
Abstract
Multifunctional light sheet imaging flow cytometry of a large population of live cells at high throughput is challenging and requires new technological advancement. Existing cytometry techniques are limited due to point-based illumination that does not allow volume interrogation and biophysical parameter estimation on the go. Here, we propose a multifunctional (multichannel, multisheet and multicolor) imaging cytometry (M3IC) system that employs vertically-aligned multi-sheet array (VAMSA) illumination for interrogating cells flowing simultaneously through multiple microfluidic channels. We studied cancer cells (volume interrogation with organelle-level resolution and high signal-to-background-ratio(SBR)) at high throughput (~2500 nl/min). M3IC system demonstrates organelle-level resolution with a SBR comparable to that of confocal, especially at low flow rates. In addition, the multicolor imaging capability of the system facilitates multi-organelle investigation, determination of critical biophysical parameters, and drug (Paclitaxel) treatment studies on cancer cells. M3IC system is expected to advance the field of fluorescence microscopy, cell biophysics, disease biology and optical physics.
Similar content being viewed by others


Introduction
To be able to carry out multiple tasks (volume visualization, biophysical parameter estimation, and statistical analysis) on the go is a demanding task that potentially calls for new technological breakthrough. This is true for the field of imaging flow cytometry, which still depends on the point illumination1,2,3,4,5,6,7,8. Although imaging flow cytometry (IFC) techniques have been used to screen biological specimens but high-throughput volume interrogation of an entire cell population with organelle-level resolution and high signal-to-background ration (SBR) remains elusive. Further advance is expected with the availability of new capabilities to make it a full-fledged diagnostic system. In this respect, the arrival of light sheet has shown promise and potential to match the current expectations. The research disciplines that are most likely to get boost from the emerging light sheet technology range from clinical biology to optical imaging.
Since its first inception in the year 2013, light sheet cytometry has been quite successful for a variety of studies ranging from cell to organism biology9,10. Subsequent years have seen accelerated development in diverse disciplines ranging from biological to physical sciences. In the year 2016, Lau et al reported light sheet-based optofluidic time-stretch imaging for high throughput interrogation of MIHA cells11. An exciting development is the on-chip cytometry realized by integrating light sheet on a mirror-embedded microfluidic chip12. Other variants include scanned Bessel beam for stem cell research, and a label-free imaging flow cytometry for cell screening13,14. On similar lines, Jiang et al have demonstrated cytometry by integrating droplet microfluidics and light sheet15. In another development, successful imaging of large live organisms (such as, C elegans) is achieved by iLIFE imaging cytometry16, and SCAPE especially at high speed17. The developments continued, with our group succeeding in high speed interrogation of cells using a single light sheet18, and Ben-Yakar group using a line excitation array detection (LEAD) fluorescence microscopy for mega-Hertz line-scanning using a virtual light sheet19. Another advance has been the use of high throughput imaging cytometry system for cell imaging20. There are many such advancements made in the past few years that have consolidated and brought forward the potential of light sheet in imaging cytometry. Here, we report a full-fledged multiple light sheet imaging cytometry that can be used for a variety of studies including disease biology.
The existing cytometry techniques use point-illumination that necessitates colineating cells to enable sequential interrogation. This necessitates two fluids, of which one is the specimen flowing fluid and the second is the sheath fluid required for hydrodynamic focusing of the flowing specimens21,22. This arrangement makes the system complex and quite cumbersome to operate. The existing systems are not suitable for all kinds of specimens (with varying shapes and sizes) and need special arrangements for optimal operation. Moreover, existing point-based cytometry techniques are largely limited to counting, sorting, mixing and imaging. However, existing techniques have found several applications in diverse research disciplines ranging from cell biology (asymmetric cell division, receptor activity, autophagy, nuclear translocation and cell cycle analysis) to medical health-care (drug-discovery, disease biology and immunology)23,24,25,26,27,28,29,30,31,32. Recently reported complex multichannel cell analysis based on machine learning techniques have advanced point-illumination-based cytometry techniques33,34,35. Apart from these benefits, there are some limitations as well. One of the key obstacle with existing techniques is their inability to image internal structure in a specimen (such as, a cell) and thus are not particularly suitable for deciphering underlying biological mechanisms in live cells. Specifically in drug-discovery, it would be advantageous to interrogate a large population of cells (both healthy and diseased) with the ability to obtain organelles level information. This requires uniform illumination of cells flowing through the channels, similar to one provided by a sheet of light. The compelling properties of light sheet that stand out are, large field-of-view (suitable for cross-sectioning imaging of large specimens), selective plane interrogation, uniform illumination, high contrast and better signal-to-background ratio (SBR). Overall, the existing techniques somewhat limit its use to trivial tasks, and new technology needs to step-up to further its progress. In the present scenario, light sheet technology seems to provide such a capability as evident from recent developments. A detailed discussion of light sheet technology and its applications in diverse disciplines can be found in Refs. 36,37,38.
In this article, we propose M3IC optical system powered by vertical-aligned multi-sheet array (VAMSA) illumination for 3D imaging cytometry on a commercial Y-type microfluidic chips. The system was calibrated and tested on a known test sample (fluorescent beads in solution) for optimal operation. Subsequently, it is employed to image internal organelles (mitochondria) in HeLa cells, and healthy HEK cells. Fine sectioning enabled by sheet-based illumination has revealed the distribution of mitochondria in the cell volume for both untreated and treated cells. In addition, drug treatment studies were carried out to access its efficacy and determine critical biophysical parameters related to cell physiology.
Results
The M3IC optical system
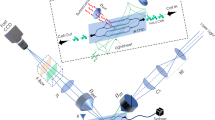
The schematic diagram of developed vertical light sheet array based M3IC system is shown in Fig. 1. M3IC has three major sub-system apart from fast data acquisition system. The illumination sub-system is at the heart of M3IC, that is comprised of three major optical components arranged in an unique configuration to generate the desired illumination PSF (VAMSA). A light of wavelength 532 nm is used as an excitation source for the specimens (fluorescent beads and fluorescently-labelled HeLa cells). The laser light is directed to a beam-stearing system (consists of a series of mirrors) for precision alignment. Subsequently the beam is allowed to pass through a specialized transmission grating beam-splitter (80 grooves/mm, Edmund Optics, Singapore) to derive a maximum of five intense beams. The beam is then subjected to the beam-expander (which is placed at the focus of the first lens L1) for 2X expansion to fill the back-aperture of cylindrical lens. The enlarged beams then goes through a combination of cylindrical and high numerical aperture objective lens to realize collinear but well-separated multiple vertically-aligned light sheets. The second sub-system consists of a fabricated microfluidic chip (containing multiple Y-type 200 × 100 μm2 sized channels) for flowing the specimen. The illumination PSF intersects the channels for optically sectioning the fluorescently-labelled cells flowing through them. During experimentation, freshly prepared cells were loaded in the reservoir and a flow-pump (New Era flow pump, NE-1002X, USA) is connected to other end for inducing flow. The pump is operated in the suction mode, and flow is computationally controlled by an interfacing software. The third sub-system is essentially an widefield 4f detection setup. The fluorescence from specimen is collected by a separate objective lens (10X, 0.30 NA Olympus, Japan) placed orthogonal to the illumination objective and focussed by the tube lens to the detector where image formation takes place. Note that, the objective and tube lens combination forms a 4f optical configuration, with a magnification of ~12X. On its way to the detector (Zyla 4.2 sCMOS camera, Andor, UK) the light is filtered by optical filters (532 nm Notch filter, ZET 532NF, Chroma Technology, USA and 550 nm Longpass filter, Thorlabs, USA) arranged in the filter box. Finally, the data (sectional images) are recorded and cell volumes are reconstructed using the available inbuilt MATLAB scripts for data handling, deconvolution and stacking.

