

Abstract
Microglia continuously survey the brain parenchyma and actively shift status following stimulation. These processes demand a unique bioenergetic programme; however, little is known about the metabolic determinants in microglia. By mining large datasets and generating transgenic tools, here we show that hexokinase 2 (HK2), the most active isozyme associated with mitochondrial membrane, is selectively expressed in microglia in the brain. Genetic ablation of HK2 reduced microglial glycolytic flux and energy production, suppressed microglial repopulation, and attenuated microglial surveillance and damage-triggered migration in male mice. HK2 elevation is prominent in immune-challenged or disease-associated microglia. In ischaemic stroke models, however, HK2 deletion promoted neuroinflammation and potentiated cerebral damages. The enhanced inflammatory responses after HK2 ablation in microglia are associated with aberrant mitochondrial function and reactive oxygen species accumulation. Our study demonstrates that HK2 gates both glycolytic flux and mitochondrial activity to shape microglial functions, changes of which contribute to metabolic abnormalities and maladaptive inflammation in brain diseases.
Similar content being viewed by others


Main
Metabolism has emerged as a key regulator in both innate and adaptive immunity1. Microglia, the primary immune cells and key guardians of brain activity in the brain, adopt different metabolic states in response to exogenous stimuli2,3,4. For example, pro-inflammatory microglia utilize aerobic glycolysis to promote synthesis of inflammatory cytokines, whereas immunomodulatory microglia in mice increase fatty acid metabolism to drive anti-inflammatory responses2,3. Moreover, metabolic dysfunction of microglia has been implicated in multiple diseases including Alzheimer’s disease (AD) and chronic demyelination disease models4,5,6. Microglia deficient in triggering the receptor expressed on myeloid cells (TREM2) or expressing TREM2 variants associated with increased risk of AD onset, exhibit curtailed anabolic metabolism and dampened phagocytic activity5. In addition, microglia surrounding the amyloid beta (Aβ) plaques exhibit elevated histone lactylation, which drives a lactylation/pyruvate kinase 2 (PKM2)/glycolysis positive feedback loop to promote AD pathology7. Thus, metabolic changes have been intimately linked to microglial function under different stimulating or pathological conditions. However, little is known about the role of cellular metabolism governing microglial function under physiological conditions.
Microglia are distributed throughout the brain with a relatively constant number and low turnover rate in the adult brain8,9. By extending and retracting their motile processes, microglia continuously survey the microenvironment to maintain brain homeostasis10,11,12. Following acute depletion or under disease conditions, microglia undergo rapid proliferation to clonally expand their number, resulting in restoration of the homeostatic pool (repopulation)13,14,15 or formation of microglial clusters surrounding disease sites (microgliosis)12. In addition, microglia quickly migrate towards the damage sites and elicit immune responses to restrain and repair the damage10,12. These dynamic features demand microglia possess a unique bioenergetic profile to facilitate their remarkable plasticity. However, little is known about the specific molecular determinants that metabolically and bioenergetically shape microglial function.
The brain predominantly uses glucose as energy fuel16,17. Hexokinase (HK), by catalysing phosphorylation of glucose to glucose-6-phosphate, is the first rate-limiting enzyme in glucose utilization18,19. Four main HK isozymes, HK1, HK2, HK3 and HK4, with different biochemical features and catalytic activities have been identified18,19. Among them, HK1 and HK2 also associate with the outer mitochondrial membrane (OMM), allowing preferential access to mitochondrial ATP to promote glycolysis18,20. While HK1 and HK2 have previously been shown to be primarily expressed in the brain or muscle and adipose tissues for quick energy supply, respectively18, recent studies have shown that they promote inflammatory response by driving glycolysis in several types of immune cells21,22,23. In addition, anchorage of HKs to the OMM was shown to prevent the opening of the mitochondrial permeability transition pore (mPTP) and release of cytochrome c and/or apoptotic mediators to assist cell survival24,25. Notably, dynamic associations of HKs with the OMM may also be involved in inflammatory responses as mitochondria are an important hub for the regulation of multiple immune signalling26,27,28,29. How HKs are involved in microglial function and brain diseases remains poorly understood.
By systematically analysing the metabolic regulator in different brain cell types, we unexpectedly found that HK2 is selectively expressed in microglia, whereas HK1 is predominantly expressed in neurons and astrocytes in the brain. HK2 is the most active HK isozyme18 and its ablation perturbs microglial glycolytic flux, resulting in an energy-deficient state, attenuated microglial surveillance and delayed microglial repopulation. HK2 is robustly elevated in immune-challenged and disease-associated microglia (DAM) to promote glycolysis in multiple disease models. In an ischaemic stroke model, however, HK2 ablation aggravated inflammatory responses and potentiated brain damages. The pro-inflammatory effects following HK2 deletion are associated with impaired mitochondrial function and reactive oxygen species (ROS) accumulation. Our study reveals that HK2 acts as a critical metabolic checkpoint to dually gate glucose metabolism and mitochondrial function to shape microglial functions and maintain brain homeostasis.
Results
Hexokinase 2 is a microglia-specific metabolic regulator in the brain
To determine whether microglia contain specific metabolic determinants, we analysed an RNA-sequencing (RNA-seq) dataset of different brain cell types (data are accessible at the NCBI Gene Expression Omnibus (GEO) database under accession GSE52564)30. We focused on metabolic genes (1,118 genes according to the KEGG database; Supplementary Table 1) and revealed distinct gene clusters enriched in astrocytes, microglia and endothelial cells using gene correlation network analysis (Extended Data Fig. 1a). Principal-component analysis (PCA) demonstrated a clear separation of microglial metabolic gene clusters, particularly in glucose metabolism, from other cell types (Fig. 1a and Extended Data Fig. 1b). Differential expression analysis between different brain cell types revealed 25 differentially expressed genes (DEGs) between microglia and other brain cells (astrocytes, neurons, oligodendrocyte progenitor cells and myelinating oligodendrocytes), with 17 genes highly enriched in microglia (Fig. 1b,c and Supplementary Table 1). Notably, HK2 and HK3, two isozymes of HK that catalyse the first step of glucose metabolism, are preferentially enriched in both mouse and human microglia (Fig. 1c,d and Extended Data Fig. 1c–f; data retrieved from GSE52564 and GSE89960)30,31. Western blot analysis demonstrated that HK2 is specifically expressed in microglia, whereas HK1 is predominantly expressed in astrocytes and neurons (Fig. 1e), consistent with the abundance of HK1 in the brain. Single-molecule fluorescence in situ hybridization (FISH) analysis on brain slices showed that Hk2 transcripts are restricted in Iba1+ microglia (Fig. 1f, g and Extended Data Fig. 2a).

a, PCA of genes in metabolic pathways from different cell types of the brain. b, Venn diagram of differentially expressed metabolic genes between microglia compared to astrocytes, neuron, OPCs and MOs. c, Heat map of 25 differentially expressed metabolic genes between microglia and other cell types including astrocytes, neurons, OPCs and MOs. d, Schematic of glycolysis highlighting the three rate-limiting reactions catalysed by HK, PFK and PK. e, Representative western blot analysis (from n = 3 independent experiments) of HK1, HK2 and actin in lysates from primary microglia, astrocytes and neurons. f,g, Single-molecule FISH of Hk2 and Iba1 staining in mouse brain slices (f) and quantification (g). White arrowheads point to Hk2+ and Iba1+ microglia. Dashed boxes show regions of higher magnification. n = 4 and 3 mice in microglia and other groups, respectively. h, Schematic of Hk2CreERT2-tdTomato mice. i, Representative micrographs of tdT and Iba1 (microglia) in the brains of Hk2CreERT2-tdTomato mice. White arrowheads point to tdT+ Iba1+ microglia. j, Quantification of tdT distribution in microglia, neurons, oligodendrocytes and astrocytes in the central nervous system. n = 3 mice per group. k, Schematic showing the expression of different HK isozymes in microglia, neurons and astrocytes. l, Representative micrographs of tdT and Iba1 (microglia) in the cortex, midbrain and cerebellum from Hk2CreERT2-tdTomato mice. m, Quantification of mean fluorescence intensity (MFI) of microglial tdT in the cortex, midbrain and cerebellum from Hk2CreERT2-tdTomato mice by flow cytometry analysis. n = 3 mice per group. MG, microglia; ECs, endothelial cells; OPCs, oligodendrocyte progenitor cells; Neu, neurons; MOs, myelinating oligodendrocytes; NFOs, newly formed oligodendrocytes; Ast, astrocytes; PFK, phosphofructokinase; PK, pyruvate kinase; F-1, 6-BP, fructose-l, 6-bisphosphate; PEP, phosphoenolpyruvate; G6P, glucose-6-phosphate. pMG, primary microglia; pAst, primary astrocytes; pNeu, primary neurons; tdT, tdTomato; UTR, untranslated region. Data are the means ± s.e.m. One-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc tests for m, *P < 0.05.
To further validate the selective distribution of HK2 in microglia, we generated a transgenic mouse line, in which a P2ACreER-P2A-tdTomato (tdT) cassette, under the control of the endogenous Hk2 promoter, was knocked in upstream of the Hk2 stop codon (Fig. 1h). This resulted in expression of tdT fluorescent protein in Hk2-expressing cells, and Cre activity after tamoxifen injection (Extended Data Fig. 3f). FISH and immunofluorescence analysis verified that tdT fluorescence is specifically present in Hk2+ and Iba1+ microglia with ramified processes and tdT expression enriched in CD11b+CD45lo microglia in the brain (Fig. 1i and Extended Data Fig. 3a–d). Conversely, non-Iba1+ cells in the brain, including NeuN-expressing neurons, Sox10-expressing oligodendrocytes and glial fibrillary acidic protein-expressing astrocytes were negative for tdT (Fig. 1j and Extended Data Fig. 3e), suggesting that microglia in the brain are selectively labelled in the Hk2CreERT2-tdTomato mice. Notably, intensities of microglial tdT fluorescence and western blot analysis of microglial HK2 in the cortex, midbrain and cerebellum remained the same, reflecting uniform expression levels of microglial HK2 in different brain regions (Fig. 1l,m and Extended Data Fig. 4a–c). Together, these data suggest that microglia express a distinct set of metabolic regulators and that HK2, a key enzyme driving the first step of glucose metabolism, is specifically expressed in microglia (Fig. 1k).
Hexokinase 2 gates glucose metabolism in microglia
To determine the function of HK2 in microglial metabolism, we selectively deleted Hk2 in microglia by tamoxifen injection into postnatal Cx3cr1CreERT2/+::Hk2fl/fl (Hk2-conditional knockout (cKO)) mice followed by primary microglial culture (schematically shown in Fig. 2a). Quantitative PCR with reverse transcription (RT–qPCR) analysis demonstrated that Hk2 was reduced by more than 95% in microglia derived from Hk2-cKO mice compared to the controls (Fig. 2b).

