
Abstract
Alzheimer’s disease (AD) and more than twenty other dementias, termed tauopathies, are pathologically defined by insoluble aggregates of the microtubule-associated protein tau (MAPT). Although tau aggregation correlates with AD symptomology, the specific tau species, i.e., monomers, soluble oligomers, and insoluble aggregates that induce neurotoxicity are incompletely understood. We developed a light-responsive tau protein (optoTAU) and used viscosity-sensitive AggFluor probes to investigate the consequence(s) of tau aggregation in human neurons and identify modifiers of tau aggregation in AD and other tauopathies. We determined that optoTAU reproduces biological and structural properties of tau aggregation observed in human brains and the pathophysiological transition in tau solubility in live cells. We also provide proof-of-concept for the utilization of optoTAU as a pharmacological platform to identify modifiers of tau aggregation. These findings have broad implications for the characterization of aggregation-prone proteins and investigation of the complex relationship between protein solubility, cellular function, and disease progression.
Similar content being viewed by others


Introduction
Alzheimer’s Disease (AD) is a multifactorial neurodegenerative disease characterized by the accumulation of amyloid-β (Aβ) plaques and neurofibrillary tangles (NFTs)1,2. Significantly, the NFT burden, but not the Aβ plaque load, more precisely correlates with the onset of cognitive impairment and regions of brain atrophy in AD patients3,4,5. NFT formation is due to the progressive pathological accumulation and aggregation of the microtubule-associated protein tau (MAPT) in degenerating neurons6,7,8,9. Although tau aggregation plays a key role in the transition from a healthy to a dying neuron, it remains a matter of debate what triggers tau aggregation in AD.
Tau is a microtubule-associated protein that is predominantly expressed in the axons of mature neurons10,11. In physiology, tau is involved in microtubule stabilization, axonal transport support, genomic DNA protection, and synaptic maintenance10. In pathophysiology, tau undergoes several post-translational modifications (PTMs; e.g., phosphorylation, acetylation, ubiquitination) that lead to protein misfolding and aggregation7,8,9,12. Specifically, tau aggregation is the gradual transition of soluble oligomeric species towards ‘irreversible’ detergent-insoluble aggregates that accumulate in AD brains10,13,14. Several tau-targeted therapies have successfully prevented tau aggregation and propagation and promoted pathological tau clearance in pre-clinical models15. However, most tau-targeted AD clinical trials have failed due to insufficient efficacy16,17,18. As such, it is crucial to identify alternative targetable mechanisms that modify tau aggregation in biologically relevant human pre-clinical models, which may facilitate and increase the efficacy of human trials.
The use of optogenetic tools to model protein aggregation is revolutionizing the study of neurodegenerative diseases19,20,21,22. Although tau aggregation is unequivocally correlated with AD symptomology, the specific tau species (i.e., monomers, soluble oligomers, and insoluble aggregates) and modifiers of tau aggregation that may induce neurotoxicity in AD are poorly understood23. Recent studies have demonstrated the utility of the self-associating cryptochrome 2 (Cry2) optogenetic protein to study pathological tau aggregation22,24. However, Cry2 is a relatively large (~75 kDa) photoreceptor, which may hinder the precise light-induced monitoring of tau aggregation. Moreover, Cry2-Tau models have not been able to distinguish between the formation of soluble and insoluble tau aggregates in live cells, which may exhibit different pathophysiological roles in disease progression. As such, refined optogenetics technologies are needed to provide light-specific control, gradual and persistent induction, and live-cell monitoring of the transition in tau solubility from a soluble monomer to an insoluble aggregate. Tools that fulfill these criteria are imperative to accurately re-capitulate disease-relevant tau aggregation and will provide a platform for basic and translational understanding of modifiers of tau biology. Importantly, to date, no optogenetic tools exist to control and visualize tau aggregation in human neuronal models.
To address this challenge, we generated an advanced optogenetic model of tau aggregation, optoTAU. OptoTAU is an inducible model of tau aggregation that provides superior light/dark selectivity, concentration-dependence, and forms irreversible tau aggregates. Further refinement of the optoTAU model permits live-cell visualization of the transition in tau solubility from a soluble oligomer to an insoluble aggregate. In addition, optoTAU reproduces disease mutation, isoform, and structural properties of tau aggregation. OptoTAU also recruits endogenous human neuronal tau into tau aggregates. Finally, inhibition of the optoTAU aggregation with a tau structure-based peptide inhibitor reduces tau aggregation and rescues neuronal viability. Collectively, we show that optoTAU reproduces physiologically relevant aggregation properties of tau and can be used as a platform to screen modifiers of tau aggregation with putative implications for the investigation of other disease-associated aggregation-prone proteins.
Results
Vivid (VVD) provides superior light-specific control of tau aggregation
Vivid (VVD) is a small photoreceptor (~17 kDa), derived from Neurospora crassa, that forms rapidly reversible, antiparallel homodimers upon light exposure25,26,27. We reasoned that the size (17 kDa) and dynamic dimerization properties of VVD would render it an invaluable tool to precisely control protein–protein interactions in living cells. To optimally induce VVD dimerization, we introduced two mutations in VVD (I74V, I85V; VVD (fast)), which allow rapid “switch-off” photo cycles that are critical for both the light-selective and reversible dimerization properties of VVD25,27. First, we introduced the VVD (fast; I74V, I85V)26 mutant to the N- or C-terminus of full-length (2N4R) tau and used a doxycycline (Dox)-inducible Tet-On® 3G system to control the levels and temporal induction of tau expression in HEK293 cells (Fig. 1A). The C-terminal VVD tag (Tau-VVD), herein referred to as optoTAU, provides superior light-specific control of high molecular weight (HMW) tau formation relative to Tau-Cry2, Cry2-Tau, VVD-Tau, and untagged tau (negative control) (Fig. 1B–D).

