
Abstract
Research on prebiotic chemistry and the origins of nucleic acids and proteins has traditionally been focussed on only one or the other. However, if nucleotides and amino acids co-existed on the early Earth, their mutual interactions and reactivity should be considered explicitly. Here we set out to investigate nucleotide/nucleoside formation by simple dehydration reactions of constituent building blocks (sugar, phosphate, and nucleobase) in the presence of different amino acids. We demonstrate the simultaneous formation of glycosidic bonds between ribose, purines, and pyrimidines under mild conditions without catalysts or activated reagents, as well as nucleobase exchange, in addition to the simultaneous formation of nucleotide and nucleoside isomers from several nucleobases. Clear differences in the distribution of glycosylation products are observed when glycine is present. This work demonstrates that reaction networks of nucleotides and amino acids should be considered when exploring the emergence of catalytic networks in the context of molecular evolution.
Similar content being viewed by others


Introduction
Nucleotides and amino acids are vital building blocks in biology1, but although nature has mastered their synthesis and polymerisation2,3, their synthesis under mild, prebiotically plausible and simple conditions, without activation, in the laboratory is challenging4. In this context, the uncatalysed synthesis of nucleosides and nucleotides from their precursors has been widely investigated. For instance, Orgel and co-workers synthesised small amounts of adenosine and guanosine nucleosides in separate reactions via the dehydration reaction of ribose together with the corresponding purine base, adenine or guanine, in the presence of inorganic polyphosphate salts5. They also showed that nucleoside yields were improved by the addition of salts to the reaction solution, which can help drive dehydration reactions. However, the synthesis of canonical pyrimidine cytosine, thymine, and uracil nucleosides is difficult, and they have not been synthesised directly from the base and ribose. Without careful control, the inherent reactivity of the different amine groups leads to non-canonical nucleosides being the preferred products. Currently, the state-of-the-art synthesis requires the stepwise addition of the components reacting under unique conditions, as well as a final photoanomerisation step6,7,8. Non-canonical nucleoside/nucleotide formation from nucleobase analogues and (5′-phosphorylated) ribose has been successfully achieved9,10. This is interesting as the synthesis presents a possibility for alternative pathways to genetic polymers during a “pre-RNA world”, but it would be interesting to consider how amino acids and peptides might feature in such a world. This is because the formation of peptides has been found to be possible under mild conditions via simple hydration-dehydration cycles by heating solutions of amino acids to >90 °C11,12,13,14,15,16,17,18,19,20. Recently, a mixture of amino acids and ribonucleotides in the presence of an activating agent (i.e. carbodiimide, ethylimidazole, or magnesium chloride) has been shown to lead to the formation of mixed polymers of nucleotides and amino acids20, 21,22 in addition to the formation of oligo-dipeptide backbones using thioester derivatives as mediators23.
Here, we study the co-reactivity of amino acids and nucleotide building blocks under simple dehydration conditions (90 °C for 5 h) (Supplementary Figs. 1, 2). Through simple one-pot dehydration reaction of an aqueous mixture (pH 2.5) containing nucleotide building blocks (ribose, phosphate and nucleobase) without additional activated or catalytic agents, simultaneous formation of nucleotide and nucleoside isomer structures from both purines and pyrimidines are obtained. We also observe an exchange of nucleobases within and between nucleoside and nucleotide compounds, indicating a dynamic environment of early-forming nucleic acid monomers (Fig. 1a). Furthermore, we observe a clear isomeric selection on glycosylation products when we incorporate amino acids into our reactions (Fig. 1b). This indicates that amino acids may have had the capacity to direct the chemistry of prebiotic nucleoside/nucleotide synthesis, further supporting the hypothesis of nucleic acid/amino acid coevolution. Our combined results suggest complex, possibly indirect pathways to a stable reservoir of nucleic acid monomers, which were almost certainly subject to dynamic isomeric exchange and interaction with neighbouring small molecules.

