
Abstract
High quality label-free imaging of oocytes and early embryos is essential for accurate assessment of their developmental potential, a key element of assisted reproduction procedures. To achieve this goal, we propose full-field optical coherence microscopy (FF-OCM), constructed as a compact module fully integrated with a commercial wide-field fluorescence microscope. Our system achieves optical sectioning in wide-field, high in-plane resolution of 0.5 µm, and high sensitivity to backscattered light. To demonstrate its imaging capabilities, we study live mouse oocytes and embryos at all important stages of meiotic maturation and early embryogenesis. Our system enables visualization of intracellular structures, which are not visible in common bright-field microscopy, i.e., internal structure of nuclear apparatus, cytoskeletal filaments, cellular cortex, cytoplasmic protrusions, or zona pellucida features. Additionally, we visualize and quantify intracellular dynamics like cytoplasmic stirring motion, nuclear envelope fluctuations and nucleolus mobility. Altogether, we demonstrate that FF-OCM is a powerful tool for research in developmental biology that also holds great potential for non-invasive time-lapse monitoring of oocyte and embryo quality in assisted reproduction.

Similar content being viewed by others


Introduction
Label-free imaging of the internal structure and dynamics of living cells provides important insights into cellular function1,2 and may facilitate accurate assessment of oocyte and embryo quality in assisted reproduction3,4,5,6. The established standards for non-invasive imaging in embryology are bright-field (BF) and differential interference contrast (DIC) microscopy7,8. The contrast in BF microscopy arises from the attenuation of the transmitted light. Despite its simplicity and widespread use, the method suffers from high-intensity background, compromised image sharpness and poor optical sectioning as the out-of-focus light cannot be rejected. Conversely, DIC microscopy relies on the interference between two slightly shifted light beams passing through the sample, while the contrast arises from the gradient of refractive index. This mainly enhances the edges of highly transparent cells and organelles, which appear darker or brighter than the uniform gray background. The contrast in DIC is primarily generated within the focal plane of the objective, which notably improves the optical sectioning with respect to BF microscopy, however, with no rejection of out-of-focus light.
In developmental biology research, live-cell confocal fluorescence microscopy has emerged as the leading microscopy technique due to its specificity and optical sectioning9,10. The former is enabled by the specific binding of a fluorescent label to the molecule of interest or by the expression of a fluorescent protein in a living cell. The latter is enabled by a confocal pinhole that rejects the out-of-focus fluorescence light. This results in excellent image contrast of bright sample features on the black background and enables volumetric reconstructions of the specimen. However, both irradiation and fluorescent labeling in confocal fluorescence microscopy hinder embryo development after imaging, rendering it inherently invasive11,12 and effectively inapplicable from a clinical perspective. Additionally, fluorophores are susceptible to photo-bleaching, which limits their use for longitudinal monitoring of embryo development.
Microscopy-based quality assessment of oocytes and embryos is crucial for the effectiveness of assisted reproduction procedures, particularly in vitro fertilization. Microscopic visualization may be conducted either in several selected time points or through a time-lapse imaging. Currently, it relies mostly on cell features visible in standard BF and DIC microscopes, such as shape, size and number of cells, number of nuclei in each cell, presence of cytoplasmic fragments (so-called fragmentation), cytoplasmic granularity or zona pellucida thickness and evenness13,14,15. Additionally, spindle shape and zona pellucida birefringence as other biomarkers of oocyte quality, can be assessed with polarized light microscopy16. All these features, although helpful in the evaluation of oocyte/embryo quality, are not sufficiently reliable to predict oocyte/embryo ability to undergo full-term development13,15,17. To extend the repertoire of oocyte/embryo quality biomarkers, a number of advanced microscopy techniques for non-invasive imaging have been proposed, including quantitative phase microscopy18, holographic microscopy8, dynamic light scattering19 and optical coherence tomography (OCT)20,21. In particular, it has been demonstrated that OCT is well-suited to in vivo volumetric imaging of mouse embryos22 and in vivo monitoring of oocytes/embryos transported through the oviduct23. OCT relies on the highly sensitive interferometric detection of light backscattered in the sample volume, which enables optical sectioning and volumetric imaging. OCT typically provides lateral and axial resolution of a few micrometers and imaging depth of several millimeters. Optical coherence microscopy (OCM) is a high-resolution variant of OCT, providing spatial resolution of <2 μm, and imaging depth of several hundred micrometers. OCM has been demonstrated for imaging mouse, bovine and porcine oocytes and preimplantation embryos24,25,26. Furthermore, it has been demonstrated that the method is non-invasive, i.e., the optical power used for imaging does not harm mouse oocytes and embryos27,28,29.