A Schematic of the blue light-inducible optoTAU model. The photoreceptor Vivid (VVD), fused to the C-terminus of 2N4R tau, initiates the light-specific, tau-driven aggregation of optoTAU. B Schematic of the experimental comparison between the photoreceptors Cry2 and VVD. C, D Representative immunoblot images (C) and densitometric analysis (D) of total protein homogenates from HEK293 cells following expression of tau alone (negative control), VVD-tau, tau-VVD, Cry2-tau, and tau-Cry2. A lower (upper panel) and higher (middle panel) exposure are shown for the representation of monomeric and HMW tau (C). E Schematic of the optoTAU time-course experimental paradigm. F–H Representative immunoblot images (F) and densitometric analysis (G, H) of the temporal accumulation of soluble HMW and insoluble optoTAU (F; upper panel) and monomeric optoTAU (F; middle panel) with or without (24 h DARK) light stimulation for 1, 4, and 24 h. I–L Representative immunoblot images and densitometric analysis of soluble (I, J) and insoluble (K, L) optoTAU in HEK293 cells following treatment with 50, 250, or 1000 ng/ml of Dox with or without light stimulation. The pan-tau antibody Tau5 was used for immunoblot detection of tau. Densitometric analysis of HMW tau was normalized to monomeric tau (C). OptoTAU monomer levels are normalized to total protein (Ponceau S; lower panels) (C, F, I). Densitometric analysis of soluble HMW optoTAU (>100 kDa) was normalized to monomeric optoTAU (<75 kDa) (F, I) and total insoluble tau represents the monomer+HMW tau (F, K). Each data point represents the average of two technical replicates for 3–4 independent experiments. Ordinary one-way ANOVA analysis followed by Tukey’s multiple comparisons. Mean ± SEM (error bars). ****P < 0.00001, ***P < 0.0001, **P < .001, *P < .05, ns not significant.
OptoTAU induces temporal and concentration-dependent control of tau aggregation
Tauopathies are characterized by the gradual accumulation of detergent-insoluble protein aggregates10,13,14. Accordingly, protein aggregation is generally described as a time- and concentration-dependent process that proceeds toward “irreversible” insolubility under pathological conditions. To determine whether optoTAU is a model of the time- and concentration-dependent aggregation of tau, we performed light exposure time-course and concentration curve experiments. OptoTAU displays a gradual time- (Fig. 1E–H) and concentration-dependent (Fig. 1I–L) increase in Sarkosyl28 soluble HMW tau oligomers and insoluble tau aggregates. These findings strongly establish optoTAU as an advanced optogenetic model with light-specific temporal control of tau aggregation.
AggFluors permit live-cell visualization of tau aggregation
Currently, no method exists to reliably visualize and distinguish between soluble HMW tau oligomers and insoluble tau aggregates in live cells. As such, we combined the optoTAU system with the AggTag method29,30, which utilizes a self-labeling protein tag, e.g., HaloTag, and covalently-bound viscosity-sensitive fluorescent probes, i.e., AggFluors, that display quenched fluorescence until a folded protein begins to misfold and aggregate. As such, AggFluors with high rotational barriers fluoresces in lower viscosity environments (soluble oligomers) relative to fluorophores with low rotational barriers (insoluble aggregates), allowing live-cell visualization of protein aggregation and measurement of aggregation kinetics (i.e., rate of transition from a soluble oligomer to an insoluble aggregate). We attached the HaloTag to optoTAU, creating optoTAU-Halo, and used two AggFluor probes, P1h and P18h, which permit a dual color imaging platform to monitor the formation of soluble oligomers (P1h red; Ex/Em: 450/520 nm) and insoluble aggregates (P18h, green; Ex/Em: 540/640 nm) in live cells (Fig. 2A). Specifically, we generated a stable, Dox-inducible optoTAU-Halo-expressing HEK293 cell line for utilization with the “turn-on” AggFluor probes P1h and P18h29. We first confirmed the light-selective biochemical detection of soluble HMW and insoluble optoTAU-Halo (Supplementary Fig. 1). We then determined that optoTAU-Halo forms pathologically phosphorylated aggregates (Supplementary Fig. 2a) that adopt disease-relevant conformations (Supplementary Fig. 2b) and amyloid-like β-sheet structures (Supplementary Fig. 2c, d). Finally, we performed longitudinal live-cell time-lapse imaging of cells expressing optoTAU-Halo (Fig. 2B). We observed a rapid increase in soluble tau oligomers (P1h fluorescence; Fig. 2B, C), which is followed by a progressive accumulation of insoluble tau aggregates (P18h fluorescence; Fig. 2B, D). Taken together, these results establish the first tau aggregation model that permits live-cell visualization of the temporal changes in tau solubility and aggregation.

A Schematic of the optoTAU-Halo model, the viscosity-sensitive AggFluor dyes P1h (red; soluble oligomers) and P18h (green; insoluble aggregates). B Schematic of the time-lapse live-cell imaging experimental paradigm. C Representative immunocytochemical analysis of soluble (P1h; red), insoluble (P18h; green), and the overlay of both dyes. Scale bar = 10 µm. D, E Average soluble (D) and insoluble (E) fluorescence per cell. Data is an average of n = 3 independent experiments (90–125 cells from n = 6 fields of view per condition). F Schematic of the experimental paradigm to investigate the persistence of optoTAU aggregation. G–J Representative immunoblot images and densitometric analysis of the accumulation of soluble HMW (G, H) and insoluble (L, J) optoTAU in HEK293 cells following four different light-stimulation paradigms, i.e., 24 h dark alone (negative control), 12 h light exposure followed by 12 h under dark condition (persistence test), 12 h dark condition followed by 12 h light exposure, and 24 h light exposure (positive control). The pan-tau antibody Tau5 was used for immunoblot detection of tau. HMW optoTAU images (G, I) are higher exposure to clearly show HMW tau detection. Soluble optoTAU monomer levels are normalized to total protein (Ponceau S; lower panels) (G). Densitometric analysis of soluble HMW optoTAU (>100 kDa) was normalized to monomeric optoTAU (<75 kDa) (G) and total insoluble tau represents the monomer+HMW tau (I). Each data point represents the average of two technical replicates for 3–4 independent experiments. K Schematic of the experimental paradigm to visualize the progressive and persistent accumulation of optoTAU-Halo with the AggFluor probes P1h (soluble oligomers) and P18h (insoluble aggregates). L Representative live-cell images of soluble (red; row 2), insoluble (green; row 3), and the overlay of both dyes (row 1) in cells prior to light stimulation (A), following dark conditions alone for 24 h (B), or following light stimulation for 24 h alone (C) and subsequent dark conditions for 24 h (D). M, N Average soluble (M) and insoluble (N) fluorescence per cell. Soluble and insoluble fluorescence was normalized to total nuclei number per field of view (n = 3 independent experiments; 70–120 cells from n = 2 fields of view/condition). Ordinary one-way ANOVA analysis followed by Tukey’s multiple comparisons. Mean ± SEM (error bars). ***P < 0.0001, **P < 0.001, *P < 0.05, ns not significant.