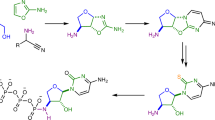
Schematic abstract. Scheme showing a the network of reactions involving the simultaneous one-pot formation of glycosylation products (nucleotide, cyclic nucleotide and nucleosides) with three nucleobases (adenine, cytosine and guanine), and b the isomeric effect of glycine on the isomeric distribution of glycosylated structures. Arrows indicate preferred positions of reactivity. All reactions were carried out in equimolar conditions and heated at 90 °C for 5 h (pH = 2.5)
Results
Simultaneous formation of glycosylation products
The formation of nucleoside and nucleotide structures from simple precursors was achieved simultaneously with three nucleobases by dehydration reactions. This is in contrast to previous work in this field, where conditions were optimised to favour specific products24,25,26. In a typical glycosylation experiment, adenine and P-ribose were heated at 90 °C for 5 h at pH 2.5. We observed the formation of adenine monophosphate (AMP) nucleotide (and its isomers) when adenine and P-ribose were combined. The extracted ion chromatograms (EICs) obtained from HPLC-MS analysis of the products revealed several peaks with m/z matching [AMP + H]+ and [2(AMP) + H]+ (Supplementary Figs. 13–15). Comparision to standards confirmed that the AMP corresponds to the peak found at RT = 4.2 min (Supplementary Figs. 5, 6); other major peaks might be consistent with AMP isomers, such as the N6-ribosylated isomer. In order to confirm this, a hydrolysis reaction in the presence of NaOH (0.1 M) was performed, in an attempt to differentiate between the different isomers, considering that the N6-ribosylated isomer is more prone to hydrolysis. However, a slight disturbance in the peaks can be observed, which makes it difficult to confirm the identity of N6-ribosylated isomer. We then analysed two pure standards of two different isomers (adenosine 3′- monophosphate and adenosine 5′-monophosphate monohydrate) individually and in a mixture in order to elucidate the elution profile of the isomers (Supplementary Figs. 148, 149). A shift in the retention time can be observed for the standard mixture providing a better correlation with the elution of these compounds in the real sample. Although it is difficult to differentiate between isomers, we can confirm the presence of at least two isomers within the AMP products by comparing the elution profile of the standard mixture to the real sample. It is now clear that the formation of different isomers (Supplementary Fig. 12) is a possible reason for eluting nucleotides with the same masses at different retention times. Furthermore, MS/MS analysis of our reaction products reveals fragmentation of AMP (and its isomers) to adenine (m/z = 136.0617 ± 0.01) (Supplementary Fig. 20); this is consistent with fragmentation of the canonical AMP standard. Our proposed reaction mechanism consists of the formation of a glycosidic bond between a 1′-OH group of ribose and an amino group of adenine (see Supplementary Fig. 12).
Time-course reactions show that water evaporation is the main driving force for glycosylation products formation, showing a significant increase in the formation of nucleoside and nucleotide structures in the time period between 2 and 4 h (Fig. 2a). This corresponds to the time range when the sample volume is drastically decreased, and reagents are extremely concentrated. After 5–6 h of reaction, the sample reaches dryness and the rate of reaction (followed by intensity in HPLC-MS measurements) stabilises. In addition to AMP, glycosidic bond-containing products, including cyclic nucleotides (i.e. cAMP)27, and nucleosides (i.e. adenosine), were detected using RP-HPLC-MS, MS/MS, and tested for 1,N6-etheno derivative formation28,29, confirmed by comparison to standards (Fig. 2, Supplementary Figs. 7–11, Supplementary Figs. 23–27). The retention times of 2′,3′-cAMP and 3′,5′-cAMP canonical standards did not correspond with the retention times of the EIC peaks in the sample. However, the mass distributions ([cAMP + H]+; [2cAMP + H]+) and fragmentation patterns ([adenine + H]+) were identical (Supplementary Figs. 16–21). These results show that while cyclic structures are formed, canonical cAMP is not the main product. Comparison to an adenosine analytical standard also shows that adenosine, together with a number of isomeric species, is formed in the condensation reaction of P-ribose and adenine (Supplementary Figs. 18–22). While the canonical forms of AMP and adenosine were confirmed in these experiments, they were not the main products in the dehydration reaction.