a, Schematic of the experimental timeline for the analysis of microglia cultures. b, RT–qRCR analysis of Hk2 in primary microglia from Hk2fl/fl and Hk2-cKO mice. n = 3 or 5 independent cultures in Hk2fl/fl or Hk2-cKO groups, respectively. c, Schematic of the Seahorse XF Glycolysis Stress Test. d,e, ECAR measurement of primary microglia from Hk2fl/fl (untreated or pretreated with 2-DG) and Hk2-cKO mice. n = 6, 4 or 5 independent cultures in Hk2fl/fl, 2-DG or Hk2-cKO groups, respectively. f,g, ECAR measurement of human microglia (untreated or pretreated with 2-DG or 3-BrPA). n = 5, 7 or 6 independent cultures in PBS, 2-DG or 3-BrPA groups, respectively. h, Schematic of the Seahorse XF Mito-stress test. i,j, OCR measurement of primary microglia from Hk2fl/fl (untreated or pretreated with 2-DG) and Hk2-cKO mice. n = 5, 7 or 6 independent cultures in Hk2fl/fl, 2-DG or Hk2-cKO groups, respectively. k,l, OCR measurement of human microglia (untreated or pretreated with 2-DG or 3-BrPA). n = 10, 9 or 5 independent cultures in PBS, 2-DG or 3-BrPA groups, respectively. m, Schematic of the Seahorse XF Mito fuel flex test. n, Dependency of primary microglia on glucose, glutamine and fatty acid oxidation. n = 7, 9 or 8 independent cultures for glucose, glutamine or fatty acid groups, respectively. o–q, Dependency on glucose (o), glutamine (p) and fatty acid (q) oxidation of primary microglia from Hk2fl/fl and Hk2-cKO mice. n = 7, 10 or 10 (o and p) independent cultures, 6, 9 or 9 (q) independent cultures in Hk2fl/fl, 2-DG group or Hk2-cKO group, respectively. r,s, Lactate (r) and ATP production (s) in Hk2fl/fl and Hk2-cKO microglia. n = 5 or 6 (r), 7 or 5 (s) independent cultures in Hk2fl/fl or Hk2-cKO groups, respectively. TAM, tamoxifen; TCA, tricarboxylic acid; Glc, glucose; Olig, oligomycin; FCCP, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; Antim, antimycin A; Rot, rotenone; αKG, α-ketoglutarate. Data are presented as means ± s.e.m. Two-tailed Student’s t-tests for b, r and s. One-way ANOVA followed by Bonferroni’s post hoc tests for e, g, j, i and n–q, *P < 0.05, **P < 0.01 and ***P < 0.001.
We analysed the metabolic states of microglia using Metabolic Flux assays. In the glycolytic stress assays, Hk2-deficient mouse primary microglia, along with cells treated with 2-deoxy-d-glucose (2-DG) and 3-bromopyruvate (3-BrPA), both HK inhibitors, exhibited reduced basal and maximal extracellular acidification rates (ECARs; Fig. 2c–e and Extended Data Fig. 5a,b). Furthermore, 2-DG and 3-BrPA also reduced maximal ECAR in human microglia, indicating a block in glycolytic influx in the inhibition/absence of HK2 (Fig. 2f,g). Notably, primary microglia did not show a change in oxygen consumption rate (OCR) in regular Mito-Stress assays, in which OCR was measured in the medium containing glucose, pyruvate and glutamine (Extended Data Fig. 5e–g). However, removal of pyruvate, a direct product of glycolysis that may compensate for HK2 deficiency, reduced basal and maximal OCR in Hk2-deficient and 2-DG/3-BrPA-treated microglia (Fig. 2h–j and Extended Data Fig. 5c,d). Human microglia also reduced maximal OCR after pharmacological inhibition of HK2 by 2-DG and 3-BrPA treatments (Fig. 2k,l). These data suggest that HK2 deletion reduces glycolysis-derived oxidative respiration in both mouse and human microglia.
We also used the Mito Fuel Flex Test (schematically shown in Fig. 2m) to determine if microglial dependency on different fuels, such as glucose, glutamine and fatty acids, changes in the absence of HK2. Under basal conditions, wild-type (WT) microglia exhibited similar mitochondrial oxidation of glucose, glutamine and fatty acids (Fig. 2n). Hk2-deficient or 2-DG-treated microglia dramatically reduced their dependency on glucose oxidation, likely due to the reduced glycolytic flux resulting from HK2 deletion/inhibition. Interestingly, HK2 deletion or inhibition did not affect microglial dependency on oxidation of glutamine or fatty acids, indicating that HK2 ablation suppressed microglial glycolysis with no apparent compensation of glutamine or fatty acid oxidation (Fig. 2o–q). Consistent with these data, levels of cellular lactate and ATP were significantly reduced in Hk2-deficient microglia, reflecting a reduced glycolytic and bioenergetic flow (Fig. 2r,s). Collectively, these data suggest that HK2 plays a key role in regulating glucose metabolic flux in microglia, and ablation of HK2 leads to an energy-deficient state in microglia.
Hexokinase 2 regulates microglial dynamics
Microglia continuously monitor the microenvironment and rapidly respond to damage in the brain parenchyma, both of which are highly energy-demanding processes. Because HK catalyses the first step of glucose metabolism and is important for energy production19, we tested if HK2 regulates microglial dynamics, including basal motility and damage-sensing migration. We selectively ablated HK2 in microglia by administering tamoxifen to adult Cx3cr1CreERT2/+::Hk2fl/fl mice (8 weeks) and sorted microglia (CD11b+CD45lo) from mouse brains by fluorescence-activated cell sorting (FACS) 4 weeks later (Fig. 3a). HK2 reduction was verified by RT–qPCR (Fig. 3b) and western blot analysis (Fig. 3c). To allow real-time tracing of microglial motility in vivo, we crossed these mice with Ai14 mice to generate Cx3cr1CreERT2/+::Ai14fl/+ (control) and Cx3cr1CreERT2/+::Hk2fl/fl::Ai14fl/+(Hk2-cKO) mice, in which microglia are labelled by tdT after tamoxifen induction. By two-photon microscopy, we tracked the movement of microglial processes in the primary somatosensory cortex (Fig. 3d). At basal states, we observed reduced microglial motility (control, 0.794 ± 0.096; Hk2-cKO, 0.642 ± 0.074; n = 5 mice per group, P < 0.05) and process velocity in Hk2-cKO mice, compared to controls (control, 0.580 ± 0.065 μm min−1; Hk2-cKO, 0.469 ± 0.063 μm min−1; n = 5 mice per group, P < 0.05; Fig. 3e–i). Following laser-induced microlesion (1 μm in diameter), control microglia surrounding the lesion site quickly extended their processes towards the injury, and by 30 min, their processes had reached the damaged spot and converged to form a narrow spherical containment. In contrast, Hk2-deficient microglia showed significantly slower responses to the injury, and their processes were still approximately 18 μm away from the damage centre within the same 30-min time window (Fig. 3j–m). These data suggest that HK2 ablation impairs the basal motility and directional migration of microglia towards the site of injury.

a, Schematic of the experimental timeline for two-photon imaging. b, RT–qRCR analysis of Hk2 mRNA in FACS-sorted microglia from adult Hk2fl/fl and Hk2-cKO mice. n = 3 or 4 mice in Hk2fl/fl or Hk2-cKO groups, respectively. c, Representative western blot analysis (from n = 2 independent experiments) of HK2 and actin in lysates from FACS-sorted microglia from Hk2fl/fl and Hk2-cKO mice. d, Schematic of in vivo imaging of microglia. e, Representative micrographs obtained at different time points by two-photon imaging. f–i, Quantification of extension index, retraction index, motility index and mean process velocity in microglia from Cx3cr1CreERT2/+::Ai14fl/+ and Hk2-cKO::Ai14fl/+ mice. n = 5 mice per group (68 cells in total). j, Representative micrographs of damage-sensing responses of microglia at indicated time points after a focal laser-induced lesion through a cranial window. Asterisks indicate the sites of laser ablation. k, Quantification of the number of microglial processes entering into the inner area (35 mm in radius) over 30 min. l,m, Quantification of microglial response measured at the 30-min time point after laser ablation (l) and the total areas under curve analysis (m). n = 5 mice per group. TAM, tamoxifen; D, day. Data are the means ± s.e.m. Two-tailed Student’s t-tests for b, f–i and l, *P < 0.05, **P < 0.01. Mann–Whitney U test for m, *P < 0.05, **P < 0.01.
Hexokinase 2 deficiency delays microglia repopulation
We next tested if HK2 is required for microglia homeostasis under regular conditions. We examined the density and morphology of microglia at 1 and 4 weeks after Hk2 ablation in adult mice, but did not observe substantial differences (Fig. 4b and Extended Data Fig. 6a–l). RNA-seq and qPCR analyses confirmed Hk2 knockdown in microglia with no compensatory elevation of Hk1 and Hk3 in the brain tissue, microglia or elevation of Hk2 in non-microglial cells (Extended Data Fig. 7a–g), suggesting that HK2 is dispensable for steady-state microglia homeostasis.

a, Schematic of the experimental timeline for the analysis of microglial repopulation from Hk2fl/fl and Hk2-cKO mice. b, Representative micrographs (from n = 3 independent experiments) of Iba1+ microglia in adult Hk2fl/fl and Hk2-cKO mouse brains 4 weeks after tamoxifen administration. c, Representative micrographs showing the total number of microglia and proliferative (BrdU+) microglia at indicated time points after removal of PLX5622. White arrowheads point to BrdU+ Iba1+ microglia. d,e, Quantification of microglial number (d) and percentage of BrdU+ microglia (e) from Hk2fl/fl and Hk2-cKO mice. n = 3 or 3 mice at D0, 4 or 4 mice at D1, 3 or 5 mice at D3, 3 or 4 mice at D5, 5 or 5 mice at D7 in Hk2fl/fl or Hk2-cKO groups, respectively. f, Schematic of the experimental design for microglia (CD11b+CD45lo cells) sorting and scRNA-seq analyses. g,h, t-distributed stochastic neighbor embedding (t-SNE) profiles of individual microglia from Hk2fl/fl and Hk2-cKO mice by unsupervised clustering. Cells were coloured by either sample groups (g) or annotated cell types (h). i, Selected markers used for microglia subcluster annotation are presented. j,k, A pseudotime trajectory of microglia on the minimum spanning tree coloured by subtypes (j) or sample (k) using Monocle 2. l, Percentage of different cell subsets in Hk2fl/fl and Hk2-cKO microglia. m, Expression pattern of selected DEGs in different microglia subclusters between Hk2fl/fl and Hk2-cKO mice. n = 1,481 or 264 cells for HomoMG, 162 or 264 cells for Precursor MG, 121 or 91 cells for G1/S MG, 200 or 108 cells for G2/M MG in Hk2fl/fl or Hk2-cKO groups, respectively. PLX5622, PLX5622-formulated diet; TAM, tamoxifen; D, day; homoMG, homeostatic microglia; G1/S MG, microglia at G1 and/or S phase of the cell cycle; G2/M MG, microglia at G2 and/or M phase of the cell cycle; precursor MG, precursor microglia; MФ, macrophage; Mono, monocyte; NPs, neutrophils. Data are the means ± s.e.m. Two-tailed Student’s t-tests for d and e. Wilcoxon rank-sum test and Bonferroni correction for m, *P < 0.05, **P < 0.01 and ***P < 0.001.
Microglia maintain a relatively low amount of self-renewal in adulthood but re-enter cell cycle and quickly proliferate after acute depletion or during diseases12,13,14,15. Inhibition of colony-stimulating factor 1 receptor (CSF1R) induces acute depletion of microglia, which rapidly repopulate and restore normal density within 1 week when inhibition is released13,14. To test if HK2 is involved in microglial repopulation, we fed WT and Hk2-cKO mice chow containing PLX5622, a selective inhibitor of CSF1R for 2 weeks, followed by normal chow (control diet, CD) to allow repopulation of microglia (Fig. 4a). One day after CD feeding (the repopulation phase), the density of microglia in WT was approximately 12 cells per mm2. By day 3, it reached nearly 135 cells per mm2, with approximately 48% of microglia being proliferative (5-bromo-2′-deoxyuridine positive, BrdU+). In contrast, the number of microglia in Hk2-cKO mice was significantly reduced at both day 1 and day 3 of the repopulation phase (Fig. 4c–e). At day 3, only 29.2% of Hk2-deficient microglia were proliferative and the total number of microglia was approximately half that of WT, suggesting an inhibition in microglia repopulation after HK2 removal. By day 5, the number of proliferating microglia in WT mice normalized back to a minimal level (1.7%), whereas 27.3% of Hk2-deficient microglia remained proliferative (Fig. 4c,e). This allowed the Hk2-deficient microglia to eventually recover to the regular number, albeit at a later stage.