The major subsystems comprise multiple vertical light sheet illumination, a microfluidic chip-based specimen flow platform, and a widefield 4f detection system. The proposed multifunctional system comprises three major units i.e, multibeam generation unit, multisheet generation unit and microfluidic chip-based cell flow unit. The illumination subsystem consists of two key units that include optical components such as transmission grating, beam-expander, cylindrical lens for realizing vertical spaced light sheets, and a high numerical aperture objective lens to generate diffraction-limited multiple vertical light sheet PSF. The inset (red box) shows a schematic realization of one of the light sheets sectioning the channel. The second subsystem is a pre-fabricated microfluidic chip-based cell flow unit with Y-type channel geometry. The inset shows cells flowing through the channels which is transected by one of the light sheet. The third sub-system is essentially a fluorescence-based 4f widefield detection system equipped with a filter box for recording the sectional images of flowing cells. Details related to the actual system and chip fabrication can be found in Supplementary Notes 1 and 2.
The Illumination VAMSA PSF
The next critical stage in the M3IC system development is the characterization of illumination PSF and flow-induced detection PSF, along with other calibrations (related to flow-rate and detector exposure time). Figure 2a shows the schematic of the optical setup used for characterizing the illumination PSF. To obtain a 3D map of the illumination PSF (hill), a CCD camera is placed in the beam-path of illumination sub-system, at and about the focus. The intensity field at every z-position is recorded by scanning the camera (placed on moving platform) and the images are recorded. To obtain the 3D field distribution, the images are then stacked together and the illumination 3D PSF is reconstructed (see, Fig. 2a). An average size of light sheet in the multi-sheet array is about ≈211.0μm (full-width at half maximum) which can section the entire channel (size ~100 μm). Specifically, the illumination PSF shows 5 prominent peaks (corresponding to 5 channels) for a specific grating-beam-expander distance, D = 10 mm. However, the M3IC system allows one to adaptively choosing the number of light sheets by just altering the distance between transmission grating and beam-expander (see, Supplementary Note 4). Practically, this is accomplished by placing the grating on a precision linear translator. Figure 2b shows three, four and five light sheets for D = 10, 20, 25 mm, respectively. This brings in an additional flexibility when designing microfluidic chips with the desired number of light sheets and user-defined inter-channel spacings. Since flow induces optical aberration (blur), this has a direct effect on the recorded images. To minimize this effect, flow-variant detection PSFs are obtained by flowing point sources (such as fluorescent beads of size ~ 1μm) and the same is recorded in the detector as shown in Fig. 2c. Calibration and analysis show a change of PSF from 4.410μm to 5.726μm for flow rates 100 to 2000 nl/min. In addition, PSF versus fluid velocity shows a near-linear increase in PSF in the velocity range, 2−12 nm/s. This is crucial since it allows better estimation of detection PSF for subsequent processing of images. The PSF is used to deconvolve recorded cross-sectional images and reconstruct cell volume as detailed in materials and methods section.

a The system PSF is characterised by scanning a CCD camera along the beam path about the focus. Corresponding 2D and 3D system PSFs are also shown along with the intensity plots. b The change in system PSF at varying grating-beam-expander distance, D is also shown, suggesting adaptive change in the number of light sheets. c The detection PSF at varying flow-rates from 100 nl/min to 2000 nl/min. The corresponding intensity plots along with PSF barplot (PSF vs. fluid velocity) shows the PSF-broadening at large flow-rates. These flow-variant PSFs are subsequently used for deconvolving the recorded sectional images. The details related to actual optical system used for VAMSA-PSF characterization is discussed in Supplementary Note 3. In addition, flow-variant PSF at each flow-rate is detailed in Supplementary Note 5. Raw data of fluorescemt beads flowing at varying flow-rate is shown in Supplementary Movie 1.
Cell Counting Statistics
In cytometry, the quantification of specimen count statistics with flow-rate is critical. At a relatively moderate flow-rate of 1000 nl/min, the sCMOS captures a maximum of 5 high quality sectional images, whereas high flow-rate (>2000 nl/min) result in <5 images of low quality. This is predominantly due to limited photon budget at large flow-rate. Both beads and cell specimens are flown and the corresponding count statistics is estimated. The count statistics for cell is shown in Fig. 3a, while bead statistics can be found in Supplementary Note 6. To determine the flow-variant PSF, fluorescent beads are used that act as a point source. Subsequently, the PSF is used to deconvolve the recorded images. In the present study, three types of chip geometries are used, one with a straight single channel and the others with Y-type two and four channels. It can be inferred from cell flow data (see, Fig. 3a), the count doesn’t follows a linear increase with flow. However, bead flow data shows close to linear increase in the count with flow (see, Supplementary Note 6). This is due to many reasons including, cell accumulation at the corners of channels and the adhesive nature of the cell that tend to make cluster. In addition, low cell count can be attributed to its relatively large mass. It may be noted that we have used cell culture medium for flowing cells in the present study, and no additional chemicals (such as, Tripsine) are used in order to prevent clustering. Numerically, we got a maximum count of 1421 cells per minute at 2000 nl/min as compared to bead count for which a count of >2000 beads per minute is estimated. In addition, the system facilitated counting of live and dead cells as shown in Fig. 3b, c. This is possible due to the high-resolution high-throughput screening capability of M3IC system.

a Both the number of HeLa and HEK cells as counted by the M3IC system at varying flow-rates (500 - 2000 nl/min.) through the Y-type multichannel system. b, c The system is further configured to selectively count live, and dead cells passing the specimen flow system. The fluorescent beads used for characterizing flow in microfluidic channels and the corresponding count-rate is discussed in Supplementary Note 6. Raw data of cells flowing through two and four channels can be found in Supplementary Movies 3 and 2. Here, error bars represent the standard deviation. The statistical analysis is based on a total of 15 cells from 5 separate experiments.