In a typical OCT or OCM configuration, the imaging beam is focused and scanned across the sample so that only a single sample point is illuminated at any given time. The depth profile of the sample is retrieved from the spectral interference pattern for each point separately and assembled in post-processing to generate a volumetric image. The imaging depth is primarily determined by the spectral resolution. However, such configurations require an imaging beam with a depth of focus larger than the imaging depth, hence, only low numerical aperture (NA) objectives are suitable. This in consequence hinders high lateral resolution imaging. This problem is addressed in full-field OCM (FF-OCM), in which the sample is illuminated in a wide-field configuration and a two-dimensional interference pattern is recorded on a camera30. As such, one 2D optical cross-section in the en face plane is generated and high NA objectives can be used to achieve high lateral resolution31. The cross-section depth is selected by adjusting the sample position in the z direction. FF-OCM can achieve spatial resolution comparable to confocal fluorescence microscopy, allowing biological specimens, such as preimplantation mouse embryos, to be imaged in detail at the intracellular level32,33. Furthermore, the OCM signal is sensitive to the movement of scattering features, thus, providing additional information on cellular dynamics. Recently, the analysis of time-varying FF-OCM, termed dynamic FF-OCM34, allowed for functional imaging of individual cells in a tissue35,36,37.
In this paper, we present a compact FF-OCM module fully integrated with a commercial inverted wide-field fluorescence microscope. The FF-OCM module together with basic control electronics and software are the only added elements to the commercial microscope, which makes our design a cost-effective solution. We use the system to study live mouse oocytes and embryos at all important stages of meiotic maturation and early embryogenesis. We achieve high-contrast tomographic images with a high in-plane resolution of 0.5 µm. The improved imaging quality is of key importance, as it allowed us to visualize intracellular structures that are hardly visible in standard microscopy techniques or previous OCM setups24,27,32,33,38, e.g., internal structure of the nuclei and spindles, cytoskeletal filaments, cellular cortex, cytoplasmic protrusions or zona pellucida features. In contrast to the existing studies, we were able to perform state-of-the-art analysis of intracellular dynamics, i.e., cytoplasmic streaming flow and fluctuations of the nuclear envelope, with no need for fluorescent labeling. Detailed label-free imaging of intracellular structures provides valuable insights into basic biological research, and shows great potential for non-invasive assessment of the developmental capability of oocytes and embryos in assisted reproduction procedures. Therefore, FF-OCM is well-suited as a complementary technique to live-cell confocal fluorescence microscopy for structural imaging, as well as a cost-effective stand-alone technique for imaging intracellular dynamics.
Results and discussion
An overview of developmental stages imaged by FF-OCM
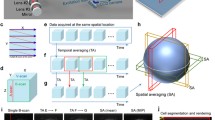
Our imaging system is based on an inverted full-field fluorescence microscope. The compact FF-OCM module is designed to fit into the space normally occupied by a filter wheel in an upper level of a two-level microscope. The FF-OCM uses the same light source and camera as wide-field fluorescence. This allows for sequential imaging of a specimen by BF, wide-field fluorescence and FF-OCM techniques. The sample is illuminated from the bottom through a coverslip (Fig. 1a).

a Schematic of the imaging geometry. The sample is illuminated from the bottom, through a coverslip and a single in-plane optical cross-section is imaged at any given time. b The comparison of bright-field (BF) and FF-OCM images of a representative GV oocyte on linear (FF-OCM) and logarithmic (FF-OCM log) scales. Characteristic cellular features are marked with arrows: (1) zona pellucida, (2) cytoplasm, (3) nucleus, (4) nucleolus. c Z-stack of optical cross-sections separated by 2.5 µm through the nucleolus of GV oocyte. Images reveal the position of the nucleolus in the nucleus marked by yellow arrows. d Stages of meiotic maturation and early embryogenesis imaged by FF-OCM and BF with representative images. White arrows indicate nuclear apparatus, red arrows indicate: (1) 1st polar body in MII oocyte, (2) cell borders in morula, (3) blastocoel cavity in blastocysts and (4) cytoplasmic strings protruding into blastocoel. All scale bars: 10 µm.