OptoTAU forms stable irreversible tau aggregates following light cessation
Given that the formation of insoluble tau aggregates is a physiologically irreversible process that persists throughout disease progression, we sought to determine whether light stimulation induces the irreversible formation of soluble HMW tau and insoluble tau aggregates following light cessation. Indeed, biochemical analysis indicates that soluble and insoluble optoTAU aggregates persist and are irreversible after light termination (Fig. 2E–I). Further immunocytochemical analysis reveals the progressive tau-driven formation of soluble and insoluble tau aggregates after light cessation (Fig. 2J–M). These results establish that light initiates the aggregation of optoTAU that persists over time in the absence of light.
OptoTAU reproduces mutation- and isoform-specific disease-relevant tau aggregation properties
In familial tauopathies, mutations in the tau gene MAPT play a causal role in the accumulation of tau pathology. Two hexapeptide regions in the repeat domain of tau (275–280, 306–311) are critically involved in the formation of disease-specific tau conformations and the nucleation of tau aggregation31. As such, we introduced a well-characterized, pro-aggregation mutation (^K280; PRO) or an anti-aggregation mutation (^K280, I277P, I308P; ANTI) into optoTAU (Fig. 3A). We show that the well-validated tau-specific pro-aggregation mutation (^K280; PRO) does not affect the accumulation of soluble HMW tau (Fig. 3B, C) but dramatically increases the accumulation insoluble optoTAU aggregation relative to optoTAU-WT (Fig. 3D, E). In contrast, the anti-aggregation (^K280, I277P, I308P; ANTI) mutations32,33 lead to the accumulation of lower levels of soluble HMW tau and insoluble tau aggregates relative to optoTAU-WT (Fig. 3B–E). These results are consistent with aggregation propensities of pro- and anti-aggregation MAPT mutations.

A Schematic of the pro- and anti-aggregation mutations in optoTAU. B–E Representative immunoblot images (B, D) and densitometric analysis (C, E) of the accumulation of soluble HMW (B, C) and insoluble (D, E) optoTAU with or without light stimulation in cells expressing optoTAU(WT), optoTAU(K280; PRO), or optoTAU(K280, I277P,I308P; ANTI). F Schematic of optoTAU isoforms. G–J Representative immunoblot images (G, I) and densitometric analysis (H, J) of soluble HMW (G, H) and insoluble (I, J) optoTAU with or without light stimulation in cells expressing 0N4R, 1N4R, or 2N4R optoTAU. The pan-tau antibody Tau5 was used for immunoblot detection of tau. HMW optoTAU images (B, D, G, I) are higher exposure to clearly show HMW tau detection. Soluble optoTAU monomer levels are normalized to total protein (Ponceau S; lower panels) (B, G). Densitometric analysis of soluble HMW optoTAU (>100 kDa) was normalized to monomeric optoTAU (<75 kDa) (C, H) and total insoluble tau represents the monomer+HMW tau (D, I). Each data point represents the average of two technical replicates for 3–4 independent experiments. Ordinary one-way ANOVA analysis followed by Tukey’s multiple comparisons. Mean ± SEM (error bars). ***P < 0.0001, **P < 0.001, *P < 0.05, ns not significant.
Tau is expressed in six different isoforms in the adult human brain10. Tau isoforms differ in the number of amino (N)-terminal inserts (0N, 1N, 2N) and contain either three or four 31–32 amino acid repeat sequences (R1–R4). NFTs in progressive supranuclear palsy predominantly contain 4R-tau and Pick disease inclusions contain mostly 3R-tau, whereas NFTs in AD contain a mixture of 4R-tau and 3R- tau34,35, suggesting that splicing is of key importance in the neuropathological process in different tauopathies and that potentially cell-specific tau isoforms assume distinct physiological roles. In sporadic tauopathies, such as AD, different tau isoforms dictate aggregation propensity (i.e., 2N4R < 1N4R < 0N4R)36,37,38. Accordingly, we show that optoTAU isoforms exhibit a similar pattern of insoluble optoTAU accumulation as observed in human AD brains with a corresponding depletion in soluble HMW tau (Fig. 3F–J). These findings indicate that light-induced optoTAU aggregation is a faithful model of disease-causing mutation and isoform-specific aggregation properties of tau. In addition, these results establish that while light initiates optoTAU aggregation, this process is subsequently driven by the distinguishing features of MAPT mutations and tau isoforms.
Human neurons form optoTAU aggregates that seed the aggregation of endogenous tau and form amyloid-like β-sheet structures
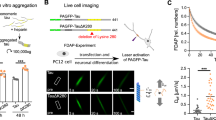
Insoluble tau aggregates deposit in amyloid-like β-sheet structures in neurons of AD patients39 and assume “prion-like” behavior, capable of seeding tau monomers into insoluble aggregates10,13,14. To determine whether optoTAU-Halo is a model for tau self-assembly in human neurons, we used the human neural progenitor cell (NPC) line, ReNcell® VM, an established platform for modeling AD pathogenesis40. First, we generated stable, Dox-inducible VVD-V5 (control) and optoTAU-Halo-expressing ReNcell® NPC lines. We then differentiated the NPCs into cortical-like (MAP2 + , 3R and 4R Tau + ) mature neurons (Supplementary Fig. 3a) and biochemically established the light-selective detection of soluble and insoluble optoTAU-Halo in neurons (Supplementary Fig. 3b–d). We then utilized the amyloid-sensitive dye, Amytracker41,42, to show the formation of amyloid-positive optoTAU-Halo aggregates (Fig. 4A–C), which is associated with reduced neuronal viability (Fig. 4D). Taking advantage of the fact that ReNcell neurons express both 3R- and 4R-tau40, we used a 3R-tau isoform-specific antibody that does not detect optoTAU-Halo (2N4R) (Supplementary Fig. 3e) to show that optoTAU-Halo recruits endogenous 3R-tau into insoluble amyloid-positive tau aggregates after light exposure (Figs. 2F and 4E, F). Together, these results demonstrate that optoTAU-Halo adopts a biologically relevant amyloid structure and is a model of endogenous tau seeding in human neurons.