Sectional Imaging and Volume Reconstruction
The quality of sectional images and the reconstructed cell volume are central to the newly developed M3IC system. Figure 4a shows the simultaneous interrogation capability of the system (considering all 4 channels) along with the ability to reconstruct cell volume in near real-time. The sectional images of the HeLa cells (spherical in shape during flow) displaying the distribution of organelle (mitochondria labelled with Mitotracker Orange) are recorded by the sCMOS camera during flow (at 1000 nl/min). Five high-quality planes of a sample HeLa cell (cell #1-5) are captured, which are then deconvolved using flow-variant PSF and stacked together to reconstruct the cell volume. A few cell samples along with 5 sectional cross-sections and reconstructed cell volumes are shown in Fig. 4b (cell 1-4). The distribution of mitochondria inside a live HeLa cell can be ascertained from the volume images. In addition, high resolution facilitates organelle count during flow (see, Fig. 4c). More details for both HEK and HeLa cells at varying concentration can be found in Supplementary Note 8. This is advantageous considering the availability of organelle-level statistics for an entire cell population, making this as a new feature in imaging flow-cytometry. The corresponding raw data of the cells passing through all the 4 channels can be found in Supplementary Movie 2. Overall, this demonstrates organelle-level resolution at high throughput of M3IC system.

a A snap-short of fluorescently-labelled cells with Mitrotracker Orange) flowing through the microfluidic channels as captured by the sensitive sCMOS camera. Several planes of the HeLa cells (indicated by white box) simultaneously passing through channels in the microfluidic chip are shown. b The corresponding cell volumes reconstructed from the sectional images are also displayed. c High resolution of M3IC system allowed organelle count during flow (flow-rates, \(\sim 500-2000\,nl\min\)). The raw experimental data is shown in the Supplementary Movie 2. More details related to image deconvolution along with volume reconstruction can be found in Supplementary Note 7 and 10. A slow-motion movie (frame-by-frame) of cells flowing through microfluidic channel and few cell volumes are shown in Supplementary Movie 4 and Supplementary Movie 5, respectively.
The developed M3IC system is compared with state-of-the-art point-illumination-based imaging cytometry system and confocal microscopy, both in terms of performance and estimation of biophysical parameters as shown in Table 1. The study confirms the fact that, although traditional imaging cytometry is 10 times faster but M3IC system has the distinct ability to reconstruct 3D volumes of an entire cell population. The system facilitates organelle count in a cell volume which is largely due to its sub-cellular resolution. Moreover, the proposed light sheet-powered system outperforms traditional imaging cytometry in terms of signal-to-background ratio (SBR). With the available computing power, the system is capable of reconstructing ~121 cell volumes per minute (at a flow-rate of 1000 nl/min). In addition, a comparison of M3IC and the existing light-sheet cytometry is provided in Table 2. All these advantages makes M3IC quite unique as far as diversity is concerned. Moreover, the availability of multiple light sheets, organelle-level resolution, and non-requirement of hydrodynamic focusing, and the real possibility of miniaturization (due to microfluidic chip-based specimen flow platform) makes M3IC as the future of imaging flow cytometry.
Multicolor imaging of organelles (Mitochondria and Lysosomes) in a HeLa cell
The ergonomic design of the M3IC system allows easy adaptation for multicolor imaging. This is a great advantage for studying multiple organelles and understanding their roles in a specific biological process. For enabling multicolor interrogation, the output fluorescence (emanating from organelles labeled with distinct fluorophores) is separated in the detection sub-system using a dichroic mirror. We chose to study two key organelles, mitochondria and lysosomes in HeLa Cells, which were labeled using mitotracker red FM and lysotracker (Thermofisher, USA), respectively. This is based on recent studies that indicate the roles of mitochondria and lysosomes in a variety of diseases and metabolic disorders, including cancer39. It may be noted that Mitotracker orange is used for multisheet single color imaging, whereas, Mito tracker red FM and Lysotracker green is used for multicolor imaging. For multicolor imaging, significant change in diffraction angles is not observed as the difference in the angle (for the first and second order splitting) between the combined beam was very small, specifically when the distance between the grating and beam-expander is small (≤10 mm). Consider a grating of pitch-distance (distance between the grooves) of d. Given mth order of diffraction for an incident light of wavelength λ, the angle of diffraction is given by, sinθm = mλ/d. In the small angle approximation, sinθm≈θm = mλ/d. For different wavelength (say, λ1 and λ2), the respective diffraction angles are given by, θ1 = mλ1/d and θ2 = mλ2/d, respectively. So, the difference between the angles is, Δθ = (θ1−θ2) = mΔλ/d. For our case, d = 100 μm/80 = 12.5 μm, m = 1 and Δλ = 532 nm−488 nm = 44 nm, giving, Δθ = 3.52 × 10−3 and Δθ = 7.04 × 10−3 for order 1 and order 2, respectively. Figure 5 shows the cross-sectional images of cells flowing at 1000 nl/min. The raw sectional images are filtered using the maximum likelihood technique, which is then correlated, corrected for lateral shift, and finally merged together to reconstruct multicolor cell volume (see Fig. 5). The process is repeated for multiple cells (at a rate of > 100 cells/min) flowing through all the channels in parallel. Cell volumes show the distribution of two different organelles (mitochondria and lysosomes) in HeLa cells during flow. The sectional images and volume of other cells (cell 1 and cell 2) is shown in Supplementary Note 9 and the corresponding 360o view of the cell volume is displayed in Supplementary Movie 9. These cell volumes indicate disruption of the mitochondrial network and its accumulation at the cell membrane. A similar effect is noted for lysosomes. The exact reason for disruption and accumulation is not known, and needs additional investigation and targeted studies. From the perspective of technological advancement in imaging cytometry, M3IC system with multicolor imaging capability at high throughput and organelle-level resolution paves the way for better investigation of an entire cell population.

The M3IC system allows imaging of more than one organelle during flow. The organelles (here, mitochondria and lysosome) labelled by two different dyes are flown through the microfluidic channel array. Then the signals are separated by using a dichroic mirror and focussed on to the camera. The images thus formed are correlated, shifted and merged together to generate the multicolor image of treated HeLa cells. These multicolor sectional images are then stacked to reconstruct the cell volume which is then subjected to further analysis. The corresponding sectional images and reconstructed volumes are shown in Supplementary Note 9 and Supplementary Movie 9, respectively.