FF-OCM can visualize intracellular structures which efficiently backscatter the incident light. Therefore, in combination with optical sectioning, FF-OCM provides high contrast between cellular components appearing bright in the images and the non-scattering medium appearing as a black background (Fig. 1b). The obtained scattering signal may also be mapped on a logarithmic scale in image processing. This approach, common in OCT imaging, facilitates visualization of weakly scattering structures placed in the vicinity of highly scattering ones without overexposure. It is particularly useful in visualizing the nuclear apparatus (Fig. 1b) as the scattering potential of the nucleus in mammalian cells is typically an order of magnitude lower than cytoplasm39. Our system enables the user to select an arbitrary in-plane optical cross-section of the specimen, termed the imaging plane, and to monitor it over time. One may also create a z-stack of cross-sections to visualize selected structures in 3D (Fig. 1c).
We used our FF-OCM system to visualize intracellular architecture of mouse oocytes and embryos at different stages of their meiotic maturation and preimplantation development (Fig. 1d). All features characteristic for each stage are readily visible, e.g., nucleus and nucleolus in oocytes arrested in prophase of the 1st meiotic division (so-called GV oocytes), 1st polar body and metaphase spindle in oocytes arrested in metaphase of the 2nd meiotic division (MII oocytes), pronuclei in zygotes or blastocoel cavity in blastocysts. Notably, such features are difficult to observe in standard BF images (Fig. 1d). Additionally, the individual cell borders in morulae and cytoplasmic strings protruding into blastocoel in blastocysts are readily distinguishable while not visible in BF images. Both these features have been indicated as potentially valuable markers of embryo quality29,40,41,42,43,44. Representative examples of intracellular dynamics in oocytes and embryos imaged by FF-OCM are presented in Supplementary Videos 1–7, respectively.
FF-OCM can visualize the detailed architecture of nuclear apparatus in oocytes and embryos
Figure 2 illustrates the capabilities of FF-OCM in imaging nuclear apparatus of mouse GV oocytes. First, FF-OCM can discern non-surrounded nucleolus (NSN) from surrounded nucleolus (SN) GV oocytes, as well as visualize the transition from NSN to SN states (Fig. 2a). In essence, the transition consists of reconfiguration of chromatin in oocyte nuclei strictly related to the cessation of their transcriptional activity. The chromatin in transcriptionally active NSN oocytes is dispersed in the nucleoplasm and fully condensed around the nucleolus for transcriptionally inactive SN oocytes45. For the NSN oocytes, we observe a non-central location of the nucleus in the oocyte, non-spherical nucleolus shape, and relatively bright, uniform signal from the nucleoplasm. This uniform signal decreases when the chromatin starts to condense and form a characteristic brighter ring around the nucleolus typical for the SN state. We observed that SN nucleolus is often located off-center, in close proximity to the nuclear envelope. The differences in signal level from chromatin and corresponding ring formation are clearly visible in the angularly averaged intensity profile shown in Fig. 2b. The NSN–SN transition in mouse oogenesis is one of the main prerequisites for embryo development. SN oocytes show significantly higher competence to complete 1st meiotic division, respond to fertilization, and undergo further embryonic development46,47,48. Such differences in chromatin configuration in GV oocytes and the corresponding developmental potential have also been reported for other mammalian species49. Therefore, the ability to visualize chromatin conformation may improve the assessment of oocyte quality, particularly in assisted reproduction protocols involving in vitro maturation of GV oocytes.

a Bright-field (BF) and OCM (OCM log) images of GV oocytes with different types of chromatin configuration: NSN non-surrounded nucleolus, SN surrounded nucleolus, NSN/SN transitional stage. b Angularly averaged pixel values of OCM log in full rotation about the center of the nucleolus marked by the asterisk in (a). The averaging procedure is schematically shown for the NSN/SN oocyte in (a). c Two types of fibrous structures in the nucleus of GV oocytes: (1) single long strands in the nucleoplasm (cyan arrow) and (2) bright and highly dynamic structures in the proximity of nuclear envelope (magenta arrows). d–f Dynamics of germinal vesicle breakdown (GVBD). d Oocyte in GV state 3 h before the transition. e Selected images from a time-lapse imaging of nucleolus dissolution dynamics during GVBD. f Oocyte 3 h after the GVBD transition. All FF-OCM images are presented on a logarithmic scale. Cropped region marked with white square. All scale bars: 10 µm.