A Schematic diagram of the experimental paradigm to detect optoTAU-Halo amyloid structure in human neurons. B Representative immunocytochemical analysis of AmytrackerTM alone (red; lower panels) and V5/AmytrackerTM overlay (upper panels). C Quantification of the total percent area occupied by AmytrackerTM relative to optoTAU-Halo (V5). D LDH cytotoxicity assay using culture supernatants from VVD-V5 and optoTAU-Halo expressing human neurons. E Schematic of the experimental paradigm using the “always-on” Oregon Green HaloTag dye to investigate optoTAU-Halo expression and colocalization with endogenous 3R-tau in human neurons. F Representative immunocytochemical analysis of optoTAU-Halo (Oregon green), endogenous 3R-tau, and AmytrackerTM. Colocalization of optoTAU-Halo and AmytrackerTM (top right), optoTAU-Halo and 3R-tau (middle right), and 3R-tau and Amytracker (lower right). G Schematic of the live-cell imaging experimental timeline in human neurons. H Representative immunocytochemical analysis of soluble (P1h; red; upper panels), insoluble (P18h; green; middle panels), and the overlay of both dyes (lower panels) with or without W-MINK (10 µM) treatment. I–K Average soluble (I) and insoluble (J) fluorescence normalized to nuclei count for Control and W-MINK-treated cells. Data points are an average of n = 3 independent experiments (90–125 cells from n = 6 fields of view per condition). K Total nuclei count with or without W-MINK treatment. Scale bar = 10 µm. Ratio paired t test (I–K), ordinary one-way ANOVA (C, D) followed by Tukey’s multiple comparisons (C, D). All mean ± SEM (error bars). ****P < 0.00001, ***P < 0.0001, **P < 0.001, *P < 0.05.
A tau inhibitor rescues neuronal viability induced by optoTAU aggregation
The formation of tau aggregates leads to a decrease in neuronal survival (Fig. 4D). As such, we sought to determine whether we could rescue the reduced neuronal viability with a tau structure-based peptide inhibitor, W-MINK31. First, using live-cell imaging (Fig. 4D), we observe a significant accumulation of soluble tau oligomers and insoluble tau aggregates in optoTAU-Halo-expressing neurons (Fig. 4H). Significantly, W-MINK treatment lowers levels of soluble oligomers and insoluble aggregates of optoTAU-Halo (Fig. 4H–J). Furthermore, W-MINK treatment prevents neuronal loss induced by optoTAU-Halo aggregation (Fig. 4K). Collectively, these results demonstrate that the AggFluor probes permit direct, live-cell visualization of temporal changes in tau solubility and aggregation in neurons and, importantly, provide a screening platform to test tau-specific aggregation inhibitors.
Discussion
Postmortem neuropathology studies and cross-sectional and longitudinal positron emission tomography (PET) imaging reveal the stepwise accumulation of Aβ and tau pathology but preclude a mechanistic understanding of how tau aggregation dictates disease progression in AD. Efforts to translate these findings into cellular and murine models of tau dysfunction have also been challenging due to the use of overexpressed mutated MAPT and species-specific differences. Furthermore, although several tau-targeted therapies have successfully prevented tau aggregation and propagation and promoted pathological tau clearance in pre-clinical models15, most tau-targeted AD clinical trials have failed due to insufficient efficacy16,17,18. As such, it is crucial to identify alternative targetable mechanisms that modify tau aggregation in biologically relevant human pre-clinical models, which may facilitate and increase the efficacy of human trials.
Optogenetics has revolutionized the field of protein activation, protein–protein interaction, and protein aggregation43,44,45,46. It has become an invaluable technique to model neurodegenerative proteinopathies such as ALS22,47, PD21, and AD19,20. To address the research challenges associated with investigating tau aggregation, we developed an optogenetic platform, termed optoTAU, to model tau aggregation in vitro. OptoTAU reproduces some of the biological and structural properties of tau aggregation observed in the human brain. We further developed optoTAU-Halo to visualize the multi-step transition in tau solubility during aggregation, given that understanding the relationship between protein solubility and its role in physiological or pathological processes is also a significant research challenge. Nevertheless, the optoTAU-Halo model does have limitations, e.g., the insertion of the VVD::V5::Halo domains may sterically alter the conformations adopted by tau and impact tau phosphorylation and aggregation. Although we show with pathological phosphorylation and conformational antibodies (i.e., PHF1 and MC1, respectively) and dyes (i.e., Thioflavin S and Amytracker™) that optoTAU-Halo adopts biologically relevant phosphorylation and conformational features of tau, these results and future studies should be interpreted and pursued with the awareness of these caveats of the model. Finally, we provide proof-of-concept that optoTAU-Halo and the AggFluor technology provide a powerful scalable platform to investigate modifiers of tau pathobiology, model the consequences of tau aggregation, and serve as a drug discovery platform for modifiers of tau solubility and aggregation. Broadly, these tools may be applied to other aggregation-prone proteins and used to characterize complex relationships between protein solubility, cellular function, and disease progression.
Methods
Plasmid generation
All protein expression plasmids generated in this work were constructed using restriction enzyme (NEB) digested backbone vectors and PCR-generated DNA fragments (Q5® High-Fidelity Polymerase, NEB) assembled with Gibson Assembly (HiFi DNA Assembly Master Mix, NEB). To generate DNA expression constructs that allow for the option of both transient and stable protein expression and doxycycline-inducible expression, a complete bi-directional, Tet-On® 3G sequence from (pCMV-Tet3G (Clontech) was assembled within 5’ and 3’ PiggyBac transposon-specific inverted terminal repeat sequences (ITR) of the PiggyBac(PB)-CAG-eGFP plasmid using EcoRV and SpeI restriction sites. All plasmids were constructed using the expression vector PB-pCMV-Tet3G (PB-Tet3G) by PCR fragment assembly using Ecor1 and Not1 restriction enzyme sites. An N-terminal truncated version of VVD (residues 37–186, 17.1 kDa) (Addgene #58689) was used for these studies. VVD mutations (I74V, I85V) were introduced by site-directed mutagenesis with Gibson Assembly to generate the VVD (fast) sequence. The addition of a V5 tag sequence to tau (2N4R) and VVD(fast) plasmids was constructed using PCR-generated fragments from 2N4R tau (Addgene #92204) and V5 (Addgene #107596) and inserted into the PB-pCMV-TET3G plasmid to generate tau (tau(2N4R))-V5, and VVD (I74V, I85V (fast))-V5. The following optogenetic plasmids: VVD-Tau(2N4R), Tau(2N4R)-VVD-V5, Cry2olig-Tau(2N4R), and Tau(2N4R)-Cry2olig, were constructed using PCR-generated fragments from the VVD plasmid and cryptochrome 2 (Cry2Olig) (Addgene #60032) plasmids and inserted into the described PB-Tet3G plasmid. Additional optoTAU isoforms (1N4R, 0N4R) were constructed using the original tau (2N4R)-VVD-V5 sequence and two-fragment Gibson assembly to remove one or both N-terminal tau inserts and insert fragments simultaneously into EcoR1 and Not1 sites of the PB-Tet3G plasmid. OptoTAU point mutations (^K280) and (^K280, I277P, I308P) were generated through site-directed mutagenesis with Gibson assembly and inserted into the PB-pCMV-TET3G plasmid. OptoTAU-Halo was generated using a pCMV-HaloTag® plasmid (Gift from Dr. Xin Zhang’s lab) to construct tau (2N4R)-VVD-V5-Halo into the PB-Tet3G plasmid. All plasmids were confirmed by Sanger sequencing (Genewiz).
