparum and Plasmodium vivax infections in a low-transmission area of Ethiopia. Sci. Rep. 10, 1–10 (2020).
Pacheco, M. A. et al. Malaria in Venezuela: changes in the complexity of infection reflects the increment in transmission intensity. Malar. J. 19, 1–15 (2020).
Mobegi, V. A., Herren, J. K., Touray, A. O., Wamunyokoli, F. & Butungi, H. Prevalence of asymptomatic P. falciparum gametocyte carriage among school children in Mbita, Western Kenya and assessment of the association between gametocyte density, multiplicity of infection and mosquito infection prevalence. Wellcome Open Res. 5, 1–38 (2021).
Imwong, M. et al. Molecular epidemiology of resistance to antimalarial drugs in the Greater Mekong subregion: an observational study. Lancet Infect. Dis. 20, 1470–1480 (2020).
Auburn, S. et al. Genomic analysis of a pre-elimination Malaysian Plasmodium vivax population reveals selective pressures and changing transmission dynamics. Nat. Commun. 9, 1–12 (2018).
Haasl, R. J. & Payseur, B. A. Multi-locus inference of population structure: a comparison between single nucleotide polymorphisms and microsatellites. Heredity 106, 158–171 (2011).
Baldeviano, G. C. et al. Molecular epidemiology of Plasmodium falciparum malaria outbreak, Tumbes, Peru, 2010–2012. Emerg. Infect. Dis. 21, 797–803 (2015).
Isozumi, R. et al. Improved detection of malaria cases in island settings of Vanuatu and Kenya by PCR that targets the Plasmodium mitochondrial cytochrome c oxidase III (cox3) gene. Parasitol. Int. 64, 304–308 (2015).
Ross, A. et al. The incidence and differential seasonal patterns of Plasmodium vivax primary infections and relapses in a cohort of children in Papua New Guinea. PLoS Negl. Trop. Dis. 10, 1–18 (2016).
Koepfli, C. et al. Evaluation of plasmodium vivax genotyping markers for molecular monitoring in clinical trials. J. Infect. Dis. 199, 1074–1080 (2009).
Masatoshi, N. Molecular Evolutionary Genetics (Columbia Univ. Press, 1987).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Haubold, B. & Hudson, R. R. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics 16, 847–848 (2000).
Langella, O. Population 1.2.32: population genetic software (individuals or populations distances, phylogenetic trees). (2011).
Takezaki, N. & Nei, M. Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics 75, 389–399 (1996).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587 (2003).
Hubisz, M. J., Falush, D., Stephens, M. & Pritchard, J. K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 9, 1322–1332 (2009).
Earl, D. A. & vonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Jombart, T. Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Lum, J. K., Kaneko, A., Taleo, G., Amos, M. & Reiff, D. M. Genetic diversity and gene flow of humans, Plasmodium falciparum, and Anopheles farauti s.s. of Vanuatu: inferred malaria dispersal and implications for malaria control. Acta Trop. 103, 102–107 (2007).
Chan, C. W. et al. Plasmodium vivax and Plasmodium falciparum at the crossroads of exchange among Islands in Vanuatu: implications for malaria elimination strategies. PLoS ONE 10, 1–13 (2015).
Caminade, J., Koshimura, S. & Moore, A. L. Vanuatu earthquake and tsunami cause much damage, few casualties. Trans. Am. Geophys. Union 81, 641–647 (2000).
Koepfli, C. et al. A large Plasmodium vivax reservoir and little population structure in the South Pacific. PLoS ONE 8, 1–9 (2013).
Benavente, E. D. et al. Distinctive genetic structure and selection patterns in Plasmodium vivax from South Asia and East Africa. Nat. Commun. 12, 1–11 (2021).
Gruenberg, M. et al. Plasmodium vivax molecular diagnostics in community surveys: pitfalls and solutions. Malar. J. 17, 1–10 (2018).
Wilson, R. J. & Williamson, D. H. Extrachromosomal DNA in the Apicomplexa. Microbiol. Mol. Biol. Rev. 61, 1–16 (1997).
Joste, V., Guillochon, E., Clain, J., Coppée, R. & Houzé, S. Development and optimization of a selective whole-genome amplification to study Plasmodium ovale Spp. Microbiol. Spectr. 10, e0072622 (2022).
Teyssier, N. B. et al. Optimization of whole-genome sequencing of Plasmodium falciparum from low-density dried blood spot samples. Malar. J. 20, 1–8 (2021).
Early, A. M. et al. Detection of low-density Plasmodium falciparum infections using amplicon deep sequencing. Malar. J. 18, 1–13 (2019).
Reda, A. G. et al. Amplicon deep sequencing of five highly polymorphic markers of Plasmodium falciparum reveals high parasite genetic diversity and moderate population structure in Ethiopia. Malar. J. 22, 1–7 (2023).
Osborne, A. et al. Drug resistance profiling of asymptomatic and low-density Plasmodium falciparum malaria infections on Ngodhe island, Kenya, using custom dual-indexing next-generation sequencing. Sci. Rep. 13, 1–10 (2023).
Neafsey, D. E., Taylor, A. R. & MacInnis, B. L. Advances and opportunities in malaria population genomics. Nat. Rev. Genet. 22, 502–517 (2021).
Neafsey, D. E. & Volkman, S. K. Malaria genomics in the era of eradication. Cold Spring Harb. Perspect. Med. 7, 1–12 (2017).
Noviyanti, R. et al. Hypnozoite depletion in successive Plasmodium vivax relapses. PLoS Negl. Trop. Dis. 16, 1–9 (2022).
Chan, C. W. et al. Surveillance for malaria outbreak on malaria-eliminating islands in Tafea Province, Vanuatu after Tropical Cyclone Pam in 2015. Epidemiol. Infect. 145, 41–45 (2016).
Acknowledgements
We want to express our sincere gratitude to the participants in this study. We are also grateful to Mayumi Fukui, Ikuko Kusuda, Satoko Naito, Tomomi Kuwana and all the supporting staff in the Ministry of Health in Vanuatu for their assistance. This project was supported by grants from the Swedish Research Council (2008-3097; 2009-3233 and 2022-04055), Formas (2022-02360), the Japanese Society for the Promotion of Science (24390141; 22406008; Asia-Africa Science Platforms; 21K02817), Health Labour Sciences and the Global COE Programme at Nagasaki University.
Funding
Open access funding provided by Karolinska Institute.
Author information
Authors and Affiliations
Contributions
C.W.C. and Akira Kaneko designed the study. M.K., S.Y., H.I. and Akira Kaneko conducted surveys and obtained samples used in this study. S.S. and C.W.C. performed microsatellite genotyping data analyses. S.S. and C.W.C. wrote the first draft of the manuscript, and S.S., C.W.C., M.K., S.Y., H.I., G.T., A.KC., W.K., Y.K. and Akira Kaneko reviewed, revised and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks Bimandra Djaafara and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sekine, S., Chan, C.W., Kalkoa, M. et al. Tracing the origins of Plasmodium vivax resurgence after malaria elimination on Aneityum Island in Vanuatu. Commun Med 4, 91 (2024). https://doi.org/10.1038/s43856-024-00524-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43856-024-00524-9
- Springer Nature Limited