Adenine nucleotide products. Time course of a reaction of P-ribose with adenine at 90 °C; products were analysed by RP-HPLC-MS. a Representation of the total peak areas in the Extracted Ion Chromatogram (EIC) for the masses of adenine glycosylation products. b EICs for adenine glycosylation products over time. Each data point represents the average of three replicates ± standard deviation
When phosphate was supplied separately as pyrophosphate and reacted with adenine and ribose, a compound with the mass of adenosine was detected and a low amount of AMP and cAMP were detected, and adenosine still formed when no phosphate source was present (Supplementary Figs. 33–41). It worth noting that we have intentionally focussed our studies on identifying phosphorylated products (AMP isomers and cAMP isomers) and paid less attention to identifying phosphorylated products that are not AMP isomers and cAMP isomers30,31,32. We propose the formation of a glycosidic bond between the hydroxyl group of ribose and an amino group of adenine, which is triggered by the loss of a water molecule by evaporation. The relative reactivity of the primary and secondary amine groups in adenine is well studied33 and without activation or the presence of a protecting group, glycosidic linkage at the primary amine is normally preferred. Therefore, the canonical isomer of adenosine/AMP is not expected to be a major product, as it would require reaction exclusively at the secondary amine. However, the reactivity is sufficiently high at the secondary amine site for the canonical isomers to be formed, though not as the major product. In other nucleobases, such as guanine, there are even more accessible amine groups, and the potential for isomeric products is greater. Reactivity of ribose is likely to be predominantly through the anomeric position, leading to fewer possible isomers, though some other minor products may be observed.
The reactivity of other canonical nucleobases (cytosine, guanine and thymine) with P-ribose was also investigated. Masses corresponding to nucleoside and nucleotide structures were detected following the dehydration reaction of guanine and cytosine with P-ribose (Fig. 3 and Supplementary Figs. 50–64). Guanine glycosylation structures were formed to a relatively low extent, likely due to the limited solubility of guanine at low pH. The product quantities measured for 5-methyluridine monophosphate (m5UMP) and 5-methyluridine (thymine nucleoside) were even lower than their equivalents from guanine and cytosine (Fig. 3 and Supplementary Figs. 65, 66). These results can be explained by the presence of a primary amino group in guanine and cytosine, which is lacking in thymine. While all three nucleobases have secondary amine groups, these are less reactive in glycosidic bond formation. Hence, the formation of nucleoside and nucleotide structures through a secondary amine reaction, as is required to form canonical glycosylation products, is disfavoured in nucleobases where primary amines are available. This has an interesting implication for the adoption of nucleic acid chemistry in the origin of life, as it suggests that the canonical nucleotides may have been initially unsuitable until further biochemical machinery had emerged to enhance selectivity towards the correct isomers. Therefore, as expected, whilst canonical nucleotide and nucleoside products of cytosine and guanine were formed in our experiments, they did not correspond to the main peaks observed (Supplementary Figs. 42–49). By combining these two observations (different retention times but same mass distribution as canonical standards in the EICs), we can conclude that the nucleotide and nucleoside species formed from the dehydration reaction of guanine/cytosine with P-ribose were mainly isomeric species of the canonical nucleotides and nucleosides (some possible structures are shown in Supplementary Figs. 50, 51).

Other products from P-ribose and nucleobase. All aqueous reaction mixtures consisting of 25 mM P-ribose + 25 mM nucleobase were heated at 90 °C for 5 h, and products were analysed by RP-HPLC-MS. Representation of the total peak areas of the EIC for the masses of cytosine, guanine and thymine glycosylation products
Typically, the formation of nucleotide structures has been performed under specific conditions depending on the nucleobase used, targeting a specific reaction product. In an alternative approach, we decided to include multiple nucleobases simultaneously in the reaction with P-ribose. Our aim was to determine whether product formation with multiple nucleobases under the same reaction conditions would yield a mixture of products, or be dominated by one. This reaction was carried out by including two or three nucleobases (adenine, guanine and cytosine) simultaneously in the reaction vessel, together with P-ribose. A mixture of glycosylation products was obtained, comprising nucleotides (AMP, GMP and CMP) as well as the respective cyclic nucleotide (cAMP, cGMP and cCMP) and nucleoside products (adenosine, guanosine and cytidine) (Supplementary Figs. 67–95). Guanine glycosylation products were formed in a lower yield than those of adenine and cytosine, as expected due to the low solubility of guanine under acidic conditions.