To define the potential molecular mechanisms underlying the differences between WT and Hk2-cKO microglia during repopulation, we used single-cell RNA sequencing (scRNA-seq) to analyse the transcriptomes of repopulated microglia at day 3 (Fig. 4f). We clustered microglia into different subpopulations, including homeostatic, precursor and proliferating microglia according to the expression of different marker genes (Fig. 4g–i). All microglial clusters showed prominent knockdown of Hk2 (Fig. 4m and Extended Data Fig. 7c). Notably, precursor microglia, which sit at the root of a pseudotime trajectory, contained substantially more Hk2-deficient cells (Fig. 4j–l and Extended Data Fig. 8a–c). Analyses of DEGs in different subpopulations (Supplementary Table 2) uncovered significant upregulation of Cdkn1a (encoding p21), a cell cycle arrest inducer32, and Slfn5, an inhibitor of DNA replication33, along with downregulation of Cx3cr1, a microglia homeostatic marker11, in Hk2-deficient microglia (Fig. 4m and Extended Data Fig. 8d). These data suggest that absence of HK2 induced cell cycle arrest and impeded microglial proliferation and maturation during repopulation.
Hexokinase 2 is upregulated in activated microglia
HK elevation has been associated with increased glycolysis in tumour cells and activated immune cells23,34,35,36. To test how microglial states may affect HK2 levels, we analysed RNA-seq data from different sources (accessible at the NCBI GEO) database, accessions are listed in Supplementary Table 3) and found that Hk2 was markedly elevated in mouse microglia after lipopolysaccharide (LPS) stimulation, in the aging brain (12-month-old mice), or from AD and Parkinson’s disease (PD) models, as well as a subset of human microglia in coronavirus disease (COVID)-infected brains and brain tumours (Fig. 5a, b).

a, Analysis of Hk2 expression in microglia from public bulk RNA-seq data. n = 3 (PBS) or 3 (LPS) samples for in vitro LPS model, 4 (PBS) or 5 (LPS) samples for in vivo LPS model, 11 (2 month) or 10 (1 year) samples for aging model, 8 (WT) or 8 (AD) samples for 4-month AD model, 7 (WT) or 6 (AD) samples for 6-month AD model, 8 (WT) or 7 (AD) samples for 8-month AD model. b, Analysis of Hk2 expression in microglia from public single cell RNA-seq data. n = 15,414 (homoMG) or 309 cells (DAM) for AD model, 14,236 (VV-GFP) or 13,316 cells (AVV-SYN) for PD model, 1,036 (Ctrl) or 1,207 cells (COVID) for COVID-infected model, 510 (periphery) or 537 cells (tumour) for GBM model. c, Representative micrographs (from n = 3 independent experiments) showing HK2 in Iba1+ microglia surrounding the Aβ plaques in the hippocampi of hAPP-J20 mice (AD models). Dashed boxes show regions of higher magnification. d, Quantification of HK2 fluorescence in DAM in the AD models. n = 3 mice per group. e, Representative micrographs (from n = 3 independent experiments) showing HK2 in Iba1+ microglia in the SNc in the PD models. Dashed boxes show regions of higher magnification. f, Quantification of HK2 protein fluorescence in DAM in the PD models. n = 3 mice per group. g, Representative micrographs (from n = 3 independent experiments) showing HK2 in Iba1+ microglia in glioma. Dashed boxes show regions of higher magnification. Dashed lines show regions of glioma in brain. h, Representative micrographs (from n = 3 independent experiments) showing tdT-labelled HK2 in Iba1+ microglia after saline or LPC injection in CC by Hk2CreERT2-tdTomato mice. Dashed lines indicate CC. HomoMG, homeostatic microglia; GBM, glioblastoma; TAM, tamoxifen; CC, corpus callosum. Data are the means ± s.e.m. Two-tailed Student’s t-tests for a, d and f, Wilcoxon rank-sum test and Bonferroni correction for b, *P < 0.05, **P < 0.01 and ***P < 0.001.
By immunofluorescence analyses, we further show that HK2 is selectively elevated in microglia surrounding Aβ plaques in hAPP-J20 mice (a mouse model of AD), also referred to as DAM, and those activated in the substantia nigra pars compacta (SNc) in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD models (Fig. 5c–f). Using a spontaneous glioma mouse model (Ng2CreERT2-tdTomato::Trp53fl/+::Nf1fl/fl), in which tumour suppressor genes (TSGs) p53 and neurofibromatosis 1 (Nf1) are specifically inactivated in adult oligodendrocyte progenitor cells, we observed that HK2 was significantly upregulated in activated microglia in the glioma (Fig. 5g). Likewise, tdT fluorescence, reflective of HK2 expression, was also increased in the callosal microglia of lysophosphatidyl choline (LPC)-induced demyelination models in Hk2CreERT2-tdTomato mice (Fig. 5h). Together, these data suggest that HK2 is sensitive to microglial status and upregulation of HK2 is a common feature of activated microglia, likely contributing to the enhanced glycolysis. Our data also indicate that Hk2CreERT2-tdTomato mice might be an excellent tool for dynamically assessing glycolytic and bioenergetics responses of microglia in vivo, when combined with real-time imaging.
Hexokinase 2 deletion potentiates brain damage and behavioural deficits in stroke models
Pro-inflammatory microglia switch oxidative respiration to glycolysis to promote inflammation2,3. Because HK2 is prominently elevated in activated microglia and HK2 ablation impairs glycolysis, we hypothesize that HK2 boosts inflammation by promoting glycolysis and HK2 ablation likely reduces inflammation by reducing microgliosis and inflammation in diseases. As stroke involves rapid microglial activation, migration and proliferation at the site of infarction37, we chose a photothrombotic stroke model for further analyses. First, using the HK2-tdT reporter line (schematically shown in Fig. 6a), we verified elevated tdT fluorescence levels in individual microglia in the peri-infarct zone, from day 1 to day 7 after stroke (Fig. 6b,c), reflecting rapid upregulation of HK2 in activated microglia after stroke.

a, Schematic of photothrombotic stroke. b, Representative micrographs showing tdT expression at indicated time points after stroke in Hk2CreERT2-tdTomato mice. Dashed lines indicate the border of ischaemic core. c, Quantification of tdT fluorescence in individual microglia in the peri-infarct zone relative to the contralateral area at indicated time points. n = 3 mice per group. d, Representative micrographs of microglia (Iba1+) and proliferative microglia (Iba1+BrdU+) in the peri-infarct zone at 3 and 7 Dpi. White arrowheads point to BrdU+ Iba1+ microglia. e,f, Quantification of microglial number (e) and percentage of BrdU+ microglia (f) in the peri-infarct zone at indicated days after stroke. n = 4 or 3 mice at 1 Dpi, 3 mice per group at 3, 7, 14 and 21 Dpi in Hk2fl/fl or Hk2-cKO groups, respectively. g,h, Representative micrographs showing the infarct size (g) and quantification (h) from Hk2fl/fl and Hk2-cKO mice at 7 Dpi. Area within the dashed lines indicate the ischaemic core. n = 7 or 6 mice in Hk2fl/fl or Hk2-cKO groups, respectively. i,j, Behavioural analyses of Hk2fl/fl and Hk2-cKO mice by cylinder (i) and grid walk (j) tests. n = 10 or 8 mice in Hk2fl/fl or Hk2-cKO groups, respectively. k, RT–qPCR analysis of Tnf, Il6 and Il1b levels in the infarcted tissues at 3 Dpi. n = 3 mice per group for sham-operated animals, 4 mice per group for ischaemic animals. l, RT–qPCR analysis of Ifitm3 and Stat1 levels in the infarcted tissues at 3 Dpi. n = 5 mice per group for sham-operated animals, 7 or 6 mice for ischaemic animals in Hk2fl/fl or Hk2-cKO groups, respectively. BL, baseline. Data are the means ± s.e.m. Two-tailed Student’s t-tests for c, e, f and h. Two-way ANOVA followed by Bonferroni’s post hoc tests for i–l, *P < 0.05, **P < 0.01, ***P < 0.001.
Next, we selectively ablated Hk2 in microglia by tamoxifen administration 4 weeks before the photothrombotic model, and observed significantly attenuated density of microglia within the peri-infarct zone at 3 and 7 d post-ischaemia (Dpi; 560 versus 314 cells per mm2 at 3 Dpi; 958 versus 746 cells per mm2 at 7 Dpi between WT and Hk2-cKO animals, n = 3–4 mice per group, P < 0.01), along with reduced BrdU labelling (42.5% versus 29.7% at 3 Dpi; 15.4% versus 11.8% at 7 Dpi between WT and Hk2-cKO animals, n = 3–4 mice per group, P < 0.05; Fig. 6d–f and Extended Data Fig. 9a). However, HK2 ablation significantly increased infarct volume (3.7 versus 5.0 mm3 between WT and Hk2-cKO animals, n = 5–7 mice per group, P < 0.01; Fig. 6g, h), suggesting that HK2 ablation may have aggravated the damage.
To further examine whether HK2 deletion affects functional recovery following ischaemia, we measured motor control of the mice by cylinder and grid walk tests38. Under basal states, WT and Hk2-cKO mice showed no significant differences in motor activities (Extended Data Fig. 9b–e). However, following stroke, Hk2-cKO mice exhibited more severe behavioural deficits with increased forelimb bias and foot faults in the cylinder and grid walk tests (Fig. 6i,j). Measurements of inflammatory cytokines in ischaemic tissues detected increased levels of tumour necrosis factor (TNF), interleukin (IL)-6 and IL-1β, along with enhanced type 1 interferon response molecules, including Ifitm3 and Stat1, in Hk2-cKO mice, suggesting that HK2 ablation also enhanced neuroinflammation (Fig. 6k,l and Extended Data Fig. 9i,j). Additionally, Hk2-cKO microglia contained a significantly increased volume of CD68+ puncta with increased cell volume at 3 d after stroke, indicating that ablation of microglial HK2 promoted microglial activation and phagocytic activity (Extended Data Fig. 9f–h).
Hexokinase 2 deletion affects mitochondrial function and promotes inflammatory responses
Reduced glycolysis is usually associated with alleviated inflammation21,22,39. Because HK2 is elevated in DAM and its ablation promotes rather than suppresses inflammation in stroke models, we hypothesized that HK2 may act as a critical metabolic checkpoint in microglia. Changes in HK2, ablation or elevation, in disease/challenged states may thus disrupt metabolic homeostasis to drive aberrant inflammatory responses. To test this hypothesis, we first verified that HK2 is prominently induced in LPS-primed pro-inflammatory microglia (Fig. 7a–d). Next, to examine how HK2 ablation affects the inflammatory responses under immune-challenged states, we treated WT and Hk2-deficient microglia with LPS and poly(I:C) to mimic bacterial or viral infections. As expected, LPS and poly(I:C) induced pro-inflammatory and interferon responses in WT microglia (Fig. 7e–h). However, Hk2-deficient microglia demonstrated more robust inflammatory responses than WT controls, with much higher levels of TNF, IL-6 or interferon responses after LPS or after poly(I:C) stimulations, (Fig. 7e–h). Moreover, Hk2-deficient microglia exhibited higher rates of microparticle uptake (Fig. 7i,j), reflective of enhanced phagocytosis.