In addition, FF-OCM allows us to visualize two types of fibrous structures contained in the nucleus of most GV oocytes imaged, as shown in Fig. 2c. First, we observed single long strands floating in the nucleoplasm (Fig. 2c cross-section 1). Notably, Scheffler et al.50 have shown that prominent nuclear actin-filament structures in the form of strands are frequently found in the GV nuclei and that their presence correlates with meiotic developmental capacity. Second, we can also visualize bright and highly dynamic structures located in the proximity of the nuclear envelope (Fig. 2c cross-section 2 and Supplementary Video 8). We hypothesize they are cross-sections through a wrinkled nuclear lamin sheets, as they closely resemble lamin layer imaged previously in GV oocytes51.
Finally, we used time-lapse FF-OCM to visualize the dynamics of meiosis resumption in GV oocytes (so-called germinal vesicle breakdown (GVBD)). Figure 2d–f shows the oocyte before and after the GVBD transition. We observed an abrupt process of nucleolus dissolution (Fig. 2e and Supplementary Video 9). Simultaneously, the signal from the nucleoplasm becomes notably elevated. Rapid decrease of the nucleolus diameter from initial 11 µm to complete dissolution lasted for ~12 min.
The optical sectioning of FF-OCM allows us also to visualize the location of spindle in MII oocytes, while the high optical resolution gives insights into the spindle structure. Depending on the orientation of the spindle in the cell, the long axis of the spindle is inclined with respect to the imaging plane. Figure 3a depicts a spindle positioned approximately perpendicular to the imaging plane, while Fig. 3b shows two closely spaced cross-sections through a spindle positioned approximately parallel to the imaging plane. The microtubule bundles forming the spindle are readily visible for both orientations, most prominently for long time signal averaging of 100 s. The two longitudinal sections closely spaced in axial direction (Fig. 3b) visualize the location of spindle poles. The spindle structure imaged by FF-OCM is remarkably well collocated with the position of chromosomes imaged by wide-field fluorescence. We hypothesize that dark regions visible at the spindle equator plane indicate the exact position of individual chromosomes. This hypothesis can be verified with further technical development of our imaging method toward higher spatial resolution.

Bright-field (BF) and OCM (OCM log) images of MII oocytes presenting a cross-section and b longitudinal sections through the MII spindle in comparison with the wide-field fluorescent image of DNA stained with Hoechst 33342 (HOE). FF-OCM visualizes microtubule bundles forming the spindle (yellow arrows, no. 1). Long time signal averaging of 100 s (OCM log 100 s) reveals a more detailed fibrous structure of the spindle. Two closely spaced sections in (b) reveal the location of spindle poles (red arrows, no. 2). Dark regions between bundles visible at the spindle equator plane (blue arrows, no. 3) indicate the possible location of individual chromosomes. c Bright-field (BF) and OCM (OCM log) images of MII oocytes aged in vitro for 24 and 48 h indicating disrupted MII spindles (magenta arrows, no. 4). Cropped region marked with white square. All scale bars: 10 µm.
It has been proposed that the shape and structure of the MII spindle can serve as an indicator of oocyte quality in assisted reproduction52,53,54,55. To verify whether FF-OCM can depict abnormalities of the spindle shape, we imaged mouse MII oocytes aged in vitro for 24 and 48 h. The aged cells display spindles that are shrunk and abnormally shaped compared to spindles visualized in fresh MII oocytes (Fig. 3c). Indeed, it has been reported previously by numerous groups that this type of aging leads to deterioration of the spindle structure56,57.
Nuclear apparatus is also readily distinguishable in FF-OCM images of embryos (Fig. 1d), e.g., in zygotes, high contrast enables observation of pronuclei. The contrast between the cytoplasm and nuclei gradually decreases from the zygote to blastocyst stage, which we attribute to chromatin remodeling related to the resumption of transcriptional activity58,59. The ability to visualize nuclei in embryos may also be helpful in embryo quality assessment. It has been shown that number, size and location of nuclei in human zygotes are associated with the embryo developmental potential4,60,61. Moreover, visualization of nuclei may help detecting multinucleation in blastomeres, which negatively correlates with human embryo quality15,62.