Optical Screening of HEK and HeLa Cells (both Control and Treated)
To elucidate the potential of M3IC system for large-scale cell population studies, we have carried out drug treatment studies on cancerous HeLa cells and normal HEK cells. Live cells were passaged few times before proceeding with the drug treatment studies. The cells were incubated with Paclitaxel drug for 24 hrs and then washed with PBS. The cells were washed with PBS, paletted and resuspended in PBS for carrying out cytometry studies. To study the effect of drug concentration on the mitochondrial structure and ultimately cell viability, the concentration of drug is varied from low (75 nM) to high (5 μM). Subsequently, the cells were labelled with Mitotracker orange (λexci = 551 nm; λemi = 575 nm) using the developed protocol as discussed in methods section. Analysis shows that the mitochondria in HeLa is distributed throughout the cell volume at low drug concentration, whereas increase in concentration caused fragmentation of mitochondrial network that appear as loop. This is consistent with the confocal images of mitochondria distribution, both at low and high concentrations for HeLa cells (see, Supplementary Note 7). The looping effect in HeLa cells is further conformed using line intensity plots that exhibit U-shaped distribution (see, Fig. 6). This strengthens the fact that effect of Paclitaxel in disrupting mitochondrial network followed by its accumulation at the cell-membrane for HeLa cells (see, Fig. 6b). For comparison, control experiments were also performed on untreated cells that exhibit normal mitochondria distribution in a cell volume. To understand the effect of drug on normal cells, we have carried out treatment studies on HEK cells as shown in Fig. 6a. It is clear that the mitochondria is distributed throughout the cell when treated with varying concentration of Paclitaxal drug. Random multi-peak distribution is evident from intensity plots that indicate throughout distribution of mitochondria in the cell volume. This is further confirmed by the confocal studies. The control experiments on untreated HEK cells are also shown (see, Fig. 6a). Note that, the characteristic U-shaped distribution is missing for HEK cells (see, Fig. 6c). The distribution of mitochondria can be alternately evaluated by entropy metric, which is the measure of randomness. Figure 6d plot shows the degree of randomness in the cell volume. The entropy plot indicates fragmentation of mitochondria and its accumulation in a specific pattern (loop) at the inner cell membrane, whereas HEK cells display random distribution thereby increasing entropy. This illustrates the distribution of mitochondria in a cell for both healthy HEK and cancerous HeLa cells post treatment.

a Normal human embryonic kidney (HEK) cells treated with Paclitaxel at varying concentration ranging from low (75 nM) to high (5μM). M3IC images show mitochondria distributed throughout the cell cytoplasm. This is further confirmed by intensity plots (along white line across the cell) that suggests the presence of multiple peaks. The observation is further confirmed by confocal images of treated cells and the associated intensity plots. Corresponding images of control i.e, non-treatment cells are also shown. b M3IC and confocal images of cancerous HeLa cells at varying concentration of Paclitaxel (75 nm−5μm). Images suggest disrupted mitochondria and its accumulation at the inner cell membrane, forming a loop pattern. This is better demonstrated by well-like pattern in the intensity plots. Corresponding images for control cells are also shown indicating regular throughout distribution of organelles. c The intensity plots indicate a well-shaped pattern for treated HeLa cells whereas, healthy HEK cells show multi-peak pattern, suggesting the presence of mitochondria at the cell-membrane for HeLa cells. d The entropy based statistics suggests throughout random distribution of mitochondria for HEK cells at large drug concentration, whereas patterned distribution is noted for treated HeLa cells, suggesting loop pattern. The study related to organelle (mitochondria) count at varying concentration, and dynamic resolution at varying flow-rates can be found in Supplementary Note 8. Reconstructed cell volumes related to control (confocal and M3IC), and treated cells (at varying concentrations ranging from 75 nm to 5 μm) are shown in Supplementary Movies 6 and 7, respectively. e Mortality statistics suggests better viability for healthy HEK cells as compared to cancerious HeLa cells. f, g The statistics for live and dead cells (HEK and HeLa). Specifically, the effect of drug cause enhanced apoptosis of HeLa cells at high concentration (5 μM) of Paclitaxal drug, whereas HEK cells seems to inhibit apoptosis even at high concentration. For comparing more than two groups, we have used one-way ANOVA with the Kruskal-Wallis test. The corresponding statistical significance (P-values) is indicated as, ns p > 0.05, *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001. The statistical analysis is based on a total of 15 cells from 5 separate experiments for each group.
One of the key aspect of drug-treatment study is to determine the viability of cells post treatment. In the present study, both healthy and cancerous cells were exposed to Paclitaxel drug, and population study was carried out. Cell mortality statistics shows enhanced apoptotic nature of HeLa cells with increasing concentration. Specifically, a maximum of upto 80% cell mortality is observed at 5μM for HeLa cell, whereas healthy cells show ~11% cell apoptosis (see, Fig. 6e). The corresponding mortality statistics for live and dead cells (HEK and HeLa) is shown in Fig. 6f, g. This is in line with the observations reported in other studies40,41. We do see a small fraction of cells that represent neither and we classified them as ‘unknown’. We further note peripheral accumulation of mitochondria in apoptotic cells, whereas the distribution is throughout the cell cytoplasm for live cells. This is further evident from the reconstructed volumes for dead cells. Additional volumes of treated cells at low and high throughput is shown in Supplementary Movie 8. Overall, the proposed M3IC system has enabled organelle-level understanding of an entire cell population. This has facilitated better visualization of organelle distribution in several cell volumes during flow.
Conclusion & Discussion
The ability to image, count, analyze multiple organelles, and visualize cell volume with organelle-level resolution at high-throughput, all on a single platform is the need of future diagnostics. A newly developed multifunctional imaging cytometry system (M3IC) is demonstrated and successfully tested on cells. The system is primarily based on diffraction-limited VAMSA illumination PSF. The selectivity of the number of vertical light sheet and the adaptive change of inter-sheet distance makes the system versatile, and ensures optimal performance, both in terms of selectivity and resolution. VAMSA PSF has the distinct advantage of cross-sectional imaging and simultaneous interrogation of several cells in-parallel. In addition, a flow variant detection PSF ensures high-quality volume reconstruction with an increase in both SBR and resolution. The system enables rapid volume visualization at high throughput (>1400 cells per minute) which is a distinct feature of M3IC system. Studies on cancer cells reveal healthy count rate and instant volume reconstruction with a sub-cellular resolution. The effective spatial resolution after deconvolution is found to be 2.53μm, which is far from the resolution of confocal images (~0.95 μm). Moreover, M3IC has the potential to carry out a variety of studies. We anticipate the system to vastly advance the field of fluorescence microscopy, bioinstrumentation and optical physics.
At the heart of the developed M3IC system is the VAMSA PSF, which is versatile due to its ability to adaptively alter the number of light sheets (see, Fig. 2b and Supplementary Note 4). This is beneficial in applications that require imaging through multiple channels of variable shape, size and inter-channel distances, thereby making it commercially viable. However, one of the hurdle is PSF-broadening, which is predominantly due to motion-blur introduced by flow. This hampers the quality of sectional images recorded at high flow rate. To reduce the artifact, image deconvolution is employed. Specifically, M3IC system use flow-variant PSF for deconvolving raw sectional images and reconstructing cell volume (see, Supplementary Notes 5 and 7). It may be noted that the light sheets have different peaks/intensities, which is due to the optical properties of the grating (as specified by the manufacturer). This may cause SBR to vary in different locations while exciting with different sheets. To overcome this issue, we have used image processing techniques. The images are normalized, followed by deconvolution to improve the overall image quality and reduce the SBR variation (see Fig. S10 in Supplementary 10). In addition, we have used relatively low intensity for illumination to avoid photobleaching/saturation. To minimize the effect of rotational motion of flowing cells, a laminar or near-laminar flow is ensured during experimentation. However, the flow may become turbulent at flow rates > 3000 nl/min. Since the flow rate is maintained in the range 500−2000 nl/min, cell rotation is not observed. Moreover, the interaction time of the cell during its stay in the light sheet is minimal, which ensures that the effect due to rotational motion is minimal. During sample preparation, we calibrated the illumination intensity with the concentration (100 nM to 250 nM with a step of 25 nM) of MitoTracker Orange. Specifically, a concentration of 175 nM is found to be nominal (a good balance between a strong signal and minimal background noise) for all the light sheets. However, for > 225 nM, we noted a proportionate increase in both saturation effect and background. Overall, this did not affect the performance of M3IC system.