a, A experimental schematic of primary microglia cultures. b–d, RT–qPCR analysis (b) and western blot analysis (c and d) of HK2 in primary microglia after LPS stimulation (100 ng ml−1) at indicated time points. n = 4 (b) or 7 (d) independent cultures per group. e, Schematic of the experimental timeline for analysis of primary microglia cultures. f–h, ELISA analysis of IL-6 (f), TNF (g) and IFN-β (h) levels in supernatants of primary microglia from Hk2fl/fl and Hk2-cKO mice before and after LPS (100 ng ml−1) or poly(I:C) (25 μg ml−1) stimulation. n = 3 or 5 (f and g), 4 or 4 (h) independent cultures for each treatment in Hk2fl/fl or Hk2-cKO groups, respectively. i,j, Analysis of phagocytic activity in primary microglia from Hk2fl/fl and Hk2-cKO mice. n = 8 or 7 independent cultures in Hk2fl/fl or Hk2-cKO groups, respectively. k,l, Analysis of MitoSOX stained cells for the detection of mitochondrial ROS in primary microglia from Hk2fl/fl and Hk2-cKO mice. n = 6 or 7 independent cultures in Hk2fl/fl or Hk2-cKO groups, respectively. m,n, Analysis of JC-10 stained cells for the detection of mitochondrial membrane potential in primary microglia from Hk2fl/fl and Hk2-cKO mice. n = 4 or 5 independent cultures in Hk2fl/fl or Hk2-cKO groups, respectively. o, Schematic of the experimental timeline for analysis of microglial inflammatory responses using scavenger of mitochondrial ROS. p–r, ELISA analyses of IL-6 (p), TNF (q) and IFN-β (r) levels in the supernatants of microglia from Hk2fl/fl and Hk2-cKO mice after LPS (10 ng ml−1) or poly(I:C) (25 μg ml−1) stimulation in the presence or absence of Mito-TEMPO. n = 7 or 7 (p and q), 4 or 5 (r) for each treatment in Hk2fl/fl or Hk2-cKO groups, respectively. TAM, tamoxifen. Data are the means ± s.e.m. Two-tailed Student’s t-tests for b, j, l and n. One-way ANOVA followed by Bonferroni’s post hoc tests for d. Two-way ANOVA followed by Bonferroni’s post hoc tests for f–h and p–r, *P < 0.05, **P < 0.01, ***P < 0.001.
HK2 localizes to the OMM by interacting with the voltage-dependent anion channel20,40 and mitochondria are a critical power plant for inflammasomal activation28. We hypothesized that HK2 ablation likely impaired mitochondrial function to enhance inflammation in microglia. By staining with JC-10 followed by flow cytometry analyses, we observed decreased mitochondrial membrane potential after ablation of HK2 (Fig. 7m,n). Moreover, measurement of mitochondrial ROS (MitoROS) production also revealed increased MitoROS in Hk2-cKO microglia, indicating mitochondrial function is altered after HK2 ablation (Fig. 7k,l). Moreover, MitoROS levels were further potentiated, along with decreased mitochondrial membrane potential in Hk2-deficient microglia after LPS and poly(I:C) treatments (Extended Data Fig. 10a,b). Importantly, Mito-TEMPO, a mitochondria-targeted antioxidant and a scavenger of MitoROS, attenuated LPS/poly(I:C) induction of MitoROS (Fig. 7o and Extended Data Fig. 10c,d) and decreased IL-6 and interferon (IFN)-β production (Fig. 7p–r). These data suggest that HK2 deletion not only affects glycolysis but also induces mitochondrial dysfunction, which likely serves as activating signals to promote inflammation.
Discussion
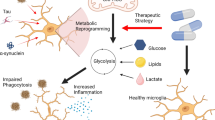
In the present study, we found that microglia specifically express HK2, a mitochondria-associated HK isoenzyme with maximal catalytic activity. HK2 ablation reduces microglial glycolysis, resulting in an energy-deficient state and impaired surveillance and damage-triggered chemotaxis. HK2 is dispensable for steady-state microglia homeostasis but is required for timely microglial repopulation after acute microglia depletion. HK2 is prominently elevated in pro-inflammatory microglia or DAM. In a model of ischaemic stroke, HK2 deficiency enhanced inflammatory responses and aggravated cerebral injury and behavioural deficits. Further analyses revealed that HK2 ablation impaired mitochondrial function and increased ROS production, thereby contributing to enhanced inflammation under immunological challenges (Fig. 8). These data demonstrate that HK2, serves as a critical metabolic checkpoint in microglia by gating both glycolytic flux and mitochondrial activity to tightly control microglial function.

Microglia specifically express HK2, whereas neurons and astrocytes predominantly express HK1. Ablation of HK2 reduces microglial glycolysis, impairs microglial surveillance and damage-triggered chemotaxis, as well as delays microglial repopulation. In response to immune challenges, HK2 deficiency boosts inflammation in microglia via dysfunctional mitochondria and accumulated ROS, resulting in aggravated cerebral injury and behavioural deficits in an ischaemic stroke model. By gating glycolytic flux and mitochondrial function, HK2 plays a dual role in shaping microglial function under different conditions.
Hexokinase 2 as a unique glycolytic gatekeeper in microglia
Acting as the first line of defence in the brain, microglia are endowed with the ability to respond to environmental stimuli and assume diverse functional states. Such characteristics demand a rapid but flexible bioenergetic supply. Similarly to other brain cells, microglia express a full set of metabolic enzymes required for glycolytic and oxidative metabolism2,41. However, HK isozymes, catalysing the first rate-limiting step in glucose metabolism, exhibited distinct distribution in different brain cell types. Different cellular distribution of HK isozymes offers the flexibility and benefit for brain cells to fully coordinate glucose utilization and to ensure normal brain function. By selecting HK2, the only isozyme containing two catalytic domains with maximal catalytic activity and readily regulatability18, microglia thus possess the strongest capacity to promote glucose metabolism in the brain. In accordance with our findings, two independent studies using positron emission tomography or fluorescence lifetime imaging have demonstrated that microglia take up and metabolize more glucose than astrocytes and neurons42,43. Therefore, HK2 is perfectly suited to function as the glycolytic gatekeeper that confers microglia the ability to rapidly tune glucose metabolism to different stimuli. It should be noted that HK2 has been detected in neurons and astrocytes in vitro or under some pathological conditions44,45,46, which might be related to its highly regulatable feature at transcriptional levels.
Microglia adopt a highly glycolytic profile in the healthy brain43. We showed that ablation of HK2 dampens routine microglial dynamics and injury-induced directed chemotaxis of microglia. In support of our data, others also observed reduced microglial processes motility and chemotaxis to the injury site, following oxygen glucose deprivation or systematic blockade of HK and glycolysis4,47. Importantly, ablation of HK2 does not affect microglial steady-state homeostasis, likely owing to the low self-renewal rate of homeostatic microglia in the adult brain with minimal demands for metabolic precursors in the anabolic pathway8,9. These data indicate that HK2-driven glycolytic flux preferentially flows towards energy production to support proper microglial surveillance to maintain brain homeostasis. Under pathological conditions, for example, microgliosis after ischaemia or repopulation after acute microglia depletion, when metabolic demands increase, HK2 ablation suppressed microglial proliferation and repopulation. Therefore, HK2 in microglia enables the potential for rapid proliferation and replenishment, in a similar manner to the reliance of cancer cells on HK2 to promote tumour growth and proliferation48.
Neurological disorders, including AD, PD and traumatic brain injury, are often accompanied by metabolic abnormalities in the brain49,50,51. Earlier studies suggested neurons as the culprits for altered metabolic profiles52. However, a recent study showed that depletion of microglia robustly decreased the glucose uptake in AD models, suggesting that microglia are the main contributors to the increased glucose uptake in AD mice42. Prominent elevation of HK2 in DAM, by promoting glycolytic flux, may help microglia meet elevated demands for energy and precursor production in driving both microgliosis and inflammation in diseases. In addition, a recent study by Leng et al.53 also showed that HK2 is elevated in microglia in both AD mouse models and patients, thus providing another angle on how HK2 is involved in the activation of microglia53. HK2 inhibition enhanced lipid metabolism to promote microglial phagocytosis, thereby facilitating the clearance of amyloid plaque and attenuating cognitive impairment in AD mice53.
Hexokinase 2, glucose metabolism and immune responses
In peripheral immune cells, upregulated HK activity and glycolysis have been associated with Toll-like receptor-induced inflammation21,22. As HK2 is significantly elevated in pro-inflammatory microglia and DAM, we thought that HK2 ablation, by reducing glycolytic flux, may alleviate inflammation and relieve disease pathology. However, we observed robustly enhanced inflammatory responses after HK2 ablation in LPS or poly(I:C)-treated microglia and potentiated brain damage in ischaemic stroke models. Moreover, by adeno-associated virus (AAV)-mediated knockdown or pharmacological blockade, Li et al.23 found that HK2 knockdown/inhibition protected brain damage in middle cerebral artery occlusion models23. Nonetheless, as microglia are refractory to viral infection54 and inhibitors suppress all HK isozymes and glucose metabolism in all brain cells, these data may be confounded. Using genetic tools to selectively delete microglial HK2 in an ischaemic stroke model, we observed deleterious brain damage, along with reduced microgliosis and enhanced inflammation. This is likely contributed by the reduced microglial migration and microgliosis, which leads to insufficient formation of the immune barrier to restrict the damage at the boundary of ischaemic injury55,56. An enlarged ischaemic core further deteriorates neuroinflammation in the damaged loci. Thus, the detrimental effects following HK2 ablation in the stroke models may arise from both the diminished microglial protection and enhanced inflammation.
While previous studies suggest increased glycolysis promotes inflammation, a recent study showed that energy deficiency in myeloid cells can drive maladaptive inflammation by increased succinate57. Other than MitoROS accumulation, HK2 deletion-resultant glycolytic inhibition may also lead to abnormal build-up of intermediate metabolites and promote inflammation in microglia.
Hexokinase 2, mitochondria and inflammation
Both HK1 and HK2 associate with the OMM via the voltage-dependent anion channel20,40. Inhibiting HK activity may induce conformational changes that promote HK dissociation from the voltage-dependent anion channel25,58, which increases mitochondrial permeability and decreases mitochondrial membrane potential25,59,60,61. Increased permeability of the OMM and opening of mPTP facilitates the release of cytochrome C, an important antioxidant in mitochondria, which may contribute to accumulated ROS in mitochondria62. Interestingly, Wolf et al.26 found that bacterial peptidoglycan binding of HK leads to its release from the OMM and triggers inflammasome activation in bone marrow-derived macrophages26. Moreover, a recent study found that HK1 dissociation from the mitochondria enhances LPS-induced inflammatory responses63 and our findings also support that HK2 absence promotes inflammation, partly through increased MitoROS. It should be noted, however, that conditional knockout of HK2 in intestinal epithelia appears to suppress cell death and dysregulate mitochondrial function to protect mice from colitis27. These counterintuitive results may reflect different functions of HK isozymes in different cellular contexts, which deserves further investigations. Our data show that HK2 plays a dual role in tightly controlling microglial functions, either by gating glycolysis to regulate microglial motility and proliferation, or by associating with the OMM to tune mitochondrial activity and immune responses under different states. Designing reagents that separately inhibit the kinase activity or the association of HK2 with the OMM may help selectively manipulate the function of HK2 in microglia with potential therapeutic usage in disease.