FF-OCM visualizes cellular cortex and details of zona pellucida structure
Another structure that backscatters light and can therefore be visualized using FF-OCM are trans-zonal projections (TZP), i.e., long extensions of cumulus cells that protrude into the zona pellucida and connect to the GV oocyte for bidirectional cellular communication63 (Fig. 4a, upper row). Notably, such structures are not visible in the corresponding BF images (Fig. 4a) nor in DIC images in the literature64. The cumulus cells appear opaque at the green LED light used in this study in the FF-OCM setup, preventing long-range penetration. This precludes imaging oocytes through the cumulus and causes a shadow in the image when an oocyte is partially occluded by a cumulus cell beneath. As TZPs are predominantly built from actin filaments63, we hypothesize that actin filaments are particularly efficient light scatterers, contributing substantial signal to the FF-OCM image. Therefore, we believe that the region indicated as zona pellucida in FF-OCM images is actually an image of a dense mesh of TZPs penetrating a transparent protein matrix65.

a Bright-field (BF) and OCM images of representative GV oocytes showing, in the upper row, trans-zonal projections (magenta arrows, no. 3) between an oocyte and cumulus cells (red arrows, no. 1), and in the bottom row, projections extending from the zona pellucida across the perivitelline space to the oocyte (cyan arrows, no. 4). Long time signal averaging of 100 s (OCM 100 s) reveals more detailed structure of trans-zonal projections. Partial occlusion of OCM signal by cumulus cell located beneath the imaging plane is marked with yellow arrow, no. 2. b Selected images from a time-lapse recording of protrusion remodeling in a representative GV oocyte subjected to a prolonged in vitro culture. Cyan and magenta arrows point to the ends of a selected protrusion in the final image frame and retrospectively to the same image pixels in previous frames for a better visual assessment of the protrusion remodeling. c Bright-field (BF) and OCM images of representative oocytes (GV, GVBD, MII) and early embryos (zygote, morula and morula/blastocyst transition) depicting remodeling of zona pellucida (blue arrows no. 1) and cell cortical region (green and orange arrows no. 2 and 3) during meiotic maturation and early embryogenesis. High signal from actin-rich cortical layer is indicated by green arrows (no. 3), low signal is indicated by orange arrows (no. 3). Red arrows indicate microlumens in morula/blastocyst transition stage. Cropped region marked with white square. All scale bars: 10 µm.
GV oocytes placed in in vitro culture tend to decrease their volume, in which case we often observed long straight protrusions extending from the zona pellucida across the perivitelline space to the oocyte (Fig. 4a, bottom row). Time-lapse FF-OCM imaging allows us to visualize the dynamics of protrusion remodeling in such oocytes (Supplementary Video 10). We observed significant elongation of the protrusions linking the zona pellucida and the oocyte, accompanying by decrease in the oocyte volume and decrease in FF-OCM signal in the cell cortex. The calculated average growth rate of the protrusions was 50 nm/s.
Moreover, FF-OCM allows us to investigate changes in the structure of zona pellucida during oocyte and embryo development, as shown in Fig. 4c. We observe no significant differences in the zona pellucida between GV, GVBD and MII stages. However, our data indicate that fertilization coincides with the gradual dissolution of the putative TZPs in the zona pellucida, which start to appear in FF-OCM images as bright dots rather than a mesh. The properties of the zona pellucida have been considered as one of the potential markers of oocyte and embryo quality in humans13,14, thus FF-OCM images may also be used for this purpose.
The FF-OCM signal in the cortical layer of the cell also changes between developmental stages. In GV and GVBD oocytes, we observed a bright ring at the border between the zona pellucida and the cytoplasm, which disappears in MII oocytes. We attribute the high OCM signal in the cortical layer to the high content of actin. However, we did not observe characteristic cortical polarity in fresh MII oocytes, presumably, because the overall content of cortical actin is significantly lower in MII cells than in GV and GVBD cells66, which might be below the sensitivity of our FF-OCM system. Cell cortex was also observed in apical parts of outer blastomeres in morulae (Fig. 4c), most likely reflecting the accumulation of actin filaments in this regions67. Additionally, at the morula/blastocyst transition, FF-OCM reveals the separation of the neighboring blastomeres and the formation of microlumens (Fig. 4c) that will subsequently merge into a blastocoel. Similar structures have been also observed by fluorescence confocal microscopy68. Visualization of cellular cortex may serve as a tool in the evaluation of oocyte or embryo developmental capabilities. The usefulness of this feature requires further research, but it is likely that the intensity and localization of the cortical signal reflects the quality of cytoskeleton, which being involved in multiple processes ranging from intracellular trafficking to cell division, may be a valuable marker of oocyte/embryo quality.