High-throughput is a key aspect of M3IC system. Given the additional requirement of high-quality volume imaging, it is imperative to quantify other parameters such as, live and dead cell count statistics. M3IC demonstrates a unique combination of high flow-rate (>1000 cells/min) and cell volume visualization (see, Fig. 4). Our results show consistent recording of 5 cell planes at a flow-rate of 1000 nl/min, and enabled near real-time volume reconstruction. Specifically, M3IC system allowed visualization of 121 cell volumes per minute in addition to organelle (mitochondria) count per cell, all at high flow rates (see, Figs. 3 and 4). The system allows easy adaptation to multicolor imaging where two different organelles of interest can be observed at high throughput. This is demonstrated by simultaneously imaging mitochondria and lysosomes in a HeLa cell (see, Fig. 5). All these factors give M3IC a distinct advantage, both in terms of adaptability and new features. We believe this as a remarkable achievement over the existing point-illumination based imaging cytometry systems since this integrates the capability of high-resolution microscopy, and imaging cytometry for large-scale population study.
To elucidate the potential of M3IC system, we carried out drug (Paclitaxel) treatment studies on cancerous HeLa cells (see, Fig. 6). Intensity plots and entropy measure show fragmentation of mitochondrial network followed by its accumulation at the cell membrane indicating cell-apoptpsis. A similar trend is observed in confocal studies. Surprisingly, this effect is minimal for healthy HEK cells. To ascertain the effect of Paclitaxel, cell viability tests were conducted on both cancerous and healthy cells (see, Fig. 6e–g). Using cytometry, we noted for the first time the effect of Paclitaxel on a large population of HeLa cells indicating cell death at large concentrations (~5 μM). Specifically, apoptosis is noted for a large fraction of HeLa cells (nearly 80%) as compared to healthy HEK cells (~11%) at moderate to large drug concentrations (see, Fig. 6e–g). Based on the respective cell volumes with organelle level resolution, M3IC can also be used for understanding underlying biological mechanism.
The availability of critical cell physiological parameters related to disease diagnosis (drug concentration study, high-throughput interrogation, volume visualization, organelle count, and live cell count.) on a single platform is the future of biomedical physics. Specifically, the system is able to count organelles (mitochondria) and access its distribution for varying drug concentration (see, Supplementary Note 7). This is in addition to regular features offered by existing flow cytometry systems. With the advantage of organelle-level resolution and high SBR (which is due to a key aspect associated with the selective nature of light sheet), and multicolor interrogation, the developed multifunctional cytometry system can be an asset for microscopy, optical imaging, clinical biology, and biomedical physics.
Materials & Methods
System design, optimization and automation
M3IC is an advanced flow-based imaging cytometry system comprises of three major sub-units: illumination, specimen flow platform and Detection.
Illumination Sub-system: The illumination (multi-sheet array) is at the heart of M3IC system that enables high-throughput optical sectioning of cells, simultaneously flowing through multiple microfluidic channels. This is achieved by a combination of transmission diffraction grating, beam-expander and high numerical aperture objective lens. The multi-sheet array illumination enables independent interrogation of cells flowing through individual microfluidic channel by a dedicated light sheet (see, Fig. 2a). In addition, the technique allows change of key parameters (light sheet size and inter-sheet spacing) as per microfluidic channel geometry/type. A laser (532nm Excel Laser, Quantum Lasers, UK) of wavelength 532nm having a beam width of 1.5mm is used as a source of light. A set of steering mirrors are employed for precision aligment in a 30 mm cage system (Thor Labs, USA). The beam is then passed to a transmission diffraction grating (80 grooves/mm, Edmund Optics, Singapore) that splits the beam into 5 intense beams (others higher-order beams are relatively weak). A beam expander (consists of two biconvex lenses of focal lengths, 25 mm and 50 mm) is employed to expand the beam by a factor of 2. Subsequently, all the 5 expanded beams are directed to the cylindrical lens to form an array of light sheets (a set of 5 light sheets). The expansion is carried out in order to fill the back-aperture of cylindrical lens (focal-length, 75 mm). Finally, a low numerical aperture objective lens (0.3NA, 10X, UPlanFL, Olympus, Japan) is placed at the focus of cylindrical lens to generate an array of diffraction-limited vertical light sheets (see, Fig. 1). The resultant multi-sheet array is generated at the working-distance (10 mm) of objective lens.
Specimen Flow Sub-system: The specimen (cells) to be investigated need to be flown through a transparent and flexible material. We choose to use PDMS for fabricating microfluidic based microchips. The process begins with the imprinting of a master-mold (containing Y-type patterns on a 4 inch silicon disc) that was carried-out in the dedicated Nanoscience Facility at Indian institute of Science, Bangalore, India (see, Supplementary Note 2). The design of the channel was made in clewin 4 software and saved in .gbr format. The master-mold is then used to fabricate PDMS chips.
Silicon elastomer and curing agent were mixed thoroughly using vortex or by spatula in a ratio of 10:1. Once mixed thoroughly, it is kept in a vacuum chamber to remove all the bubbles generated during mixing. After 2 hours, the mixture is poured on to the master-mold (silicon disc with imprinted channels) and left to dry at room temperature for 3 h. Thereafter, the chips were cut by a surgical-blade, and taken for plasma bonding with the cover-slip (No.1 thickness). The chip is baked at 60–90 degree for 2–5 min with coverslip side towards the heater so that coverslip is attached properly with the PDMS. Subsequently, it is punched to make inlet-outlet for the channels. Teflon Tubing of diameter 0.5 mm is used to connect the inlet with the reservoir (with a capacity of 0.5 μlit), and to the outlet with suction pump (New Era Flow Pump, Model No: NE-1002X). The resultant chip (PDMS attached with coverslip) is then connected to a glass slide for fitting with the chip-holder (PDMS chip Interface Starter Pack, Dolomite, UK). The PDMS holder is mounted on a XYZ-translator for necessary alignment with the multi-sheet array. The PDMS chip is placed at 45° with respect to both illumination and detection arm. The pump is controlled by the acquisition desktop computer by interface software. Needle of flow pump is attached to one end of the Microfluidic channel through the teflon tubing of diameter 0.5 mm to withdraw water from the channels. 1 ml syringe of diameter 4.80 mm is used to suck the medium containing cells.