HEK293 cell culture, transient transfection, and stable cell line generation
HEK293 cells were purchased from (ATCC). The cells were maintained in Dulbecco’s modified eagle medium (DMEM/F12) supplemented with 10% fetal bovine serum at 37 °C and 5% CO2. On day 1, cells were seeded in six-well plates at 70–80% confluency. On day 2, at ≥90% confluency, cells were transfected with cDNA using X-tremeGENE HP DNA (Millipore Sigma) according to the manufacturer’s instructions. On day 3, the cell culture medium (1.5 ml for 6-well, and 500 µl for 24-well plates) was replaced with phenol-free DMEM/F12 containing 1 μg/ml doxycycline (Millipore Sigma) and placed in a dark incubator for 24 h. On day 4, the cells were incubated in the presence or absence of blue light exposure for the indicated times. For the generation of HEK293 cells that stably express Tau-VVD-V5-Halo, cells were seeded on six-well plates at 80% confluency. The following day, cells were co-transfected with PB-Tet3G-Tau-VVD-V5-Halo plasmid (2 μg DNA; transposon) and the PBase enzyme (transposase) expression plasmid (0.5 μg DNA) at a 4:1 transposon: transposase ratio using X-tremeGENE HP DNA. On the following day, fresh medium containing 5 µg/ml puromycin (Millipore Sigma) was added to the cell culture medium. Once confluent, the cells were trypsinized in 0.25% Trypsin-EDTA (ThermoFisher) and plated in a T75 flask in the presence of 5 μg/ml puromycin selection for up to 2 weeks.
ReNcell culture, differentiation, and stable cell line generation
The ReNcell® VM (Millipore) neural progenitor cell (NPC) line was maintained and differentiated as previously described40. Briefly, cells were maintained on MatrigelTM-coated flasks in proliferation medium (DMEM/F12 supplemented with 1× B27 (ThermoFisher), 2 μg/ml heparin (Millipore Sigma), 20 ng/ml bFGF (Millipore Sigma), and 20 ng/ml hEGF (Millipore Sigma) and filtered through a 0.2-μm PES filter (Fisher Scientific)). For stable expression of PB-Tet3G-Tau-VVD-V5-Halo, and PB-Tet3G-VVD-V5, cells were seeded on six-well MatrigelTM-coated plates at 80% confluency in proliferation medium. On day 2, cells were co-transfected with appropriate PB-Tet3G plasmid (2 μg DNA) and the PBase (500 μg DNA) using the jetOPTIMUS® (Polyplus transfection) transfection reagent according to the manufacturer’s instructions. On day 3, fresh proliferation medium supplemented with 1 μg/ml puromycin was added to the cell culture medium. When the cells reached confluency, they were treated with Dispase (1U/ml, StemCell Technologies), scraped, and transferred to T75 flasks in proliferation medium supplemented with 1 μg/ml puromycin for 2 weeks.
Differentiation
ReNcell® VM neural progenitor cells were seeded on MatrigelTM coated 6-well plates, 24-well coverslips, or 24-well glass imaging plates in differentiation medium (DMEM/F12 supplemented with 1× B27 and 2 μg/ml heparin and filtered through a 0.2-μm filter). Cells were allowed to differentiate for 25 days with medium changes every 3 days.
Peptide inhibitor preparation
The previously described W-MINK inhibitor31 with the following amino acid sequence (DVWMINKKRK) was synthesized by Genscript with a minimum purity of 90%. The stock peptide was dissolved in sterile DPBS to a final stock concentration of 5 mM, aliquoted for single use, and stored at −20 °C. For each experiment, W-MINK was diluted to 10 μM in phenol red-free medium.
Blue light stimulation
Chronic blue light exposure was performed in 24- or six-well plates using a custom-built LED stage (5 μM, 465 nm) housed in a cell culture incubator (37 °C and 5% CO2). For multiday Confocal imaging experiments, light-exposed cells were transferred from the cell culture incubator to a preheated (37 °C and 5% CO2) microscope stage top incubator and allowed to equilibrate for 10 min prior to imaging. For the 16 h time-lapse imaging experiments, a custom-built LED stage (5 μM, 465 nm) designed for the Confocal stage top incubator allowed simultaneous light exposure and imaging. A dark cell culture incubator was used for the control dark condition.
LDH cytotoxicity assay in HEK293 and ReN cells
Cell culture medium was collected from six-well plates after the respective blue light exposure experiment. For each experimental condition, 50 μl of the cell culture medium was added to 3 wells of a 96-well plate to perform the lactate dehydrogenase (LDH) release assay using the CyQUANT™ LDH Cytotoxicity assay kit (ThermoScientific) according to the manufacturer’s instructions. Briefly, 50 μl of the Reaction Mixture was added to each sample. The plate was protected from light and incubated for 30 min at room temperature (RT). In total, 50 μl of Stop Solution was then added to each well. The absorbance was measured at 490 nm and 680 nm using a Synergy microplate reader (Biotek). To determine LDH activity, the 680 nm absorbance value (background signal) was subtracted from the 490 nm absorbance value (LDH signal).