In summary, we show that HK2 is a genetically programmed metabolic signature in microglia. By utilizing HK2, microglia are able to achieve maximal glycolytic flux and adaptive capacity to instruct cellular metabolism and support diverse functions in response to different stimuli. Moreover, HK2 also plays a role in dynamically regulating mitochondrial function and fine-tuning microglial immune responses under different states. Our study reveals an important mechanism, by which microglia control intracellular metabolism and immune responses through HK2 to adapt to environmental stimuli. Our data also provide mechanistic insights into the altered metabolic patterns observed in multiple neurodegenerative diseases and may shed light on therapeutic strategies with potential metabolic interventions.
Methods
Animals
Cx3cr1CreERT2, Ai3 and Ai14 mice were purchased from the Jackson Laboratory. Hk2 floxed (Hk2fl/fl) mice and Hk2CreERT2-tdTomato mice were generated by Biocytogen Company (China). Hk2fl/fl mice were crossed with the Cx3cr1CreERT2/+mice to generate the Cx3cr1CreERT2/+::Hk2fl/fl animals. The hAPP-J20 mice, expressing human APP with the Swedish (p.Lys670Asn and p.Met671Leu) and Indiana (p.Val717Phe) mutations under the control of the platelet-derived growth factor beta-chain promoter64, were from the Mutant Mouse Resource and Research Center. The glioma mice were provided by C. Liu (Zhejiang University, China)65. All animals were housed under a 12-h light/dark cycle with food and water available. Animal experiments were conducted following the Guidelines for the Care and Use of Laboratory Animals of Zhejiang University.
Tamoxifen administration
For induction of the Cre recombinase in adult mice, two doses of tamoxifen citrate (TAM, 10 mg per mice, Meilunbio, dissolved in 0.9% sodium chloride solution) were given to the animals by oral gavage at 48-h intervals. For induction of Cre recombination in postnatal mice, 100 μg tamoxifen (Sigma) dissolved in corn oil was injected intraperitoneally for 2 consecutive days at postnatal days 1–2.
Primary culture of microglia and astrocytes
Cells were dissociated from the cortices, plated on flasks, and cultivated in minimum essential medium (MEM, Fisher Scientific) supplemented with 10% FBS (Fisher Scientific) and 1% penicillin–streptomycin (Fisher Scientific) at 37 °C. After 12–14 d of culture, mature microglia were separated from mixed glial cultures by quick and gentle shaking, resuspended in MEM supplemented with 3% FBS, and plated onto plates. The remaining mixed glial cells were passaged 2–3 times and shaken at 200 r.p.m. for 6 h. The supernatants were discarded and the remaining adherent cells were collected as astrocytes. Cells were allowed to settle down for 2–3 h for further experiments. For drug treatments, microglia were incubated with LPS or poly(I:C) (Sigma) for different time periods as indicated. To scavenge mitochondrial superoxide, 250 μM Mito-TEMPO (Sigma) was added to medium 1 h before LPS/poly(I:C) stimulation.
Primary neuronal culture
Hippocampi were dissected from mice at postnatal days 1–2, trypsinized and dissociated into single cells by gentle trituration. Cells were resuspended and cultured in DMEM (Fisher Scientific) containing 10% nutrient mixture F-12 (Fisher Scientific) and 10% FBS and 1% penicillin–streptomycin at 37 °C. After 12–14 d of culture, mature neurons were collected.
Measurements of lactate and ATP
Primary microglia were homogenized, and lactate and ATP were measured using lactate quantification colorimetric assay (BioVision) and ATP bioluminescent assay kit (Sigma). The contents of lactate and ATP were normalized by the total protein.
Metabolic extracellular flux analysis
Real-time changes in the ECAR and OCR of primary microglia were determined using the XF-96 Seahorse extracellular flux analyzer (Seahorse Bioscience) as described previously2. Briefly, microglia were plated onto XF-96 cell culture plates in the presence or absence of 2-DG (5 mM) or 3-BrPA (50 μM) for 3 h. Microglia were rinsed and analysed in the XF Running buffer A (XF assay medium containing 10 mM glucose, 1 mM pyruvate sodium and 2 mM glutamine) or XF Running buffer B (XF assay medium containing 10 mM glucose and 2 mM glutamine) for OCR measurements, or in the XF Running buffer C (XF assay medium, 2 mM glutamine) for ECAR measurements. After completion of the measurements, the number of cells within the plate was counted based on DAPI staining. Data were obtained as changes in pH and oxygen consumption (picomoles) per min in the medium and normalized to the cell number.
In the Mito Fuel Flex Test, the import of three major metabolic substrates, pyruvate, glutamine and fatty acids, was inhibited by adding UK5099 (inhibitor of mitochondrial pyruvate carrier), BPTES (inhibitor of glutaminase) and etomoxir (inhibitor of carnitine palmitoyl-transferase 1A), respectively, according to the manufacturer’s instructions. Dependency was calculated with the following formula: dependency (%) = ((baseline OCR − target 1 inhibitor OCR) / (baseline OCR − all 3 inhibitor OCR)) × 100. To compare the Mito fuel flex between microglia from Hk2fl/fl and Hk2-cKO mice, the fuel oxidation was measured in the XF Running buffer B without additional pyruvate.
Flow cytometry and microglial sorting
Mice were anaesthetized with sodium pentobarbital and perfused with ice-cold 0.9% sodium chloride solution. Brains were rapidly dissected and transferred to a pre-chilled dish with cold PBS on ice. The brains were minced and dissociated in MEM media containing 0.5 mg ml−1 collagenase IV (Worthington-Biochem) and 10 U ml−1 DNase I (Roche). Myelin and tissue debris were removed by centrifuging through a 37–70% (vol/vol) Percoll (GE Healthcare BioSciences) gradient density. Mononuclear cells containing microglia were collected and washed in PBS. Cells were then incubated with mouse Fc block (CD16/CD32, eBioscience) for 10 min on ice, washed in PBS, and stained with antibodies conjugated with fluorophores (CD11b-APC and CD45-PE, BioLegend) for 30 min on ice followed by wash and resuspension in FACS buffer (sterile-filtered PBS containing 2% FBS). Microglia were purified as the CD11b+CD45lo population on a BD FACSAria II using the 85-μm nozzle.
Magnetic-activated cell sorting of human microglia
Human microglia were acquired by magnetic-activated cell sorting as previously described66. Participants were informed regarding the experimental use of brain tissues and voluntarily provided consent. The radiologically healthy brain tissue samples were then derived from participants undergoing brain surgery for epilepsy. Sample collection and data analysis were approved by the Institutional board of the Second Hospital affiliated to Zhejiang University (Protocol 2021-0291) and carried out in compliance with national and provincial regulations.
The human prefrontal cortex brain tissues (from a male and a female both at 35 years of age) were dissociated using the Adult Brain Dissociation Kit (Miltenyi Biotec) according to the manufacturer’s protocol. Microglia were isolated using CD11b MicroBeads (Miltenyi Biotec) and cultured in microglial medium (ScienCell) with 5% FBS, 1% penicillin, 1% streptomycin and microglial growth supplement for 7 d.
RT–qPCR
Total RNAs from cells were extracted using RNeasy micro kit, and those from brain tissues were extracted using TRIzol (Takara Bio). RNAs were reverse transcribed using PrimeScript RT Master Kit (Takara Bio). Real-time PCR was performed using the TB Green Premix Ex Taq (Tli RNaseH Plus) on a CFX96 Real-Time PCR Detection System (Bio-Rad). Primer sequences are provided in Supplementary Table 4.
In vivo two-photon imaging
Microglia were visualized by two-photon microscopy imaging, through a small craniotomy at the somatosensory cortex of mice as described previously, with minor modifications10. Mice were anaesthetized by ketamine (100 mg per kg body weight) and xylazine (10 mg per kg body weight) in 0.9% sodium chloride solution and their eyes were protected with ointment. The skull area over the somatosensory cortex was shaved and cleaned by iodine tincture and a customized metal head plate was attached to the exposed skull using tissue glue (3M Vetbond). A round cranial window (3 mm in diameter) was made on the skull above the somatosensory cortex using a high-speed drill under a dissecting microscope. A drop (~2 μl) of hydrogel (acquired from Blafar) was applied onto the exposed region for the duration of the experiment. The open skull window was enclosed by a small coverslip with dental cement. Mice were transferred to the microscope after the surgery and kept under anaesthesia, with body temperature maintained at 37 °C ± 1 °C on a thermostatic heating system for the entire imaging period. The Olympus FVMPE-RS laser-scanning system with a ×25 water-immersion objective lens (1.05 NA XLPN25XWMP2, Olympus) was used for in vivo imaging. Microglia (tdTomato-labelled) were visualized using a laser with an excitation wavelength of 1,000 nm. Images were acquired at least 50-μm deep, primarily between 100–300 μm, into the slice surface to exclude any surgical artifacts. z-axis micrographs were recorded with a z-stack step size of 1 μm (31 steps) at a resolution of 512 × 512 pixels every 2 min for 20 min for basal microglial dynamics, followed by 30 min imaging for microglial damage-sensing responses.
For quantification of microglial dynamics, maximum Z-projections were created for each time point. Lateral motion artifacts were corrected by ImageJ using StackReg and TurboReg functions and background variability were normalized by thresholding. Overlays in RGB format were created for images obtained from different time points (representing changes over 4 min). Motility, retraction or extension indexes were calculated from all RGB overlays using custom R (colour distance) packages according to a previous study67.
The velocity of microglial processes movement was analysed by ImageJ using ProMoIJ functions68. In brief, images were cropped and aligned, and individual microglial processes were selected for automated motility analysis by reconstructing the three-dimensional (3D) skeleton and calculating the absolute difference of processes length between two consecutive time frames.
For tissue ablation, a region of interest (1 μm in diameter) was ablated with an 860-nm laser at 2.48 W for 0.2 s. To quantify the extent and speed of microglial responses to laser-induced injury, the number of microglial processes entering from the outer area Y (70 mm in radius) into the inner area X (35 mm in radius) surrounding the ablation was measured by ImageJ. The number of pixels corresponds to the region covered by processes within the area X, and its increase over time provides a measure of the microglial response. The microglial response at any time point (R(t) is therefore given by R(t) = (Rx(t) – Rx(0))/Ry(0).
Immunofluorescence analysis
Brains were fixed in 4% paraformaldehyde, transferred to 30% sucrose solution, embedded in optimal cutting temperature compound, and cut by a cryostat microtome (Leica CM1860 UV) at a thickness of 30 μm. Antigen retrieval was perfromed in the sections using citrate buffer (10 mM sodium citrate, 0.05% Tween-20, pH 6.0), and sections were blocked with 10% (wt/vol) BSA and incubated with primary and secondary antibodies. Nuclei were stained with DAPI (Beyotime) and slides were mounted using anti-fade reagents (Millipore). Images were captured by an FV-1200 confocal microscope (Olympus). For quantification, images were obtained from at least three areas for each slice and three sections for each animal.
Single-molecule fluorescence in situ hybridization
Single-molecule FISH was carried out using the RNAscope Multiplex Fluorescent Reagent Kit. Briefly, brain slices were incubated in RNAscope hydrogen peroxide, target retrieval and protease III solution. Slices were then hybridized with the probe (mouse HK2, ACDbio) and incubated in amplification solutions. Signals were detected using TSA Plus FITC and horseradish peroxidase. For post-hybridization immunofluorescence analyses, slices were incubated with primary antibodies and the following procedures were carried out as described above but with light protection throughout the procedure. The number of average Hk2 mRNA copies per cell was counted according to the manufacturer’s instructions.
5-Bromo-2′-deoxyuridine labelling
The adult mice received intraperitoneal injection of BrdU (50 mg per kg body weight, Sigma) 24 h before euthanasia. For immunofluorescence analysis, brain sections were obtained by antigen retrieval in citrate buffer and sequentially denatured with 2 N HCl for 1 h, followed by immunofluorescence analysis as indicated above.