Nucleobase exchange
Nucleobase exchange was observed when Na+AMP was heated for 5 h at 90 °C in acidic aqueous media with cytosine or guanine. Nucleobase exchange resulted in the formation of nucleotide (CMP or GMP), cyclic nucleotide (cCMP or cGMP) and nucleoside (cytidine or guanosine) structures (see Supplementary Figs. 96–102, and Supplementary Table 1 for semi-quantitative yields). In this experiment, CMP and cytidine showed an increasing trend in intensity, whereas cCMP reached a maximum intensity when [cytidine] was 12.5 mM and then dropped until an intensity of 4.0 × 104 AU (Supplementary Figs. 102a and 155–158). At a cytosine concentration of 37.5 mM, the compound with the higher intensity was found to be CMP and the intensities measured for cCMP and cytidine were almost the same. Two main isomeric species were observed in the EICs of CMP and cCMP. [cytosine + H]+, [CMP + H]+ and [2CMP + H]+ masses were detected in the case of CMP, while cCMP showed [cytosine + H]+, [cCMP + H]+ and [2cCMP + H]+ within their respective mass distributions. In the case of cytidine, [cytidine + H]+ was the main detected peak. These mass distributions matched that observed in the standards. However, the retention time of the main isomers did not correspond with the canonical CMP and cytidine. When AMP was reacted with increasing concentrations of guanine (Supplementary Fig. 102b), all three glycosylation products (GMP, cGMP and guanosine) reached a maximum value when the concentration of guanine was 2.5 mM (Supplementary Figs. 160–162). These results were the consequence of the limited guanine solubility at acidic pH, thus, even if more guanine was added to the reaction vessel, the effective concentration in the solution was the same. Following the maximum value, the intensity of cGMP and guanosine were constant, due to poor solubility of guanine. Only a small increase in the intensity of all three guanine glycosylation products was observed when [guanine] = 37.5 mM, which might be related to a larger presence of guanine in the solution due to the high concentration added. In the EIC of GMP, a broad area without well-defined peaks was observed, although two main peaks stood out. [GMP + H]+ was the only chemical species, related to guanine compounds, detected in the mass distribution on its EIC. Four peaks, grouped in pairs, were observed when the EIC of cGMP was extracted from the MS data and the mass distribution showed [cGMP + H]+ and [2cGMP + H]+ species. On the other hand, the EIC of guanosine presented three peaks, the most intense of which showed the presence of [guanosine + H]+ in its mass distribution.
The formation of cytosine and guanine glycosylation products demonstrated that cleavage of the AMP glycosidic bond occurred under our reaction conditions. When the EICs of AMP, cAMP and adenosine were analysed, several peaks were observed in each chromatogram, supporting the theory that glycosidic bonds undergo dynamic hydrolysis/formation during the dehydration reaction34. The reactions of cytosine and guanine nucleotides with adenine were also investigated. In the case of the dehydration reaction of CMP and adenine, no products corresponding to nucleobase exchange could be observed (Supplementary Figs. 103–108/a). However, adenine glycosylation products were clearly detected in the reaction of GMP with adenine (Supplementary Figs. 105–108/b). This is because the hydrolysis of glycosidic bonds in GMP is more facile than for CMP, under the same reaction conditions34. We have also carried out the nucleobase exchange reaction with UMP and adenine, in order to compare with the direct pyrimidine analogue of adenine (Supplementary Fig. 109). Results were more similar to the CMP yields shown in Supplementary Fig. 108, than with the GMP yields, obtaining a very low yield for the adenine glycosylation products in the UMP reaction which is not sufficient to enable detection of AMP and cAMP.
Amino acids effect on glycosylation products distribution
As previously mentioned, amino acids, nucleotides and their building blocks could have been present on the early Earth at the same time. Therefore, products of a co-polymerisation reaction, or even products resulting from some catalytic effect of one type of polymer over the other, could have occurred under a prebiotic environment. To study the co-reactivity of nucleotide building blocks and amino acids in a one-pot dehydration, glycine, the simplest amino acid, was included in the dehydration reactions of P-ribose and respective nucleobases. The incorporation of glycine had a clear effect on the formation of glycosylation products, causing the overall yield of products with the mass of AMP isomers, cAMP isomers and adenosine (Fig. 4a and Supplementary Figs. 28–32) to decrease. This indicates that glycine plays a role in either consuming nucleotide building blocks (P-ribose and/or adenine) through a side reaction, or that it becomes attached to the product structure, changing its mass. EIC analysis for these reactions reveals peaks corresponding to the mass of glycine adducts (i.e. AMP-Gly, cAMP-Gly, adenosine-Gly and adenine-Gly) (Supplementary Figs. 115–122), though these side products are not formed in sufficient quantity to account for all the changes observed. Glycine adducts were also confirmed by using deuterated glycine as starting material together with P-ribose and adenine, which caused changes in the isotopic distribution of the adduct masses (Supplementary Figs. 123, 124). A maximum semi-quantitative yield of 59% was obtained for the formation of glycosylation products (AMP isomers, cyclic AMP isomers and adenosine) in the reaction of P-ribose and adenine; however, only 46% yield was obtained when glycine was also present in the reaction medium (Supplementary Fig. 144). Quantifying the yields for all possible individual isomers is technically difficult, but semi-quantitative yields of some isomers could be determined using pure standards: Adenosine 5′-monophosphate and adenosine 2′,3′-cyclic monophosphate were found at 38.7% and 18.2%, respectively, in the absence of glycine, while the yield in the presence of glycine was found to be significantly lowered (<2%) for both isomers.