Detergent solubility assay (SarkoSpin)
All detergent solubility assays were performed from HEK293 and ReN cells plated on 6-well dishes. Detergent solubility assays were performed according to the SarkoSpin28 method with minor modifications. For the stable optoTau-Halo HEK293 cell line and transient transfection experiments, cells were washed with RT DPBS following removal of the cell culture medium. The cells were then scraped in 170 µl of RT 1× homogenization-solubilization (HS) buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 0.1 mM EDTA,1 mM dithiothreitol, complete EDTA-free protease inhibitors (Roche) and phosphatase inhibitors (Millipore Sigma), and 0.5% Sarkosyl), followed by the addition of a mixture of 2 mM MgCl2 and 25U (per well) Benzonase (Millipore Sigma) before cell scraping. For sample solubilization, 52 μl 1× HS buffer (no Sarkosyl) and 178 μl 2× HS buffer with 4% Sarkosyl were added to aliquots of the 170 μl lysate for a final concentration of 2% sarkosyl in 400 μl of lysate. Alternatively, for ReNcell cultures, three wells from a six-well plate were combined in a total of 400 μl of lysis buffer. After a 3 min of incubation at RT with vortexing every 10 min, the samples were diluted by adding 200 µl of 1× HS buffer. For fractionation, the lysate was centrifuged at 21,200 × g on a benchtop centrifuge (Eppendorf) for 45 min at RT. Supernatants (soluble fractions) were collected in a fresh tube. The insoluble pellet was washed with 250 µl of wash buffer (1× HS buffer, 1.5% Sarkosyl). The pellet was briefly vortexed and then centrifuged at 21,200 × g for 30 min at RT. The wash buffer was completely removed prior to freezing the insoluble pellet. Both the soluble fraction and insoluble pellet were frozen (−20 °C) prior to western blot analysis.
Western blot analysis
For gel loading, the soluble fractions were mixed with loading sample buffer (4× Bolt™ LDS Sample buffer (ThermoFisher) containing 4% β-mercaptoethanol (BioRad). Insoluble pellets were resuspended in 30 μl of 1× HS buffer containing 0.5% Sarkosyl and 10 μl of loading sample buffer (4× Bolt™ LDS Sample buffer (ThermoScientific). The samples were then heated for 10 min at 70 °C. Soluble and insoluble (20 µl, 50% of total pellet) samples were loaded onto NuPAGE™ 3–8% Tris-Acetate,1.0 mm gels (ThermoFisher). The samples were run at 50 V for 1 h followed by 100 V for 1 h using Novex NuPAGE Tris-Acetate SDS Running Buffer (ThermoFisher). Gels were equilibrated in fresh Towbin transfer buffer (25 mM Tris-base, 192 mM glycine, 20% (v/v) ethanol, 0.05% SDS (pH 8.3)) for 10 min. Protein was then transferred to 0.45 µm nitrocellulose membranes (BioRad) using a wet transfer method (Invitrogen™ Mini Blot Module) at 10 V for 90 min. After the transfer, the membranes were rinsed for 5 min in distilled water, stained with Ponceau S (0.1% (w/v) in 5% acetic acid; Sigma Aldrich) for 5 min, and rinsed with distilled water to remove excess Ponceau S. For total protein measurement, Ponceau S-stained membranes were imaged on an Amersham ImageQuant 800 imager (GE Healthcare). Membranes were then rinsed for 10 min and blocked in 1× TBS (Tris-buffered Saline) + 5% non-fat milk powder for 30 min at RT, and then briefly rinsed with 1× TBS-T (1× TBS + 0.1% Tween 20) and incubated with primary antibodies diluted in 1× TBS-T + 5% milk overnight at 4 °C on a shaker. Membranes were washed with 1× TBS-T (3× 10 min) and then incubated with a species-specific IgG (H + L) HRP-conjugated secondary antibody (BioRad) in 1× TBS-T, 5% milk for 1 h at RT. Membranes are then washed with 1X TBS-T (3× 10 min) and 1× TBS (3× 5 min). Western blots were developed using Western Lightning Plus-ECL (PerkinElmer) or SuperSignal™ West Femto substrate (Fisher Scientific) and imaged on an Amersham ImageQuant 800 imager. Quantification was performed using ImageLab software 6 (BioRad). For both soluble and insoluble fractions, background-adjusted band densities were obtained for the total tau monomer (75 kDa) and the total HMW tau signal (>100 kDa). For soluble fractions, tau protein was normalized to total protein (Ponceau S), and a HMW/Monomer tau ratio was calculated from a single non-overexposed image. All HEK293 data presented was from at least n = 3 independent biological experiments using two technical replicates per experimental condition. All ReN data presented is from at least n = 3 independent biological experiments with three technical replicates/wells pooled together during cell processing.
Immunocytochemical analysis
Subcellular distribution and colocalization of optoTau(2N4R)-V5-Halo with tau antibodies and amyloid dyes were investigated by multi-labeling immunocytochemical analysis of fixed optoTau-V5-Halo expressing HEK293 cells or ReN cells grown on MatrigelTM-coated coverslips. Following the respective experimental protocol, cells were rinsed with 1× DPBS at RT and then fixed with freshly made 4% paraformaldehyde (PFA) for 15 min at RT. Cells were washed with 1× DPBS (3× 5 min) before incubation with a permeabilization/blocking solution (0.1%Triton X-100, 0.1% Tween 20, 5% normal goat serum in 1× DPBS) for 30 min (3× 10 min) at RT. Cells were incubated with primary antibodies overnight at 4 °C, washed with 1× DPBS (3× 10 min) at RT, followed by incubation of Alexa Fluor-conjugated secondary antibodies for 1 h at RT in the dark. All antibodies were diluted in a permeabilization/blocking solution. After primary and secondary antibody incubations, cells were stained with the amyloid Amytracker 680 dye (1:1000; Ebba Biotech) for 30 min at RT followed by sequential washes with 50% ethanol/1× DPBS (2× 3 min), 30% ethanol/1× DPBS (2× 3 min), 10% ethanol/1× DPBS (3× 1 min), and 1× DPBS (3× 5 min). 4”6’-diamino-2-final-indol (DAPI), diluted in 1× DPBS, was then added to the cells for nuclei counterstaining followed by 1× DPBS washes (3× 5 min). The coverslips were mounted with ProLong® Diamond Antifade mounting medium (ThermoFisher) and stored in the dark. For fixed-cell HaloTag fluorescence imaging, the HaloTag-specific dye, Oregon Green (Promega), was added at 1 µM to phenol-free DMEM/F12 containing 1 μg/ml doxycycline for 24 h before light stimulation followed by cell fixation, antibody/nuclei staining, and mounting as described above.