Three-dimensional reconstruction
Confocal images were acquired using a ×60 objective lens of an FV-1200 microscope with 0.5-μm z-intervals, followed by analyses using IMARIS 9.5 software (Bitplane). Iba1+microglia and CD68+ lysosomes in the peri-infarct cortex were reconstructed, using the IMARIS function ‘Surface’ with the same thresholds for all reconstructions. For ‘Sholl’ analysis of microglial morphology, the Iba1+ microglia framework was outlined by setting the cell body as a central point and processes terminals as end points based on Iba1+ microglial reconstruction.
Western blotting
Cells were lysed in RIPA lysis buffer (Beyotime) by sonication and protein extracts were obtained from the supernatants after centrifugation. The extracts were subjected to SDS–PAGE. The membranes were blocked with 5% BSA, followed by incubation with primary antibodies and species-appropriate horseradish peroxidase-conjugated secondary antibodies. Membranes were subsequently washed in PBS-T three times and immunodetected with enhanced chemiluminescence reagents (PerkinElmer).
Antibodies
All primary and secondary antibodies with source, dilutions and validations are listed in Supplementary Table 5.
Enzyme-linked immunosorbent assay
TNF, IL-6 and IFN-β levels of supernatant of cultured microglia were determined by TNF, IL-6 ELISA kit (BD Biosciences) and IFN-β ELISA kit (BioLegend) according to the manufacturer’s instructions.
Microglial depletion treatment
To effectively ablate adult mice brain microglia, mice were fed with PLX5622-formulated AIN-76A diet (1,200 mg PLX5622 per kilogram of diet, Plexxikon) as the sole food for a consecutive 2 weeks. Control mice were fed with normal AIN-76A diet for 2 weeks (Plexxikon).
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced acute Parkinson’s disease model
For the acute MPTP model, C57BL/6 mice (male, 8 weeks of age) were intraperitoneally injected with two doses of MPTP (20 mg per kg body weight, Sigma) or saline at a 2-h interval, and euthanized 7 d later.
Glioma model
In the Ng2CreERT2-tdTomato::Trpfl/+::Nf1fl/fl mouse model, TSGs p53 and Nf1 were specifically inactivated in adult oligodendrocyte progenitor cells to induce spontaneous gliomas as previously described65. To further validate generated mutant cells, a Cre-recombinase-dependent reporter tdTomato was incorporated.
Lysophosphatidyl choline-induced demyelination model
For the demyelination model, Hk2CreERT2-tdTomato mice (male, 8 weeks of age) were anaesthetized with sodium pentobarbital (100 mg per kg body weight). Next, 1% LPC (1.5 μl, Sigma) or saline was injected into the right corpus callosum using a stereotaxic apparatus with the following coordinates (AP, 1.6 mm; ML, 1.0 mm; and DV, 2.2 mm). Mice were sutured and allowed to recover in a heating pad before being returned to their home cages. Mice were then anaesthetized 5 d later for histological analysis.
Public microglia sequencing data analysis
Microglia sequencing data were downloaded from the GEO database. Detailed information is provided in the Supplementary Table 3.
To examine the expression of metabolic regulators in different brain cell types. We compared expression profiles of 1,118 genes from nine selected KEGG-derived metabolic pathways according to the KEGG database (pathway entry IDs: ‘00010’, ‘00020’, ‘00030’, ‘00620’, ‘00190’, ‘00061’, ‘00072’, ‘00590’ and ‘00562’). Gene clusters were generated using BioLayout Express3D by plotting correlation graph with the Pearson correlation threshold r = 0.8 and 3D PCA by R package pca3d. Metabolic genes with fragments per kilobase of transcript per million mapped reads > 5 in at least one cell type (381 genes) were used to distinguish the DEGs between astrocytes, myelinating oligodendrocytes, neurons and oligodendrocyte progenitor cells versus microglia, respectively. DEGs from different comparisons with fold change > 5 and fold change < 0.2 were further intersected, which identified 25 DEGs between microglia and other brain cells (astrocytes, myelinating oligodendrocytes, neurons and oligodendrocyte progenitor cells). Data of Hk2 expression in microglia in different neurological diseases were obtained from the GEO database and analysed according to standard protocols for bulk-cell and single-cell sequencing data. The Wilcoxon test was used for comparing means and adjusted P values were calculated using Bonferroni correction.
Single-cell RNA-sequencing and data analyses
Single-cell capture and library construction was performed according to the manufacturer’s instructions of the 10X Chromium Next GEM Single Cell 3′ GEM and Library & Gel Bead Kit v3. Libraries were sequenced on an Illumina NovaSeq 6000 sequencing system (paired-end multiplexing run, 150 bp) by LC-Bio Technology at a minimal depth of 20,000 reads per cell. Raw sequencing data demultiplexing, conversion, sample demultiplexing, barcodes processing and single-cell 3′ gene counting were performed following standard the Cell Ranger pipeline. Data were aligned to the Mus musculus genome (GRCm38/mm10) and 5,696 cells were captured. The Cell Ranger output was loaded into R and the Seurat package was used for quality control and downstream analysis. To ensure the quality of data for downstream analyses, only cells with the number of genes detected between 500 and 5,000 and percent.mt < 10 were reserved. Overall, 3,914 cells passed the above cell filtration threshold and were retained for further analysis.
Count data were normalized using the LogNormalize method to eliminate confounding effects. After normalization and feature selection, 2,173 cells from Hk2fl/fl mice and 1,741 cells from Hk2-cKO mice were integrated. Linear transformation of features and PCA dimensionality reduction of cells was applied, while cell clusters were identified with a resolution of 1.5. Consequently, 30 distinct Seurat clusters were identified, and the t-SNE algorithm was used for two-dimensional visualization. According to the expression of different cellular markers, cells were annotated into seven subtypes and DEGs between different cell subtypes (min.pct, 0.5; logfc.threshold, 0.5) and DEGs between Hk2fl/fl and Hk2-cKO cells within each microglial subset (min.pct, 0.5; logfc.threshold, 0.5) were further identified.
Developmental trajectory analyses were executed by Monocle 2. To reduce noises caused by abundant genes, high-variance genes (average expression levels > 0.1, dispersion_empirical ≥ 1 × dispersion_fit) were selected for downstream DDRTree construction and cell ordering.
Measurement of mitochondrial reactive oxygen species
MitoROS was measured using MitoSOX (Invitrogen). Briefly, primary microglia were loaded with 5 μM MitoSOX at 37 °C for 20 min, and then washed by PBS two times. Cells were collected and the intensity of MitoROS fluorescence within individual microglia was analysed using flow cytometry (ACEA NovoCyte) with excitation at 510 nm and emission at 580 nm.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential was measured by JC-10 reagent (Abcam). Primary microglia were incubated with JC-10 dye-loading solution for 30 min at 37 °C, and then washed by PBS 2 times. Cells were collected and analysed using a flow cytometer (ACEA NovoCyte). Monomeric (green) and J-aggregate (red) fluorescence were measured using the FL1 and FL2 channels with flow cytometry, respectively. The ratio of FL2 (red) to FL1 (green) intensity was used to calculate the mitochondria membrane potential.
Microparticle uptake assay
Phagocytic activities of microglia were measured by analysing the uptake of microparticles (sulfate microspheres, 2 μm in diameter, Invitrogen) as previously described4. Microglial cultures were incubated with 2% (vol/vol) microsphere particles for 30 min at 37°C. After washing with PBS twice, cells were collected and analysed using a flow cytometer (ACEA NovoCyte) with excitation at 505 nm and emission at 515 nm to determine the uptake of microparticles in the cells.
Photothrombotic stroke
The mouse skull was exposed and a green light (532 nm) from a laser generator (Changchun Laser Technology, LR-GSP-532/300 mW) with a 2-mm aperture was positioned onto the skull over the right motor cortex (0.5 mm anterior to bregma and 1.5 mm lateral to the midline). Rose bengal (30 mg per kg body weight, Sigma) dissolved in sterilized 0.9% sodium chloride solution or normal saline solution was administrated through the retro-orbital sinus. The laser was turned on with a 15-mW power output for 2 min to induce photothrombosis through an intact skull. Mice were sutured and allowed to recover in a heating pad before being returned to their home cages.
Infarct volume analysis
Brain tissues were cut into 30-μm serial coronal sections. Every fifth brain slice of the ischaemic stroke region was stained with Nissl Staining Solution (Beyotime) for 30 min. Then sections were washed in PBS and dehydrated with gradient ethanol. Images were acquired using a VS120 (Olympus) under a bright-field microscope. The areas of infarction were delineated and quantified using customized macros with ImageJ. The infarct volumes were calculated by summation of the lesion areas of all slices and integrated by the slice thickness.
Behavioural tests
Pretraining
Mice were handled for 5 d and habituated to the arena. All behavioural tests were carried out and analysed by two investigators unaware of the genotypes and treatments of animals. Mice were tested before stroke induction to establish baseline performance. Next, they were tested 1, 3, 7 and 14 d after stroke. Behavioural tests were carried out at approximately the same time each day.
Grid walk test
Mice were allowed to walk on a 35 cm × 30 cm wire grid, with an 11-mm-square mesh fixed 40 cm above the ground frame for 5 min as described previously38. A camera was placed beneath the grid to record video for assessing errors of the left forelimb during the first 100 steps.
Cylinder test
For the cylinder test (spontaneous forelimb asymmetry task), mice were placed in a transparent cylinder (10 cm in diameter, 17 cm in height) and video-taped for 10 min as described previously38. All paw contacts with the cylinder wall during vertical exploration were recorded. Forelimb asymmetry index for mice was calculated as:
\((\left( {{{{{n}}}}\,{{{\mathrm{R}}}}\,{{{\mathrm{contacts}}}} - {{{{n}}}}\,{{{\mathrm{L}}}}\,{{{\mathrm{contacts}}}}} \right)/({{{{n}}}}\,{{{\mathrm{R}}}}\,{{{\mathrm{contacts}}}} + {{{{n}}}}\,{{{\mathrm{L}}}}\,{{{\mathrm{contacts}}}} + {{{{n}}}}\,{{{\mathrm{both}}}}\,{{{\mathrm{paws}}}}\,{{{\mathrm{contacts}}}})).\)
Open field test
Mice were placed individually into the centre of a square open field (45 cm length × 45 cm width × 30 cm height) and allowed to explore for 10 min. Activities of mice in the field were recorded using a movement tracking system (ANY-maze, Stoelting) connected to a camera mounted above the field.
Rotarod tests
Mice were first trained for 10 min on the rotarod for three consecutive days with gradually elevated rotational speed: 10 r.p.m. on day 1 and 20 r.p.m. on day 2 and 40 r.p.m. on day 3. On the testing day (day 4), the latency to fall was recorded at a speed of 45 r.p.m. (accelerated from 0 r.p.m. to 45 r.p.m. over a 2-min period) using a Rota-Rod test apparatus (Panlab).
Quantification and statistical analysis
Data are presented as the mean ± s.e.m. from at least three independent experiments. Statistical differences were analysed by two-tailed Student’s t-test for two groups or one-way ANOVA with Bonferroni’s post hoc tests for more than two groups. RT–qPCR, ELISA and behavioural data were analysed by two-way ANOVA with Bonferroni’s post hoc tests. P < 0.05 was considered significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA-seq data of repopulating microglia are available from the GEO under accession GSE188642. The data that support the finding of this study are available from the corresponding authors upon request. Source data are provided with this paper.