Detection Sub-system: The detection is carried out at orthogonal to the illumination system or equivalently at 45° with respect to PDMS chip (see, Fig. 1). Orthogonal detection is a necessity to record the sectional views of specimens and has many advantages including, selectivity, better contrast, and better signal to background ratio. The fluorescence from the cells were collected by a detection objective. The detection system is a 4f optical system comprising of objective lens, tube lens and the sensitive sCMOS (Zyla 4.2, Oxford Instruments, Andor, UK). A set of optical filters (high-pass (550 nm Longpass, Thorlabs, USA) and notch filters (ZET532NF, Chroma, USA)) are used to reject the excitation light and background. In addition, we have employed an iris to remove non-parallel rays that cause spherical aberration. A fast sCMOS camera of high quantum efficiency (Zyla 4.2 plus, Andor, UK) is used to record the sectional images.
Optimization : Calibration and optimisation of the system is critical for obtaining quality images of the flowing specimens through multiple channels. This include determination of illumination and detection PSF. The illumination PSF is calibrated by placing a camera in the illumination arm at the working distance of the objective (10X olympus) with the translator to record the light field for both lateral (XY) and axial (Z) axis. The images are taken at regular intervals of 10μm. Approximately 390 planes were taken at varying z-positions and stacked together to reconstruct the 3D field (see Fig. 2a). Special care is taken to align the light sheet array with the microfluidic channels (here, 4 channels). Accordingly, the FOV for each light sheet in the multi-sheet array is ~211 μm. Here, 170μm is the dimension of each light sheet along the direction of light propagation (here, z-axis). The FOV for the detection sub-system is calculated to be 2.65 mm. Specifically, the FOV, SBR and resolution of the complete system are determined to be, 1 mm × 170 μm (211μm × 170 μm for individual light sheets), 4.8 dB (post deconvolution) and 2.5 μm, respectively.
A high throughput is ensured by counting the number of cells flowing per minute through all the channels. We have varied the count from 620 to 1470 (considering all the channels). Here we targeted a maximum of 4 channels for simultaneous volume visualization and recording. We have flown the specimen (HeLa cells in medium) at varying flow-rates (100−2000 nanolits/min) and matched that with appropriate camera frame-rates during data collection. At each flow-rate which is controlled by the programmable syringe pump, the sCMOS camera is synchronized to collect data in tandem. For appropriate location matching of the light sheets with microfluidic channels, the distance between the diffraction grating and the beam-expander system is varied by placing the grating on a translating stage.
Sample Preparation
Beads Sample Preparation: Fluoro-spheres carboxylated modified microspheres (size ~1 μm, Invitrogen, USA) are used to calibrate the system. The bead is excitable at 532 nm for which the emission occurs at 575 nm. The bead stock solution is mixed with 1 ml of milli-Q distilled water and mixed thoroughly by vortexing. Diluted bead solution is loaded in the reservoir and flown at varying flow-rate to calibrate the M3IC system and also to determine the flow variant PSF.
HeLa and HEK Cell Culture: Hela and HEK (Human Embryonic Kidney) cells of Passage 5 and 14 respectively are obtained from our collaborator, Prof. Annapoorni Rangarajan, MRDG, Indian Institute of Science in a frozen form. The cells are thawn from –80 degree and centrifuged to get the palette and remove the DMSO. The palette was resuspended in 1 ml Culture media (DMEM, Gibco, Thermo Fisher Scientific (89%) + FBS, Gibco, Thermo Fisher Scientific (10%) + Antibiotic, Gibco, Thermo Fisher Scientific (1%)) and seeded in T25 flask (2 ml of culture media and 1 ml of resuspended cells media). The cells are incubated for 24 hr. in 37 degree + 5% CO2. After 24 h, the cells are washed with PBS and kept for 48 h in incubator. The cells were split and cultured for 2–3 passages before going for the experiment. Cells are seeded in 35 mm dish for a count of 105 cells per dish using Hemocytometer.
Fluorescence Labelling of Cells: Mitotracker orange is used for labelling Mitochondria. Mitotracker is a dye, it reacts with thiol groups of cysteine residues and get accumulates in the mitochondrial Matrix. It diffuses across the plasma membranes passively. Mitotracker orange is bought in a Powder form from Invitrogen (Catalog-M7510). Powder Form is diluted in DMSO to make stock concentration of 1mM. Accordingly, it is diluted to a concentration of 1mM, and further diluted in DMEM to make working concentration of 175 nM. The working stock is then used for the experiments.
Multi-organelle Labelling of HeLa Cells: Cells were seeded (105 cells) in T25 flasks 24 h before mitochondrial and lysosomal labelling. Mitotracker red FM and Lysotracker green DND-26 (purchased from Invitrogen) of 1mM concentration in DMSO was used as stock. Subsequently, 6 different working concentration of mitotracker red FM and Lysotracker green DND-26 (50, 100, 150, 200, 250, 500 nM) in DMEM was tested to find the optimum concentration with respect to the density of HeLa cell, staining efficiency and quantum yield in imaging flow cytometry. Cell with 70% confluency was taken and washed with PBS to remove cell debris. It was incubated with Mitotracker Red FM of 150 nM concentration and kept for 30 min in the incubator with 5% CO2 and 37 °C to lable mitochondrial network thereafter cells were washed with PBS and again incubated with 100 nM of Lysotracker green DND-26 and kept for 30 min in the incubator with 5% CO2 and 37 °C to lable Lysosomes. After incubation, cells were trypsinized to detach them from disc-surface and the cells were centrifuged at 2000 RPM for 5 min. Subsequently, the cells were resuspended in PBS and prepared for experiments. The cells were loaded in the sample reservoir and flown through microfluidic chip for cytometry.
Drug Preparation: The drug, Paclitaxel is bought from Sigma Aldrich in powder form (T7402). Paclitaxel (molecular Weight 853.91) is diluted in DMSO to make the final concentration of 1 M. 1 molar Paclitaxel stock solution is diluted down to the required concentration of 75, 250, 500, 1000, 5000 nM. After 24 hr of seeding (confluency is around 70%) the cells were treated with paclitaxel at different concentration and the control cells are culture in the cell media. After 24 hr post drug treatment, cells were washed 2–3 times with PBS and were stained with Mitotracker orange and then incubated for 1.15 hr at 37 degree 5% CO2. Cells were trypsinized using trypsin and then centrifused at 1400 rpm to get pallets, which is further resuspended in PBS and taken for the flow cytometry experiments.
Image Processing
Images were captured using Andor sCMOS zyla 4.2 camera (pixel size, 6.5 μm). The images are captured in .sif format in Andor software compatible with zyla 4.2 camera. Using Andor software, the captured images are saved in tiff format (batch conversion) as image sequences. The exposure time and the flow rates are adjusted such that we get a minimum of 5 cross-sections of a single HeLa cells for volume reconstruction1.