AggFluor live-cell imaging
OptoTau-V5-Halo expressing HEK293 cells were seeded on Matrigel™--coated 24-well glass-bottom plates at 80% confluency. For the 16 h time-lapse imaging experiments, on the day after plating the cells, the culture medium was removed, and the cells were incubated with DMEM/F12 supplemented with 1 μg/ml Hoechst dye (ThermoFisher) for 10 min. OptoTAU protein expression was then induced by refreshing the cell culture medium with DMEM/F12 containing the following components: 1 μg/ml doxycycline to induce protein expression, 10 µM cytosine β-D-arabinofuranoside (AraC; Millipore Sigma) to mitotically arrest cells, and both Aggfluor probes 1 μM P1h (red) and 1 µM P18h (green). For cells treated with W-MINK inhibitor, an aliquot of the above dye solutions was combined with a 10 μM W-MINK solution and added to cells. Cells were then placed in a dark incubator for 4 h to allow protein expression and optoTau-V5-Halo labeling with Aggfluor probes. Cells were then allowed to equilibrate on the preheated (37 °C and 5% CO2) stage top incubator for 10 min prior to imaging. Two fields of view were followed for both untreated and treated cells. After the initial imaging of a pre-light timepoint (time = 0 h), cells were exposed to light and imaged every two hours over a 16-h experiment. For multiday imaging, protein expression was induced by replacement with fresh DMEM/F12 containing the following components: 1 μg/ml doxycycline, 10 μM AraC, and both Aggfluor probes 1 μM P1h and P18h or 0.5 μM coumarin (Blue) HaloTag ligand (Promega). For cells treated with the W-MINK inhibitor, an aliquot of the above dye solutions was combined with a 10 μM W-MINK solution and added to cells. Cells were then placed in a dark incubator for 24 h to allow protein expression and optoTau(2N4R)-V5-Halo labeling with Halo-tag ligands. The culture medium was replaced with fresh DMEM/F12 to remove excess probes and placed in a dark incubator for 5 min. Medium was removed, and cells were incubated with DMEM/F12 supplemented with 1 μg/ml Hoechst for 10 min in a dark incubator. Then, the medium was replaced with fresh phenol-free DMEM/F12 containing 1 μg/ml doxycycline and 10 μM AraC and incubated in the dark for 1 h before live-cell imaging to capture pre-blue light conditions. After imaging, cells were treated with light for 24 h after which cells were imaged to capture post-light conditions. After imaging, cells were placed under dark conditions for 24 h and then imaged to capture a post 24 h light/post 24 h dark (aggregate persistence) condition. For stable optoTau-V5-Halo expressing ReN 25-day, differentiated cells had protein expression induced by replacement with fresh differentiation medium containing the following components: 1 μg/ml doxycycline and both Aggfluor probes P1h and P18h or coumarin HaloTag. For cells treated with the W-MINK inhibitor, an aliquot of the above dye solutions was combined with a 10 μM W-MINK solution and added to cells. Thereafter, both ReN and HEK293 cells were treated identically except for the use of the appropriate cell culture medium.
Image acquisition
Confocal images were obtained with a Nikon A1R HD25 confocal microscope using a Galvano scanner. For fixed-cell imaging: n = 6 fields of view per coverslip from at least n = 3 independent biological experiments were taken from cells under either a 40× or 60× oil-immersion objective (either 512 × 512 or 1024 × 1024 frame size; 1.39× zoom; 1.2 Airy units pinhole dimension; unidirectional scanning; Offset unchanged) with appropriate negative controls (dox-free conditions with appropriate combinations either primary/secondary antibodies and Halo dyes). For every image, we performed Z-stacks with a step size of 0.15 μm allowing imaging of an entire layer of cells. Acquisition settings (laser intensity, gain, and offset) were kept constant for all images within a staining group. For live-cell imaging: All live-cell imaging experiments were performed on a Nikon A1R HD25 laser-scanning confocal microscope outfitted with a Tokai HIT stage top incubator using a resonant scanner. Cells were allowed to equilibrate on the preheated (37 °C and 5% CO2) stage top incubator for 10 min before imaging. For 16 h time-lapse imaging: n = 2 fields of view from n = 4 independent biological experiments were taken from cells under 60× oil-immersion objective (512 × 512 frame size; 1.39× zoom; 1.2 Airy units pinhole dimension; unidirectional scanning; Offset unchanged). For every image, we performed Z-stacks with a step size of 0.3 μm allowing imaging of an entire layer of cells. Acquisition settings (laser intensity, gain, and offset) were kept constant throughout the experiment. For multiday imaging: n = 6–10 fields of view per well (two wells per condition) were taken from cells under the 60× oil-immersion objective. (512 × 512 frame size; 1.39× zoom; 1.4 Airy units pinhole dimension; unidirectional scanning; Offset unchanged). For every image, we performed Z-stacks with a step size of 0.3 μm, allowing imaging of an entire layer of cells. Timing and order of image acquisition were alternated across experiments between experimental groups. Acquisition settings (laser intensity, gain, and offset) were kept constant for all time points during a multiday imaging experiment. For both fixed- and live-cell imaging, representative images of optoTau-V5-Halo expressing HEK293 or ReN cells were acquired with higher resolution settings (Nyquist sampling) using a 60× oil-immersion objective.
Image analysis
Confocal images were post-processed using Nikon 3-D deconvolution, and MaxIP images were further processed and analyzed using Nikon’s NIS-Elements software. Intensity-based analysis was conducted using the General Analysis 3 (GA3) plugin (NIS-Elements). Before any quantification was assessed, all the channels collected within an image were corrected for background subtraction and potential uneven illumination using the “rolling ball” method. The general analysis included the counting of nuclei number (DAPI) per field of view and the total summed intensity (per cell or field of view) for the specific channels being imaged (FITC, TRITC, and Cy5).
Statistical analysis
Statistical significance was calculated with GraphPad Prism software (Version 9.3) and resulting p-values less than or equal to 0.05 were considered significant. No statistical methods were used to predetermine sample sizes. The normality of the datasets was verified with the Shapiro-Wilk normality test and equality of variances with an F test. Otherwise, data distribution was assumed to be normal. Unpaired Student t tests were used to determine statistical significance in datasets comparing two variables. One-way or two-way ANOVA followed by Tukey’s multiple comparison test was used for multiple variables. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. For details of statistical analysis, see respective figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
References
Hippius, H. & Neundörfer, G. The discovery of Alzheimer’s disease. Dialogues Clin. Neurosci. 5, 101–108 (2003).
Dugger, B. N. & Dickson, D. W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 9, a028035 (2017).
Nelson, P. T. et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381 (2012).
Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T. & Hyman, B. T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639 (1992).
Josephs, K. A. et al. Beta-amyloid burden is not associated with rates of brain atrophy. Ann. Neurol. 63, 204–212 (2008).
Iqbal, K., Liu, F., Gong, C. X. & Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 7, 656–664 (2010).
Zempel, H. & Mandelkow, E. Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 37, 721–732 (2014).
Guo, J. L. & Lee, V. M. Y. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 20, 130–138 (2014).
Alquezar, C., Arya, S. & Kao, A. W. Tau post-translational modifications: dynamic transformers of Tau function, degradation, and aggregation. Front. Neurol. 11, 595532 (2020).