Glycosylation products in the presence and absence of glycine. a Products of the reaction of 25 mM P-ribose and 25 mM adenine are shown with a solid line; products of the reaction of 25 mM glycine, 25 mM P-ribose and 25 mM adenine are shown with a dashed line. All reactions were performed by heating the starting materials at 90 °C for the indicated time in an acid aqueous media, after which samples were analysed by RP-HPLC-MS. b EIC for adenosine monophosphate (m/z = 348.0683 ± 0.01) comparing the reaction of 25 mM adenine + 25 mM P-ribose in the presence (red) and absence (black) of 25 mM glycine. c EIC for cyclic guanosine monophosphate (m/z = 346.0547 ± 0.01) comparing the reaction of 25 mM guanine + 25 mM P-ribose in the presence (red) and absence (black) of 25 mM glycine. Each data point represents the average of three replicates ± standard deviation
Glycine also affected the distribution of isomeric species, and clear differences were observed in comparison to the base peak chromatogram (BPC) from the reaction of P-ribose and adenine, in the presence and absence of glycine (Fig. 4b and Supplementary Figs. 110–114). These data indicated the presence of different chemical species and a resultant change in mass distribution between the reactions with and without glycine. Individual EICs were then analysed for each adenine glycosylation product, resulting in clear differences being observed in the peak relative intensities when glycine was added. These results clearly show that glycine has a selective effect on which isomeric species are preferentially formed. Glycine is known to react readily with other amines under dehydrating conditions12, and it is likely to react with the primary amines of nucleobases. Hybrid side products including glycine (Gly-AMP, Gly-cAMP, Gly-Adenosine, Gly-Adenine) were detected in ~1% yield (Supplementary Fig. 145), however, this small percentage has an important effect on the isomeric distribution of the adenine glycosylation products (see Supplementary Figs. 146, 147). Change in the isotopic distribution of the masses of hybrid products was detected (Supplementary Figs. 123, 124) when deuterated glycine was included in the dehydration reaction, confirming glycine inclusion in hybrid structures.
A similar effect on isomer distribution was also observed when P-ribose was reacted with cytosine/guanine in the presence of glycine (Fig. 4c, Supplementary Figs. 125–140). The maximum intensities of the different isomer species decreased (GMP, CMP, cGMP, cCMP, guanosine and cytidine), while the distribution of the relative intensities was also affected. The differences between experiments were rigorously verified using a statistical method such as cluster analysis of EIC data to segment samples, in the presence and absence of glycine, into constituent groups/clusters with common characteristics (Fig. 5). The objective of cluster analysis in this study is to group data (i.e. nucleotide and nucleoside structure formation) into constituent assemblies with shared characteristics (e.g. glycine addition versus no glycine). This analysis should demonstrate high internal homogeneity within clusters/groups and high external heterogeneity between clusters/groups. Fig. 5 displays a dendrogram with “Wards” linkage35. To identify clusters in the dendrogram, we have coloured the spectra according to the presence of glycine (glycine - red, no glycine - black, blank - blue). As can be observed, glycine containing samples cluster together. One cluster corresponds to P-ribose + adenine + glycine (three samples), which is separated from samples without glycine. A second cluster corresponds to P-ribose + guanine + glycine and P-ribose + cytosine + glycine. Samples containing adenine separate into a larger cluster that is distinguished from the other samples, indicating strong influence of adenine in the reaction. Indeed, there are a number of other possible products and reactions that might take place under the conducted reaction conditions (see Supplementary Note 1 and Supplementary Figs. 150–162 for more details).