Flow-variant PSF: Beads are flown through the channels and captured at different flow rates. The generalized bivariant Gaussian is fitted using a written script in MATLAB and the PSF at different flow rates are calculated. The central lobe of the airy-disc pattern is approximated as bivariate Gaussian function. The beads appear elongated with the flow, termed as Flow variate PSF. The size of PSF at different flow rate (ranging from 100 to 2000 nl/min) is calculated as shown in Fig. 2. The Flow variate PSF is used to remove motion-blur induced by flow using deconvolution operation. For counting cells and beads, we have used object-based tracking algorithm using kalman filter, written in Matlab. It is observed increased flow rate linearly increases the PSF as shown in Fig. 2. Beads counts increases from few hundred to few thousands as the flow rate increases by an order.
Cell Imaging: HeLa and HEK cells are loaded in the reservoir and using the pump in the suction mode it is flown through microfluidic channels. The images of stained Cells are captured using sCMOS camera at a different flow rate (500, 1000, 1500, 2000 nl/min). The cells are counted at different flow rates. It is noted that the increase in the number of cell with respect to flow rate is not linear, which is predominantly due to the fact that cells get clumped together at lower flow rates. Raw images captured by sCMOS are deconvolved using flow variate PSF. These deconvolved sectional images are then stacked together using MATLAB. The stack imaged is then combined to form a volume. The intensity-based thresholding is used while reconstructing the volume, so that the noise is eliminated from the volume.
Image Deconvolution : Images captured by M3IC system are deconvolved using a maximum likelihood algorithm using flow-variant PSF. In short, the goal of maximum likelihood estimation is to determine the parameters for which the joint probability (likelihood function, L(g∣ f )) of the observed data has the highest probability42,43 i.e,
where, g is the recorded image of the object ‘o’ flowing through the microfluidic channel with A is the convolution operator expressed as, Af = h*f. Maximization of the likelihood function fives rise to the following iterative equation:
where, we have ignored the normalizing constant.
Maximum likelihood algorithm are standard operation im Matlab and the same is used to determine the maximum likelihood estimate of the recorded sectional images of HeLa and HEK cells. Subsequently, the images are stacked together to reconstruct the 3D volume of flowing cells.
Confocal fluorescence microscopy studies
A Zeiss LSM 510 inverted microscope (Indian Institute of science, Bio Facility) was used to compare M3IC with the conventional microscope. The microscope was operated at room temperature using objective lenses, 10X, 0.40 NA and 20X, 0.70NA. The cells, prepared using the protocol mentioned in sample preparation, was trypsinated and kept in live imaging dish using pipette and was let down to settle for few minutes. Once the cells are static, images were taken. The cells were illuminated by 514 nm laser (argon multiline)and the emission fluorescence signal was captured beyond 550 nm by using long pass filter (spectral filter) using a 1 airy unit pinhole.
Multisheet and multicolor detection
The detection system is specially designed to record images from two different organelles (labeled with two different dyes) in cells flowing through all four channels in parallel. The multicolor detection is achieved in a 4f optical configuration comprising two lenses (objective and tube lenses), a set of mirrors, and optical filters. The mirrors are arranged so that the distance between the tube lens and the detector is the same for both the optical arms (see Supplementary 9, Fig. S9-1). A linear translator is used in one of the arms to ensure equal distance (i.e, focal length of tube lens) for both optical arms. On each optical arm, specific dichroic mirror (cut-off wavelength = 600 nm, Edmund Optics, Singapore) and filters (488 Notch and 532 Notch filters) are placed to ensure separation of fluorescence (from lysotracker green DND-26 labeling Lysosomes and MitoTracker red FM labeling mitochondria) to pass through and reach the detector (Andor sCMOS Zyla 4.2 camera, Orford Instruments, UK). It is ensured that both the images are formed on different regions of the sCMOS detector chip (pixel size = 6.5 μm × 6.5 μm, chip size in pixels = 2048 × 2048, Quantum Efficiency = 0.82). Two additional band-pass filters (641/80 nm, OD = 6.0, Edmund Optics, Singapore, and 525/50 nm, Chroma, USA) are introduced in the optical arm to reduce background. This ensures parity with respect to intensity fluctuations, contrast and signal-to-noise ratio. Such an optical arrangement in a 4f system with integrated dual-channel detection (two optical arms) ensures high throughput. After capturing the image data, the images from respective channels are separated, correlated, shifted, and merged to obtain a multicolor image/volume. Each image obtained from the respective microfluidic channel is processed separately. Since 5 planes of a single cell are captured, they are stacked together to reconstruct cell volume. This ensures near real-time multicolor volume imaging on the go.
Correlation and calibration
For multicolor imaging, the correlation of both red and green channel images is critical while merging to reconstruct multicolor image/volume. The process is initiated by separating the fluorescence based on their distinct wavelengths using a dichroic mirror. To determine the shifts, fluorescent beads (FluoSpheres Carboxylate-Modified Microspheres, 1.0μm, Invitrogen, USA) with broad emission spectra (550−700 nm) are flown through the microfluidic channel. This ensures that the beads are visible in both the channels. Upon recording the images, they are subjected to correlation analysis using Matlab built-in scripts. For our case, we observed a shift of ΔX = 23.46 μm and ΔY = 3.45 μm, with a correlation factor of 0.83. Subsequently, the sectional images of flowing cells were subjected to respective shifts, normalized, deconvolved, and overlapped to obtain multicolor sectional images. The images are then stacked to reconstruct cell volume.
Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
Code availability
All MATLAB codes generated for the current study are available from the corresponding author on reasonable request.
References
Shapiro, H. M. et al. Combined blood cell counting and classification with fluorochrome stains and flow instrumentation. J. Histochem. Cytochem. 24, 396–401 (1976).
Reinherz, E. L., Kung, P. C., Goldstein, G. & Schlossman, S. F. Separation of functional subsets of human T cells by a monoclonal antibody. Proc. Natl Acad. Sci. USA 76, 4061–4065 (1979).
Gray, J. W. et al. High-speed chromosome sorting. Science 238, 323–329 (1987).
Grandi, F. C. et al. Single-cell mass cytometry reveals cross-talk between inflammation-dampening and inflammation-amplifying cells in osteoarthritic cartilage. Sci. Adv. 6, eaay5352 (2020).
Basiji, D. & Gorman, M. R. G. O. Imaging flow cytometry. J. Immunol. Meth. 423, 1–2 (2015).
Cheng, S. et al. Single-cell cytometry via multiplexed fluorescence prediction by label-free reflectance microscopy. Sci. Adv. 7, eabe0431 (2021).
Cohen, A. et al. Mechanism and reversal of drug-induced nephrotoxicity on a chip. Sci. Trans. Med. 13, eabd6299 (2021).
Porichis, F. High-throughput detection of miRNAs and gene-specific mRNA at the single-cell level by flow cytometry. Nat. Commun. 5, 5641 (2014).
Raju, R., Kavya, M. & Partha, P. Mondal, light sheet based imaging flow cytometry on a microfluidic platform. Micros. Res. Tech. 76, 1101–1107 (2013).
Raju, R., Kavya, M. & Mondal, P. P. High resolution light-sheet based high-throughput imaging cytometry system enables visualization of intra-cellular organelles. AIP Adv. 4, 097125 (2014).