Wang, Y. & Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21 (2016).
Binder, L. I., Frankfurter, A. & Rebhun, L. I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 101, 1371–1378 (1985).
Qian, W. et al. PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3beta. J. Alzheimers Dis. 19, 1221–1229 (2010).
Bodea, L.-G., Eckert, A., Ittner, L. M., Piguet, O. & Götz, J. Tau physiology and pathomechanisms in frontotemporal lobar degeneration. J. Neurochem. 138, 71–94 (2016).
Long, J. M. & Holtzman, D. M. Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179, 312–339 (2019).
Limorenko, G. & Lashuel, H. A. Revisiting the grammar of Tau aggregation and pathology formation: how new insights from brain pathology are shaping how we study and target Tauopathies. Chem. Soc. Rev. 51, 513–565 (2022).
Congdon, E. E. & Sigurdsson, E. M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 14, 399–415 (2018).
Jadhav, S. et al. A walk through tau therapeutic strategies. Acta Neuropathol. Commun. 7, 22 (2019).
Cummings, J. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement 5, 272–293 (2019).
Lim, C. H. et al. Application of optogenetic Amyloid-β distinguishes between metabolic and physical damages in neurodegeneration. eLife 9, e52589 (2020).
Zhang, X. et al. The proline-rich domain promotes Tau liquid-liquid phase separation in cells. J. Cell Biol. 219, e202006054 (2020).
Bérard, M. et al. Optogenetic-mediated spatiotemporal control of α-synuclein aggregation disrupts nigrostriatal transmission and precipitates neurodegeneration. SSRN J. https://doi.org/10.2139/ssrn.3416893 (2019).
Mann, J. R. et al. RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 102, 321–338.e8 (2019).
Cowan, C. M. & Mudher, A. Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 4, 114 (2013).
Jiang, L. et al. Interaction of tau with HNRNPA2B1 and N6-methyladenosine RNA mediates the progression of tauopathy. Mol. Cell 81, 4209–4227.e12 (2021).
Kawano, F., Suzuki, H., Furuya, A. & Sato, M. Engineered pairs of distinct photoswitches for optogenetic control of cellular proteins. Nat. Commun. 6, 6256 (2015).
Zoltowski, B. D., Vaccaro, B. & Crane, B. R. Mechanism-based tuning of a LOV domain photoreceptor. Nat. Chem. Biol. 5, 827–834 (2009).
Zoltowski, B. D. & Crane, B. R. Light activation of the LOV protein vivid generates a rapidly exchanging dimer. Biochemistry 47, 7012–7019 (2008).
Laferrière, F. et al. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat. Neurosci. 22, 65–77 (2019).
Wolstenholme, C. H. et al. AggFluor: fluorogenic toolbox enables direct visualization of the multi-step protein aggregation process in live cells. J. Am. Chem. Soc. 142, 17515–17523 (2020).
Liu, Y. et al. Modulation of fluorescent protein chromophores to detect protein aggregation with turn-on fluorescence. J. Am. Chem. Soc. 140, 7381–7384 (2018).
Seidler, P. M. et al. Structure-based inhibitors of tau aggregation. Nat. Chem. 10, 170–176 (2018).
Mocanu, M.-M. et al. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J. Neurosci. 28, 737–748 (2008).
Barghorn, S. et al. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry 39, 11714–11721 (2000).
Buée, L., Bussière, T., Buée-Scherrer, V., Delacourte, A. & Hof, P. R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33, 95–130 (2000).
Espinoza, M., de Silva, R., Dickson, D. W. & Davies, P. Differential incorporation of tau isoforms in Alzheimer’s disease. J. Alzheimers Dis. 14, 1–16 (2008).
Wesseling, H. et al. Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell 183, 1699–1713.e13 (2020).
Strang, K. H., Golde, T. E. & Giasson, B. I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Invest. 99, 912–928 (2019).
Liu, F. & Gong, C.-X. Tau exon 10 alternative splicing and tauopathies. Mol. Neurodegener. 3, 8 (2008).
Fitzpatrick, A. W. P. et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190 (2017).
Choi, S. H. et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278 (2014).
Hochmair, J. et al. Molecular crowding and RNA synergize to promote phase separation, microtubule interaction, and seeding of Tau condensates. EMBO J. 41, e108882 (2022).
Ghosh, A. et al. Alzheimer’s disease-related dysregulation of mRNA translation causes key pathological features with ageing. Transl. Psychiatry 10, 192 (2020).
Fenno, L., Yizhar, O. & Deisseroth, K. The development and application of optogenetics. Annu. Rev. Neurosci. 34, 389–412 (2011).
Park, H. et al. Optogenetic protein clustering through fluorescent protein tagging and extension of CRY2. Nat. Commun. 8, 30 (2017).
Wiens, M. D. & Campbell, R. E. Surveying the landscape of optogenetic methods for detection of protein-protein interactions. Wiley Interdiscip. Rev. Syst. Biol. Med. 10, e1415 (2018).
Redchuk, T. A. et al. Optogenetic regulation of endogenous proteins. Nat. Commun. 11, 605 (2020).
Otte, C. G. et al. Optogenetic TDP-43 nucleation induces persistent insoluble species and progressive motor dysfunction in vivo. Neurobiol. Dis. 146, 105078 (2020).
Acknowledgements
The study was supported by the National Institute on Aging Grant R01 AG058851, an ADRC seed monies grant, and a grant from the Clear Thoughts Foundation to A.T. B.H. was supported by an NIH T32 (AG021885). We thank the Thathiah lab for the helpful discussions. We are especially grateful to Thais Rafael Guimarães for helpful discussions and assistance with the preparation of the manuscript. BioRender was used to generate the images in Figs. 1A, B, E; 2A, B, F, K; 3A, F; and 4A, E, G and Supplementary Figs. 1a and 3a.
Author information
Authors and Affiliations
Contributions
B.H. and A.T. designed the study. Material preparation and experiments were performed by B.H. Data collection and analysis were performed by B.H. The manuscript was written by B.H. and A.T. C.D. provided helpful discussion. X.Z. provided the HaloTag plasmid and AggFluor dyes and helpful discussion. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Abid Oueslati and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Benjamin Bessieres. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hurtle, B., Donnelly, C.J., Zhang, X. et al. Live-cell visualization of tau aggregation in human neurons. Commun Biol 7, 1143 (2024). https://doi.org/10.1038/s42003-024-06840-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06840-z
- Springer Nature Limited