References
O’Neill, L. A., Kishton, R. J. & Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565 (2016).
Hu, Y. et al. mTOR-mediated metabolic reprogramming shapes distinct microglia functions in response to lipopolysaccharide and ATP. Glia 68, 1031–1045 (2020).
Orihuela, R., McPherson, C. A. & Harry, G. J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 173, 649–665 (2016).
Baik, S. H. et al. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 30, 493–507 (2019).
Ulland, T. K. et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170, 649–663 (2017).
Nugent, A. A. et al. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105, 837–854 (2020).
Pan, R. Y. et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease. Cell Metab. 34, 634–648 e6 (2022).
Tay, T. L. et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 20, 793–803 (2017).
Lawson, L. J., Perry, V. H. & Gordon, S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience 48, 405–415 (1992).
Davalos, D. et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758 (2005).
Ransohoff, R. M. & Perry, V. H. Microglial physiology: unique stimuli, specialized responses. Annu Rev. Immunol. 27, 119–145 (2009).
Shemer, A. et al. Microglia plasticity during health and disease: an immunological perspective. Trends Immunol. 36, 614–624 (2015).
Elmore, M. R. et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82, 380–397 (2014).
Huang, Y. et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat. Neurosci. 21, 530–540 (2018).
Bruttger, J. et al. Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity 43, 92–106 (2015).
Rao, J., Oz, G. & Seaquist, E. R. Regulation of cerebral glucose metabolism. Minerva Endocrinol. 31, 149–158 (2006).
Raichle, M. E., Posner, J. B. & Plum, F. Cerebral blood flow during and after hyperventilation. Arch. Neurol. 23, 394–403 (1970).
Wilson, J. E. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J. Exp. Biol. 206, 2049–2057 (2003).
Wilson, J. E. Hexokinases. Rev. Physiol. Biochem. Pharmacol. 126, 65–198 (1995).
Pastorino, J. G. & Hoek, J. B. Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 40, 171–182 (2008).
Moon, J. S. et al. mTORC1-Induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep. 12, 102–115 (2015).
Everts, B. et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat. Immunol. 15, 323–332 (2014).
Li, Y. et al. Hexokinase 2-dependent hyperglycolysis driving microglial activation contributes to ischemic brain injury. J. Neurochem. 144, 186–200 (2018).
Pastorino, J. G., Shulga, N. & Hoek, J. B. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 277, 7610–7618 (2002).
Nederlof, R. et al. Targeting hexokinase II to mitochondria to modulate energy metabolism and reduce ischaemia-reperfusion injury in heart. Br. J. Pharmacol. 171, 2067–2079 (2014).
Wolf, A. J. et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 166, 624–636 (2016).
Hinrichsen, F. et al. Microbial regulation of hexokinase 2 links mitochondrial metabolism and cell death in colitis. Cell Metab. 33, 2355–2366 e8 (2021).
Moehlman, A. T. & Youle, R. J. Mitochondrial quality control and restraining innate immunity. Annu. Rev. Cell Dev. Biol. 36, 265–289 (2020).
Jin, H. S. et al. Mitochondrial control of innate immunity and inflammation. Immune Netw. 17, 77–88 (2017).
Zhang, Y. et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947 (2014).
Gosselin, D. et al., An environment-dependent transcriptional network specifies human microglia identity. Science 356, eaal3222 (2017).
Stott, F. J. et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 17, 5001–5014 (1998).
Gu, X. et al. Human Schlafen 5 inhibits proliferation and promotes apoptosis in lung adenocarcinoma via the PTEN/PI3K/AKT/mTOR pathway. Biomed. Res. Int. 6628682 (2021).
Sen, S. et al. Role of hexokinase-1 in the survival of HIV-1-infected macrophages. Cell Cycle 14, 980–989 (2015).
Roiniotis, J. et al. Hypoxia prolongs monocyte/macrophage survival and enhanced glycolysis is associated with their maturation under aerobic conditions. J. Immunol. 182, 7974–7981 (2009).
Nowak, N., Kulma, A. & Gutowicz, J. Up-regulation of key glycolysis proteins in cancer development. Open Life Sci. 13, 569–581 (2018).
Qin, C. et al. Dual functions of microglia in ischemic stroke. Neurosci. Bull. 35, 921–933 (2019).
Schaar, K. L., Brenneman, M. M. & Savitz, S. I. Functional assessments in the rodent stroke model. Exp. Transl. Stroke Med. 2, 13 (2010).
Soto-Heredero, G. et al. Glycolysis—a key player in the inflammatory response. FEBS J. 287, 3350–3369 (2020).
Shoshan-Barmatz, V. et al. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med 31, 227–285 (2010).
Ghosh, S. et al. Bioenergetic regulation of microglia. Glia 66, 1200–1212 (2018).
Xiang, X. et al. Microglial activation states drive glucose uptake and FDG-PET alterations in neurodegenerative diseases. Sci. Transl. Med. 13, eabe5640 (2021).
Bernier, L. P. et al. Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat. Commun. 11, 1559 (2020).
Gimenez-Cassina, A. et al. Mitochondrial hexokinase II promotes neuronal survival and acts downstream of glycogen synthase kinase-3. J. Biol. Chem. 284, 3001–3011 (2009).
Chow, H. M. et al. Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nat. Neurosci. 22, 1806–1819 (2019).
Chao, C. C. et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 179, 1483–1498 (2019).
Eyo, U. & Dailey, M. E. Effects of oxygen-glucose deprivation on microglial mobility and viability in developing mouse hippocampal tissues. Glia 60, 1747–1760 (2012).
Palsson-McDermott, E. M. & O’Neill, L. A. The Warburg effect then and now: from cancer to inflammatory diseases. Bioessays 35, 965–973 (2013).
Apostolova, I. et al. Hypermetabolism in the hippocampal formation of cognitively impaired patients indicates detrimental maladaptation. Neurobiol. Aging 65, 41–50 (2018).
Mao, C. Y. et al. Brain glucose metabolism changes in Parkinson’s disease patients with CHCHD2 mutation based on [18F]-FDG PET imaging. J. Neurol. Sci. 369, 303–305 (2016).
O’Connell, M. T. et al. Glucose metabolism in traumatic brain injury: a combined microdialysis and [18F]-2-fluoro-2-deoxy-d-glucose-positron emission tomography (FDG-PET) study. Acta Neurochir. Suppl. 95, 165–168 (2005).
Sokoloff, L. Energetics of functional activation in neural tissues. Neurochem. Res. 24, 321–329 (1999).
Leng, L. et al. Microglial hexokinase 2 deficiency increases ATP generation through lipid metabolism leading to beta-amyloid clearance. Nat. Metab. 4, 1287–1305 (2022).
Maes, M. E. et al. Targeting microglia with lentivirus and AAV: recent advances and remaining challenges. Neurosci. Lett. 707, 134310 (2019).
Bellver-Landete, V. et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat. Commun. 10, 518 (2019).
Lloyd-Burton, S. M. et al. SPARC regulates microgliosis and functional recovery following cortical ischemia. J. Neurosci. 33, 4468–4481 (2013).
Minhas, P. S. et al. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature 590, 122–128 (2021).
Pajak, B, et al. 2-Deoxy-d-glucose and its analogs: from diagnostic to therapeutic agents. Int. J. Mol. Sci. 21, 234 (2019).
Shoshan-Barmatz, V. & Golan, M. Mitochondrial VDAC1: function in cell life and death and a target for cancer therapy. Curr. Med. Chem. 19, 714–735 (2012).
Ciscato, F. et al. Hexokinase 2 displacement from mitochondria-associated membranes prompts Ca2+-dependent death of cancer cells. EMBO Rep. 21, e49117 (2020).
Cai, M., He, P. & Fang, D. L. Hypoxiainduced mitochondrial translocation of DNM1L increases mitochondrial fission and triggers mPTP opening in HCC cells via activation of HK2. Oncol. Rep. 42, 1125–1132 (2019).
Pasdois, P., Parker, J. E. & Halestrap, A. P. Extent of mitochondrial hexokinase II dissociation during ischemia correlates with mitochondrial cytochrome c release, reactive oxygen species production, and infarct size on reperfusion. J. Am. Heart Assoc. 2, e005645 (2012).
De Jesus, A. et al. Hexokinase 1 cellular localization regulates the metabolic fate of glucose. Mol. Cell 82, 1261–1277 (2022).
Mucke, L. et al. High-level neuronal expression of Aβ(1–42) in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058 (2000).
Liu, C. et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221 (2011).
Zhang, X. et al. Aged microglia promote peripheral T cell infiltration by reprogramming the microenvironment of neurogenic niches. Immun. Ageing 19, 34 (2022).
Sipe, G. O. et al. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 7, 10905 (2016).
Paris, I. et al. ProMoIJ: a new tool for automatic three-dimensional analysis of microglial process motility. Glia 66, 828–845 (2018).
Acknowledgements
We greatly appreciate A. Gitler at Stanford University for comments and suggestions on the manuscript. We thank P. Shi at the Institute of Translational Medicine of Zhejiang University for help with microglia sorting, and M. -X. Guan at the Division of Medical Genetics and Genomics of Zhejiang University School of Medicine for help with Metabolic Extracellular Flux analysis, W. Wang at the Charles Institute of Dermatology of University College Dublin and L. Chen at the First Affiliated Hospital of Zhejiang University for generously providing hydrogel, C. Liu at the School of Brain Science and Brain Medicine of Zhejiang University for generously providing transgenic mice. We are thankful to S. Fang at the Core Facilities of the School of Brain Science and Brain Medicine of Zhejiang University, H. Wu at the Center of Cryo-Electron Microscopy of Zhejiang University, Y. Huang, Y. Li and J. Wang at the Core Facilities of Zhejiang University for technical assistance. We greatly appreciate X. Zhang and A. Ping at the Second Affiliated Hospital of Zhejiang University for help with human microglia sorting. This work was supported by grants from the National Key Research and Development Program of China (2017YFA0104200 to Z.G.), National Natural Science Foundation of China (32070974 to Z.G., 82090033 to Z.G. and 82101413 to Y.H.), major programme of the National Natural Science Foundation of China (82090033 to S.D.) and China Postdoctoral Science Foundation (2020M671693 to Y.H.).
Author information
Authors and Affiliations
Contributions
Z.G., S.D., Y.H., K.C. and F.W. conceived the project and designed experiments; Y.H., K.C., F.W. and L.Q. carried out experiments; Y.H., K.C., F.W., W.W., W.M. and Y.L. analysed the data; B.S., L.S., J.Z. and J.Z. provide technical assistance. Z.G., Y.H., K.C., F.W., and W.W. wrote the paper; Z.G., S.D., Y.H., K.C., F.W., W.W., Y.L., W.G., Z.W., Y.X. and S.D. reviewed and edited the paper; Z.G., S.D. and Y.H. provided funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Metabolism thanks Marco Colonna and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Alfredo Giménez-Cassina, in collaboration with the Nature Metabolism team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 HK expression in human and mouse microglia.
(a) A correlation network graph of genes in metabolic pathways (1147 genes) expressed in different types of brain cells (data retrieved from GSE52564). Nodes represent transcripts, while edges represent the degree of correlation in expression. Heatmaps and line charts showing the expression profile of transcripts contained within 3 clusters corresponding to astrocytes, endothelial cells and microglia clusters are presented. (b) Principal component analysis (PCA) of the metabolic genes in glycolysis/gluconeogenesis in 7 cell types of the brain (data retrieved from GSE52564). (c) A comparison of metabolic gene expression in human and mouse microglia (data retrieved from GSE89960). (d-f) The expression of Hk1, Hk2 and Hk3 in mouse brain cells (data retrieved from GSE52564), n = 2 mice per group. MG, Microglia; ECs, Endothelial cells; OPC, Oligodendrocyte progenitor cells; Neu, Neurons; MO, Myelinating oligodendrocytes; NFO, Newly formed oligodendrocytes; Ast, Astrocytes. Data are presented as means ± SEM.