Analysis of FF-OCM signal fluctuation in time reveals cytoplasm motion dynamics in oocytes
Active remodeling of cytoplasm in cells is driven by cytoskeletal motor proteins69,70,71,72,73. In growing mouse oocytes, random forces predominantly generated by actin filaments and myosin-motor activity result in random stirring movements of cytoplasm which drive non-specific transport of organelles, e.g., displacement of nucleus from peripheral to center location74,75. The movement of cytoplasm in x–y directions (imaging plane) can be simply observed by time-lapse FF-OCM imaging. However, FF-OCM signal is also sensitive to the movement of scattering features in the z direction perpendicular to the imaging plane76. Therefore, the motility of the cell cytoplasm and organelles causes fluctuations of FF-OCM intensity recorded over time. By analyzing the power spectral density (PSD) of such time-varying signals we can decompose and visualize the intensity of cytoplasm motion activity at different frequency components. In particular, one can isolate the static signal from the dynamic one. The static signal appears below the minimum measurable frequency (equal to the inverse of the total measurement time), whereas the dynamic signal appears between the minimum measurable frequency and the frequency at which the PSD hits the noise level. To demonstrate these capabilities, we compared control oocytes and oocytes treated with cytochalasin D (CytoD), an inhibitor of actin-filament polymerization (Supplementary Videos 1 and 11). Figure 5a depicts the analysis of power spectrum for control and CytoD-treated GV oocytes. First, the analysis for untreated cells reveals high dynamics in the cytoplasm and little dynamics in the zona pellucida. This accords with zona pellucida being a static extracellular structure. We attribute the residual dynamic signal in zona pellucida to external vibrational noise affecting the entire system. Second, the static part of FF-OCM signal within the cytoplasm region is significantly increased for CytoD-treated oocytes resulting from inhibition of the stirring cytoplasmic movements. The PSD averaged over the cytoplasm and zona pellucida area for control and CytoD-treated oocyte is depicted in Fig. 5b. The plot shows a clear difference between the two groups mostly in the low-frequency region, i.e., below 1 Hz. Higher frequency oscillations remain similar in both cases indicating that faster movements of cell cytoplasm are less affected by CytoD, hence, are not related to actin polymerization process. The apparent peak in PSD at ~0.5 Hz, results most likely from mechanical noise generated by the equipment working in the laboratory. The peak is clearly visible for zona pellucida region which is at extracellular protein structure with virtually no internal motility.

a Comparison of cytoplasm motion dynamics in control and cytochalasin D (CytoD) treated GV oocytes divided into static (frequency < 0.01 Hz) and dynamic (frequency range 0.01–3 Hz) components of FF-OCM intensity fluctuations. b Power spectrum density of FF-OCM intensity fluctuations averaged for the whole cytoplasm and zona pellucida for control and CytoD-treated GV oocytes. c Exponent of inverse power law fitted to the power spectrum for control and CytoD-treated oocytes calculated for five oocytes in each group. Box plot: center line, median; box limits, upper and lower quartiles; whiskers, 1.5× interquartile range. Statistics: two-tailed paired sample t-test; ***p < 0.001. All scale bars: 10 µm.
It has been shown that the PSD of OCT fluctuations caused by deterministic motion in living cells follow the inverse power law dependence77: \(y=a{x}^{-b}\), which appears as linear dependence in log–log plot. It can be described by two fitting parameters: the proportionality factor \(a\) and the exponent \(b\). The proportionality factor describes static scattering potential of the samples while the exponent describes the spectral shape of PSD, hence, the characteristics of cytoplasm motion. For the same a, higher values of b indicate higher velocities of scattering features moving in the cytoplasm and oscillations distributed over a broader frequency range, hence, higher motility of the cytoplasm. As such, we observe statistically significant differences of the exponent between the untreated and CytoD-treated oocytes, as shown in Fig. 5c. Such parameter describing cytoplasm motion can be straightforwardly extracted from FF-OCM data with little computational power requirements in contrast to methods based on sequence image correlations6,78.