Dendrogram and base peak chromatograms (BPC). Cluster analysis was used to bundle the samples into constituent groups with common characteristics. Here the dendrogram displays high internal homogeneity within clusters for three replicates each of reactions performed in the presence and absence of glycine. At the same time, the method displays high external heterogeneity between clusters where adenine samples compose a larger cluster that is more distant than other nucleotides
Other amino acids were also included in the dehydration reaction of adenine with P-ribose to test if they would also have some effect on the isomeric distribution of the glycosylation products (Supplementary Figs. 141–143). The six amino acids selected for this study (arginine, glutamic acid, threonine, methionine, phenylalanine and tryptophan) have different side chains, with different chemical natures and functional groups. When the results were compared with the data obtained from the reaction of only P-ribose and adenine, changes in the isomeric distribution of AMP were observed in all the reactions, except in the case of tryptophan, which might be attributed to conformational limitations due to the presence of tryptophan's indole-based side chain. Analysing cAMP EICs, smaller changes in the relative intensity of the isomeric peaks were noticed. However, a clear difference was observed in the EIC of adenosine only for the reactions including phenylalanine and threonine.
Discussion
Our results show that glycosidic bonds can be simultaneously formed with three different nucleobases in a one-pot dehydration reaction under acidic conditions. Nucleotide products were observed upon reacting d-ribose-5′-phosphate, adenine, guanine and cytosine as the only starting materials, without the need for any mineral, catalyst or activating agents. The observed nucleobase exchange implies a dynamic equilibrium in the dehydration reaction, where bonds are broken and formed towards the enrichment of the most thermodynamically stable products. Glycosidic bond formation between P-ribose and the nucleobase is found to preferentially occur at the primary amine sites of the nucleobase (where available), leading to non-canonical nucleobases forming as major products. However, reactivity at secondary amine sites also occurs, leading to a distribution of isomeric products. Furthermore, while the reaction of ribose is most likely to occur through the anomeric position, glycosidic bond formation at other positions is also possible, further increasing the number of possible isomeric products. These results support the idea that the initial nucleoside/nucleotide structures in a pre-RNA world might not have been canonical structures, but the most stable or plausible structures preferentially formed in the environmental conditions present on the early Earth. Canonical structures might have only appeared later as a result of further evolution building on these more abundant species. Addition of amino acids was found to significantly alter the relative intensity and distribution of product isomers, and we suggest that this is partially through competition for reactive amine sites on the nucleobase, though amino acid adducts do not account for all the changes observed. Based on these results, we suggest that the Origins of Life community should not discount possible cooperative effects between peptide and nucleotide building blocks on the early Earth, with no predominance of a pure RNA-world or pure Peptide-world, but rather a combination of both. Since the conformation of the nucleobases in a nucleic acid is critical for hydrogen bonding and base pairing, future work will investigate how this selectivity may be tuned using different amino acids or reactive species, in order to promote specific nucleobase isomers.
Methods
General Protocol
In a 7-ml glass reaction vessel the below reagents were added: 1000 µl of 0.1 M glycine, 1000 µl of 0.1 M 5′-phosphate d-ribose and 1000 µl of 0.1 M adenine. The pH was adjusted to the desired value using acid (HCl) or base (NaOH) and finally the total volume was taken to 4 ml using the corresponding amount of HPLC water. Then, the hot plate (equipped with Drysyn hotplate inserts) was pre-heated at 90 °C. Glass reaction vessels were placed in the corresponding Drysyn hotplate inserts. Lids with three integrated holes were placed on each vial which facilitated the evaporation during the drying step (see Supplementary Fig. 1). The vials were kept at 90 °C for a given time, in order to evaporate the solution to complete dryness. Once a cycle was finished, the vials were taken out of the heating plate; otherwise 4 ml of HPLC water was added (depending if the reaction was run for 1 cycle or more). Once finished, products were collected for analysis by adding 8 ml of HPLC water to the reaction vial. Then, 1.5 ml sample was filtered using nylon syringe filters (cut off = 0.22 µm). 500 µl of the extracted sample were mixed with 500 µl of HPLC water for RP-HPLC-MS analysis. For detailed materials and methods see the Supplementary Methods and Supplementary Figs. 1-4.
Data availability
All relevant data are available from the authors upon reasonable request.
Change history
21 March 2019
The previously published version of this Article contained errors in Fig. 1. In Fig. 1a, an oxygen atom was omitted from one structure. In Fig. 1b, one structure was drawn as the incorrect enantiomer. These errors have been corrected in both the PDF and HTML versions of the Article.
References
Ruiz-Mirazo, K., Briones, C. & De La Escosura, A. Prebiotic systems chemistry: new perspectives for the origins of life. Chem. Rev. 114, 285–366 (2014).