Extended Data Fig. 2 HK2 is selectively expressed in microglia.
(a) Representative micrographs (from 3 independent experiments) showing single-molecule fluorescence in situ hybridization (FISH) of Hk2 mRNA with different cellular markers, NeuN (neurons), Olig2 (oligodendrocytes) and GFAP (astrocytes) in the mouse brain. White arrowheads point to Hk2+ and Iba1+ microglia. Dotted boxes show regions of higher magnification.
Extended Data Fig. 3 Characterization of Hk2CreERT2-tdTomato mice.
(a) Flow cytometry analyses of tdT+ cells and CD11b+CD45low microglia in Hk2CreERT2-tdTomato mice. (b) Representative micrographs (from 3 independent experiments) showing the co-labeling of tdT+, Iba1+and Hk2+ in brain slices from Hk2CreERT2-tdTomato mice. Dotted boxes show regions of higher magnification. (c) FISH of Hk2 and tdT staining in brain slices from Hk2CreERT2-tdTomato mice and quantification. Dotted boxes show regions of higher magnification. n = 3 mice per group. (d) Representative micrographs (from 3 independent experiments) showing the three-dimension (3D) reconstruction of a tdT+ cell in brain slices from Hk2CreERT2-tdTomato mice using Imaris. (e) Representative micrographs (from 3 independent experiments) of tdT and different cellular markers, NeuN (neurons), Sox10 (oligodendrocytes) and GFAP (astrocytes) in the brains of Hk2CreERT2-tdTomato mice. (f) Representative micrographs (from 3 independent experiments) showing full co-localization of GFP, tdT and Iba1 in brain slices from Hk2CreERT2-tdTomato::Ai3fl/+ mice after tamoxifen induction. TAM, tamoxifen; D, day; tdT, tdTomato. Data are presented as means ± SEM.
Extended Data Fig. 4 HK2 is similarly expressed in microglia in different brain regions.
(a) Representative gating strategies for flow cytometry-based measurement of fluorescence intensity of microglial tdTomato in different brain regions. (b) Representative Western blot (from 2 independent experiments) analysis of HK2 protein in lysates from FACS-sorted microglia from adult cortex, midbrain and cerebellum and quantification (c). n = 2 from 16 mice per group. Data are presented as means ± SEM.
Extended Data Fig. 5 ECAR and OCR analyses of microglia after HK2 inhibition or deletion.
(a) ECAR measurement of primary mouse microglia (untreated or pretreated with 3-BrPA). (b) Quantification of basal ECAR and maximal ECAR from a. n = 7 or 4 independent cultures in Hk2fl/fl or Hk2-cKO groups, respectively. (c) OCR measurement of primary mouse microglia (untreated or pretreated with 3-BrPA). (d) Quantification of basal OCR and maximal OCR from c. n = 5 independent cultures per group. (e) A scheme of the standard Seahorse XF Mito-stress test. (f) OCR measurement of primary microglia from Hk2fl/fl (untreated or pretreated with 2-DG) and Hk2-cKO mouse brains in regular assay media containing pyruvate and glutamine. (g) Quantification of basal OCR and maximal OCR from f. n = 5 independent cultures per group. ECAR, extracellular acidification rate; OCR, oxygen consumption rate; OXPHOS, oxidative phosphorylation; TCA, tricarboxylic acid cycle; Glc, glucose; 3-BrPA, 3-Bromopyruvate; FCCP, Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; Olig, oligomycin; Antim, antimycin A; Rot, rotenone; 2-DG, 2-Deoxy-D-glucose; Data are presented as means ± SEM. Two-tailed Student’s t-tests for b, d. One-way ANOVA followed by Bonferroni’s post hoc tests for g, *p < 0.05, **p < 0.01 and ***p < 0.001.
Extended Data Fig. 6 Ablation of HK2 did not affect microglial density and morphology in the adult stage.
(a) A scheme of the experimental timeline for the analysis of microglial density and morphology from Hk2fl/fl and Hk2-cKO mice at 1 week after TAM administration. (b) Representative micrographs (from 3 independent experiments) showing the Iba1+ microglia and proliferative (BrdU+) microglia in adult Hk2fl/fl and Hk2-cKO mouse brains at 1 week after TAM administration. (c, d) Quantification of microglial number (c) and percentage of BrdU+ microglia (d) in the cortex from Hk2fl/fl and Hk2-cKO mice. n = 3 mice per group. (e) A scheme of the experimental timeline for analysis of microglial density and morphology from Hk2fl/fl and Hk2-cKO mice at 4 weeks after TAM administration. (f, g) Flow cytometry analysis and quantification of CD11b+ CD45low microglia in adult mice at 4 weeks after TAM administration. n = 3 mice per group. (h) Representative micrographs (from 3 independent experiments) showing 3D reconstruction of microglia from Hk2fl/fl and Hk2-cKO mice in the cortex using Imaris. (i) Scholl analysis of all reconstructed cells. n = 3 mice per group (total 36 cells). (j-l) Quantification of microglial morphology in the cortex from Hk2fl/fl and Hk2-cKO mice by Sholl analysis. n = 3 mice per group (total 36 cells). TAM, tamoxifen; D, day. Data are presented as means ± SEM. Two-tailed Student’s t-tests for c, d, f, g, j-l, *p < 0.05.
Extended Data Fig. 7 Ablation of HK2 did not affect Hk1 and Hk3 levels.
(a) The expression of Hk1, Hk2 and Hk3 in FACS-sorted microglia from Hk2fl/fl and Hk2-cKO mice at adult. n = 4 or 3 mice in Hk2fl/fl or Hk2-cKO groups, respectively. (b-d) The expression of Hk1, Hk2 and Hk3 in sorted microglia during repopulation in Hk2fl/fl and Hk2-cKO mice. n = 1481 or 264 cells for HomoMG, 162 or 264 cells for Precursor MG, 121 or 91 cells for G1/S MG, 200 or 108 cells for G2/M MG in Hk2fl/fl or Hk2-cKO groups, respectively. (e) Quantitative analyses of Hk1, Hk2, Hk3 and Cx3cr1 expression in the brain tissues from Hk2fl/fl and Hk2-cKO mice. n = 4 or 3 mice in Hk2fl/fl or Hk2-cKO groups, respectively. (f) CD11b+CD45low microglia and CD11b-CD45− non-microglial cells sorting from Hk2fl/fl and Hk2-cKO mice by flow cytometry. (g) Quantitative analyses of Hk2 and Cx3cr1 expression in CD11b−CD45− non-microglial cells and CD11b+CD45low microglia from Hk2fl/fl and Hk2-cKO mice. n = 3 mice for microglia and non-microglial cells or 4 mice for microglia in Hk2fl/fl or Hk2-cKO groups, respectively. Data are presented as means ± SEM. Two-tailed student t-tests for a, e, g. Wilcoxon rank sum test and Bonferroni correction for b-d, *p < 0.05, **p < 0.01 and ***p < 0.001.
Extended Data Fig. 8 A comparison of repopulated microglia in Hk2fl/fl and Hk2-cKO mice.
(a) Pseudotime trajectory was constructed using Monocle 2 by re-ordering microglia population and single cells on the trajectory. Each microglia on the minimum spanning tree (MST) were colored by pseudotime. (b) Pseudotime trajectory was constructed and each microglia on the MST were colored by annotated microglial cell types during repopulation in Hk2fl/fl and Hk2-cKO mice. (c) The number of cells in different subsets of microglia from Hk2fl/fl and Hk2-cKO mice. (d) t-SNE plots of Hk2, Cx3cr1, Cdkn1a and Slfn5 gene expression between Hk2fl/fl and Hk2-cKO mice. n = 1481 or 264 cells for HomoMG, 162 or 264 cells for Precursor MG, 121 or 91 cells for G1/S MG, 200 or 108 cells for G2/M MG in Hk2fl/fl or Hk2-cKO groups, respectively. The color key indicates the expression levels.
Extended Data Fig. 9 Analyses of microglial number, phagocytosis, motor behavior and inflammation in Hk2fl/fl and Hk2-cKO mice before and after stroke.
(a) Representative micrographs (from 3 independent experiments) showing the density of microglia in the peri-infarct zone from Hk2fl/fl and Hk2-cKO mice at 3 and 7 Dpi. Dashed lines and dashed boxes indicate the border of ischemic core and regions of higher magnification, respectively. Rotarod (b, c) and open field (d, e) tests of adult Hk2fl/fl and Hk2-cKO mice. n = 10 mice per group. (f) Representative micrographs (from 3 independent experiments) showing CD68+ labeling in Iba1+ microglia in the peri-infarct zone from Hk2fl/fl and Hk2-cKO mice under sham and ischemic conditions (3 Dpi) and quantification of microglial volume and CD68+ volume in microglia (g, h). Dashed boxes show regions of higher magnification of microglia. n = 30 cells from 3 mice per group. RT-qPCR analysis of Tnfα, Il-6 and Il-1β levels (i) and Ifitm3, Stat1 levels (j) in the infarcted tissues at 1 Dpi. n = 5 mice per group for sham-operated animals or 6 mice per group for ischemic animals. RPM, rotations per minute; Dpi, days post ischemia. Data are presented as means ± SEM. Two-tailed student t-tests for b-e. Two-way ANOVA followed by Bonferroni’s post hoc tests for g, h, *p < 0.05, **p < 0.01 and ***p < 0.001.
Extended Data Fig. 10 Analyses of mitochondrial membrane potential and ROS levels in Hk2fl/fl and Hk2-deficient microglia upon LPS/ poly(I:C) stimulation.
(a-d) Analysis of microglial mitochondrial membrane potential and mitochondrial ROS after stimulation of LPS or poly(I:C) from Hk2fl/fl and Hk2-cKO mice in the presence or absence of Mito-TEPMO. n = 6 or 7 (a), 7 or 6 (b), 7 or 5 (c), 6 or 6(d) independent cultures for each treatment in Hk2fl/fl or Hk2-cKO groups, respectively. Data are presented as means ± SEM. Two-way ANOVA followed by Bonferroni’s post hoc tests for a-d, *p < 0.05, **p < 0.01 and ***p < 0.001.
Supplementary information
Supplementary Information
Supplementary Tables 3–5 and authors statements
Supplementary Table 1
List of metabolic genes related to Fig. 1.
Supplementary Table 2
List of genes related to Fig. 5.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 1
Unprocessed western blots.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 7
Statistical source data.
Source Data Fig. 7
Unprocessed western blots.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 4
Unprocessed western blots. gels
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 10
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hu, Y., Cao, K., Wang, F. et al. Dual roles of hexokinase 2 in shaping microglial function by gating glycolytic flux and mitochondrial activity. Nat Metab 4, 1756–1774 (2022). https://doi.org/10.1038/s42255-022-00707-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42255-022-00707-5
- Springer Nature Limited
This article is cited by
-
Emerging roles of lactate in acute and chronic inflammation
Cell Communication and Signaling (2024)
-
Emerging role of senescent microglia in brain aging-related neurodegenerative diseases
Translational Neurodegeneration (2024)
-
Focusing on mitochondria in the brain: from biology to therapeutics
Translational Neurodegeneration (2024)
-
Complex roles for mitochondrial complexes in microglia
Nature Metabolism (2024)
-
Mitochondrial respiration in microglia is essential for response to demyelinating injury but not proliferation
Nature Metabolism (2024)