Lau, A. K. S., Shum, H. C., Wong, K. K. Y. & Tsia, K. K. Optofluidic time-stretch imaging: An emerging tool for high-throughput imaging flow cytometry. Lab Chip 16, 1743–1756 (2016).
Miura, T. et al. On-chip light-sheet fluorescence imaging flow cytometry at a high flow speed of 1 m/s. Biomed. Opt. Express 9, 3424–3433 (2018).
Collier, B. B., Awasthi, S., Lieu, D. K. & Chan, J. W. Non-linear optical flow cytometry using a scanned, Bessel beam light-sheet. Sci. Rep. 5, 10751 (2015).
Mahjoubfar, A., Chen, C., Niazi, K. R., Rabizadeh, S. & Jalali, B. Label-free high-throughput cell screening in flow. Biomed. Opt. Exp. 4, 1618–1625 (2013).
Jiang, H. et al. Droplet-based light-sheet fluorescence microscopy for high-throughput sample preparation, 3-D imaging and quantitative analysis on a chip. Lab Chip 17, 2193–2197 (2017).
Chelur, R. K. et al. Integrated Light-sheet imaging and flow-based enquiry (iLIFE) system for 3D in-vivo imaging of multicellular organism. Appl. Phys. Lett. 111, 243702 (2017).
Bouchard, M. B. et al. Swept confocally-aligned planar excitation (SCAPE) microscopy for high-speed volumetric imaging of behaving organisms. Nat. Photon. 9, 113–119 (2015).
Kumar, P., Joshi, P., Basumatary, J. & Mondal, P. P. Light sheet-based volume flow cytometry (VFC) for rapid volume reconstruction and parameter estimation on the go. Sci. Rep. 12, 78 (2022).
Martin, C. et al. Line excitation array detection fluorescence microscopy at 0.8 million frames per second. Nat. Commun. 9, 4499 (2018).
Zhang, Z. et al. A high-throughput technique to map cell images to cell positions using a 3D imaging flow cytometer. PNAS 119, e2118068119 (2022).
Hiramatsu, K. et al. High-throughput label-free molecular fingerprinting flow cytometry. Sci. Adv. 5, eaau0241 (2019).
Han, Y., Gu, Y., Zhang, A. C. & Lo, Y. H. Review: Imaging technologies for flow cytometry. Lab. Chip 16, 4639–4647 (2016).
Chang, S., Serena, K., Karen, S. & Gyongyi, S. Impaired expression and function of toll-like receptor 7 in hepatitis C virus infection in human hepatoma cells. Hepatology 51, 35–42 (2010).
Maguire, O., Collins, C., OLoughlin, K., Miecznikowski, H. & Minderman, H. Quantifying nuclear p65 as a parameter for NF-B activation: correlation between ImageStream cytometry, microscopy, and Western blot. Cytometry A 79, 461–469 (2011).
Begum, J. et al. A method for evaluating the use of fluorescent dyes to track proliferation in cell lines by dye dilution. Cytometry A 83, 1085–1095 (2013).
Riordon, J. et al. Deep learning with microfluidics for biotechnology. Trends Biotechnol. 37, 310–324 (2019).
Isozaki, A. et al. AI on a chip. Lab Chip 17, 3074–3090 (2020).
Doan, M. et al. Carpenter Holger Hennig, diagnostic potential of imaging flow cytometry, trend. Biotech. 36, 649–652 (2018).
Lin, J.-R., Fallahi-Sichani, M. & Sorger, P. K. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat Commun. 6, 8390 (2015).
Lin, J.-R. et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife 7, e31657 (2018).
Goltsev, Y. et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174, 968–81 (2018).
Bodenmiller, B. Multiplexed epitope-based tissue imaging for discovery and healthcare applications. Cell Syst. 2, 225–38 (2016).
Heath, J. R., Ribas, A. & Mischel, P. S. Single-cell analysis tools for drug discovery and development. Nat. Rev. Drug Discov. 15, 204–216 (2016).
Sommer, C. & Gerlich, D. W. Machine learning in cell biology–teaching computers to recognize phenotypes. J. Cell Sci. 126, 1–11 (2013).
Caicedo, J. et al. Data-analysis strategies for image-based cell profiling. Nat. Methods 14, 849–863 (2017).
Mondal, P. P. A perspective on light sheet microscopy and imaging: Applications across the breadth of applied physics and biophysics. Appl. Phys. Lett. 119, 160502 (2021).
Mondal, P. P. Book: Light sheet microscopy and imaging, AIP Publishing, (2021).
Stelzer, Ernst,H. K. et al. Light sheet fluorescence microscopy. Nat. Rev. Meth. Prim. 1, 73 (2021).
Bonam, S. R., Wang, F. & Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Dis. 18, 923–948 (2019).
Peng, X. et al. Autophagy promotes paclitaxel resistance of cervical cancer cells: involvement of Warburg effect activated hypoxia-induced factor 1-a-mediated signaling. Cell Death Dis. 5, e1367 (2014).
Young Chi, E. et al. Tae Churl Park. Obstet. Gynecol. Sci. 56, 84–92 (2013).
Mondal, P. P., Vicidomini, G. & Diaspro, A. Markov random field aided Bayesian approach for image reconstruction in confocal microscopy. J. Appl. Phys. 102, 044701 (2007).
Mondal, P. P., Vicidomini, G. & Diaspro, A. Image reconstruction for multi-photon fluorescence microscopy. Appl. Phys. Lett. 92, 103902 (2008).
Sandison, D. R. and Webb, W. W. Background rejection and signal-to-noise optimization in confocal and alternative fluorescence microscopes, Appl. Opt. 33, 4, 603–615 (1994).
Bortolomeazzi, M. et al. A SIMPLI (Single-cell Identification from MultiPLexed images) approach for spatially-resolved tissue phenotyping at single-cell resolution. Nat. Commun. 13, 781 (2022).
Acknowledgements
The authors acknowledge financial support from parent institute (Indian Institute of Science, Bangalore, India), SERB (Department of Science and Technology, India), and Department of Biotechnology, India. The authors thank Abhishek Kumar (Marine Biological Labs, Woods-Hole, MA, USA) and Subhra Mondal (University of Nebraska - Lincoln, USA) for rigorously going through the manuscript and providing valuable inputs.
Author information
Authors and Affiliations
Contributions
P.P.M. conceived the idea. P.J., P.K., A.S. and P.P.M. carried out the experiment. P.J., A.S., P.K. and J.M.V. prepared the samples. J.M.V., P.J. and P.P.M. discussed the result and proposed the mechanism. P.P.M. wrote the paper by taking inputs from all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Physics thanks Peng Fei and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Joshi, P., Kumar, P., S, A. et al. Fluorescence-based multifunctional light sheet imaging flow cytometry for high-throughput optical interrogation of live cells. Commun Phys 7, 25 (2024). https://doi.org/10.1038/s42005-024-01522-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42005-024-01522-y
- Springer Nature Limited
This article is cited by
-
Thin light sheets image cells at organelle level
Nature India (2024)