Rentergent, J., Driscoll, M. D. & Hay, S. Time course analysis of enzyme-catalyzed DNA polymerization. Biochemistry 55, 5622–5634 (2016).
Kafri, M., Metzl-Raz, E., Jona, G. & Barkai, N. The cost of protein production. Cell Rep. 14, 22–31 (2016).
Hud, N. V., Cafferty, B. J., Krishnamurthy, R. & Williams, D. L. The origin of RNA and “My Grandfather’s Axe”. Chem. Biol. 20, 466–474 (2013).
Fuller, W. D., Sanchez, R. A. & Orgel, L. E. Studies in prebiotic synthesis. VII solid-state synthesis of purine nucleosides. J. Mol. Evol. 1, 249–257 (1972).
Sanchez, R. A. & Orgel, L. E. Studies in prebiotic synthesis. V Synthesis and photoanomerization of pyrimidine nucleosides. J. Mol. Biol. 47, 531–543 (1970).
Powner, M. W., Gerland, B. & Sutherland, J. D. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 459, 239–242 (2009).
Xu, J. et al. A prebiotically plausible synthesis of pyrimidine β-ribonucleosides and their phosphate derivatives involving photoanomerization. Nat. Chem. 9, 303–309 (2017).
Bean, H. D. et al. Formation of a β-pyrimidine nucleoside by a free pyrimidine base and ribose in a plausible prebiotic reaction. J. Am. Chem. Soc. 129, 9556–9557 (2007).
Cafferty, B. J., Fialho, D. M., Khanam, J., Krishnamurthy, R. & Hud, N. V. Spontaneous formation and base pairing of plausible prebiotic nucleotides in water. Nat. Commun. 7, 11328 (2016).
Fox, S. W. Thermal polymerization of amino-acids and production of formed microparticles on lava. Nature 201, 336–337 (1964).
Rodriguez-Garcia, M. et al. Formation of oligopeptides in high yield under simple programmable conditions. Nat. Commun. 6, 8385 (2015).
Leman, L., Orgel, L. & Ghadiri, M. R. Carbonyl sulfide–mediated prebiotic formation of peptides. Science 306, 283–286 (2004).
Forsythe, J. G. et al. Ester-mediated amide bond formation driven by wet–dry cycles: a possible path to polypeptides on the prebiotic Earth. Angew. Chem. Int. Ed. 54, 9871–9875 (2015).
Forsythe, J. F. et al. Surveying the sequence diversity of model prebiotic peptides by mass spectrometry. Proc. Natl Acad. Sci. USA 114, E7652–E7659 (2017).
Duncan, K. L. & Ulijn, R. V. Short peptides in minimalistic biocatalysts design. Biocatalysis 1, 67–81 (2015).
Nakashima, T. & Fox, S. W. Synthesis of peptides from amino acids and ATP with lysine-rich proteinoid. J. Mol. Evol. 15, 161–168 (1980).
Gorlero, M. et al. Ser-His catalyses the formation of peptides and PNAs. FEBS Lett. 583, 153–156 (2009).
Tyagi, S. & Ponnamperuma, C. Nonrandomness in prebiotic peptide synthesis. J. Mol. Evol. 30, 391–399 (1990).
Jauker, M., Griesser, H. & Richert, C. Spontaneous formation of RNA strands, peptidyl RNA, and cofactors. Angew. Chem. Int. Ed. 54, 14564–14569 (2015).
Griesser, H. et al. Ribonucleotides and RNA promote peptide chain growth. Angew. Chem. Int. Ed. 55, 1219–1223 (2016).
Griesser, H., Bechthold, M., Tremmel, P., Kervio, E. & Richert, C. Amino acid-specific, ribonucleotide-promoted peptide formation in the absence of enzymes. Angew. Chem. Int. Ed. 128, 1224–1228 (2016).
Ura, Y., Beierle, J. M., Leman, L. J., Orgel, L. E. & Ghadiri, M. R. Self-assembling sequence-adaptive peptide nucleic acids. Science 325, 73–77 (2009).
Stairs, S. et al. Divergent prebiotic synthesis of pyrimidine and 8-oxo-purine ribonucleotides. Nat. Commun. 8, 15270 (2017).
Kim, H.-J. & Benner, S. A. Prebiotic stereoselective synthesis of purine and noncanonical pyrimidine nucleotide from nucleobases and phosphorylated carbohydrates. Proc. Natl Acad. Sci. USA 114, 11315–11320 (2017).
Becker, S. et al. A high-yielding, strictly regioselective prebiotic purine nucleoside formation pathway. Science 352, 833–836 (2016).
Cassone, G. et al. Stability of 2′,3′ and 3′,5′ cyclic nucleotides in formamide and in water: a theoretical insight into the factors controlling the accumulation of nucleic acid building blocks in a prebiotic pool. Phys. Chem. Chem. Phys. 19, 1817–1825 (2017).
Kochetkov, N. K., Shibaev, V. N., Kost, A. A. & Zelinsky, N. D. New reaction of adenine and cytosine derivatives, potentially useful for nucleic acids modification. Tetrahedron Lett. 22, 1993–1996 (1971).
Bhatt, D. P., Chen, X., Geiger, J. D. & Rosenberger, T. A. A sensitive HPLC-based method to quantify adenine nucleotides in primary astrocyte cell cultures. J. Chromatogr. B 889–890, 110–115 (2012).
Gull, M. Prebiotic phosphorylation reactions on the early Earth. Challenges 5, 193–212 (2014).
Banoub, J. H., Newton, R. P., Esmans, E., Ewing, D. F. & Mackenzie, G. Recent developments in mass spectrometry for the characterization of nucleosides, nucleotides, oligonucleotides, and nucleic acids. Chem. Rev. 105, 1869–1915 (2005).
Strzelecka, D., Chmielinski, S., Bednarek, S., Jemielity, J. & Kowalska, J. Analysis of mononucleotides by tandem mass spectrometry: investigation of fragmentation pathways for phosphate- and ribose-modified nucleotide analogues. Sci. Rep. 7, 8931 (2017).
Brotzel, F., Ying Cheung Chu, Y.-C. & Mayr, H. Nucleophilicities of primary and secondary amines in water. J. Org. Chem. 72, 3679–3688 (2007).
Mungi, C. V. & Rajamani, S. Characterization of RNA-like oligomers from lipid-assisted nonenzymatic synthesis: implications for origin of informational molecules on rarly earth. Life 5, 65–84 (2015).
Jain, A. K., Murty, N. M. & Flynn, P. J. Data clustering: a review. ACM Comput. Surv. 31, 264–323 (1999).
Acknowledgements
We acknowledge financial support from the EPSRC (grant nos EP/H024107/1, EP/I033459/1, EP/J00135X/1, EP/J015156/1, EP/K021966/1, EP/K023004/1, EP/K038885/1, EP/L015668/1 and EP/L023652/1), the ERC (project 670467 SMART-POM), and the John Templeton Foundation Grant ID 60625 and Grant ID 61184. We gratefully acknowledge collaboration with RELX Intellectual Properties SA and their technical support to access Reaxys. We thank Dr. Maria D. Castro and David Doran for their help with HPLC-MS analysis, Dr. Marc Rodriguez-Garcia and Dr. Andrew J. Surman for initial experimental discussions.
Author information
Authors and Affiliations
Contributions
L.C. conceived the idea, designed the project and coordinated the efforts of the research team with I.S.-M., R.T.-M. and Y.M.A.-H. I.S.-M. developed and performed the synthetic and analytical protocols. I.S.-M. and R.T.-M. conducted data analysis, and P.S.G. carried out statistical analysis. A.O.O. helped with some experiments and S.C.S. helped with some data analysis. R.T.-M., Y.M.A.-H. and G.C. contributed to experimental discussions and paper writing. L.C., I.S.-M., Y.M.A.-H. and R.T.-M. co-wrote the paper with input from all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Suárez-Marina, I., Abul-Haija, Y.M., Turk-MacLeod, R. et al. Integrated synthesis of nucleotide and nucleosides influenced by amino acids. Commun Chem 2, 28 (2019). https://doi.org/10.1038/s42004-019-0130-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-019-0130-7
- Springer Nature Limited
This article is cited by
-
Prebiotic N-(2-Aminoethyl)-Glycine (AEG)-Assisted Synthesis of Proto-RNA?
Journal of Molecular Evolution (2024)
-
Regioselective ribonucleoside synthesis through Ti-catalysed ribosylation of nucleobases
Nature Synthesis (2023)
-
Tides over titanium
Nature Synthesis (2023)
-
Nonenzymatic Template-Directed Primer Extension Using 2′-3′ Cyclic Nucleotides Under Wet-Dry Cycles
Origins of Life and Evolution of Biospheres (2023)