FF-OCM enables quantification of nuclear envelope fluctuations and nucleolus movement in oocytes
The random forces generated by cell’s actomyosin cytoskeleton that cause cytoplasm stirring also exert random forces, i.e., ‘kicks’, on the nucleus, which in turn causes random fluctuations of the nuclear envelope51,74. Importantly, these forces are propagated into the nucleus enhancing the mobility of nucleolus and contributing to chromatin remodeling and regulation of gene expresion51,74. The high contrast on FF-OCM images between the nucleus and cytoplasm in GV oocytes enables automatic image segmentation and analysis of the nuclear envelope fluctuations over time. Figure 6a and Supplementary Videos 1 and 11 show the representative examples of control and CytoD-treated GV oocytes imaged for 300 s. Nuclei in CytoD-treated oocytes have more convoluted shape than in control cells. It has been suggested that these invaginations are caused by microtubules protruding from microtubule organizing centers (MTOCs) present in the proximity of the nuclei51. Moreover, the control oocytes show intense stirring of the entire cytoplasm resulting in strong fluctuations of the nuclear envelope, which has been demonstrated by 3D confocal fluorescence microscopy74. As we monitor only a single cross-section through the nucleus, we cannot exclude the possibility that axial movements of the nucleus greater than the depth of focus might also contribute to the apparent change of nuclear envelope shape. Nonetheless, relevant results on nuclear dynamics were previously obtained using confocal fluorescence microscopy to monitor 2D cross-sections of the nucleus51,79.

a Nuclear envelope fluctuation in control GV oocytes in comparison to oocytes treated with cytochalasin D (CytoD) imaged for 300 s. Representative bright-field (BF) and OCM images are shown. Orange and blue arrows mark regions of high and low fluctuations, respectively. Arrows in consecutive time stamps point to the same image pixel to help visually assess the change of shape. b Averaged nuclear envelope fluctuations of control and CytoD-treated oocytes shown in (a). The gray color scale shows a fraction of the measurement time for which nuclear envelope remained in a certain position. A well-defined single line in this plot indicates no significant change of shape, while fuzzy sections of the envelope show regions with high fluctuations. c Mean nuclear envelope shape and corresponding standard deviation for control and CytoD-treated oocytes presented in (a). d Nuclear envelope stability parameter calculated as a fraction of non-zero pixels in the averaged nuclear envelope fluctuations images (b) remaining after measurement time fraction \({t}_{f}\), normalized to the nuclear envelope perimeter. Stability parameter shows exponential decay with increasing \({t}_{f}\). High volatility is characterized by fast decay, i.e., low decay lifetime. A perfectly stable envelope shape would be characterized by a constant value of 1. e, f Decay lifetime \({\tau }_{v}\) and amplitude \({A}_{v}\) of the nuclear envelope stability parameter for control and CytoD-treated oocytes calculated for five oocytes in each group. Curves fitted in the linear range of log plot, i.e., 0–50% and 0–75% for control and CytoD group, respectively. Box plot: center line, median; box limits, upper and lower quartiles; whiskers, 1.5× interquartile range. Statistics: two-tailed paired sample t-test; **p < 0.01, ***p < 0.001. g Space-time trajectories of nucleus centroid and h the calculated mean square displacement (MSD) for each trajectory for control and CytoD-treated oocytes presented in (a). The slopes α of linear fit in the regions marked by solid lines are indicated. The fitting error for all regions was lower than 0.02. Cropped region marked with white square. All scale bars: 10 µm.
The stirring movements observed in our study are substantially inhibited in CytoD-treated oocytes. The difference in fluctuation intensity is clearly visible in the time average graphs (Fig. 6b), which show the fraction of measurement time for which the nuclear envelope remained in a certain position. This information allows us to extract the mean nuclear envelope shape and corresponding standard deviation, i.e., fluctuation intensity, plotted in Fig. 6c. To quantify the nuclear envelope fluctuations we introduce a stability parameter defined as non-zero pixels in the averaged nuclear envelope fluctuations images remaining after a measurement time fraction \({t}_{f}\), normalized to the nuclear envelope perimeter (Fig. 6d). Stability parameter decay exponentially with increasing \({t}_{f}\). The decay lifetime and amplitude show statistically significant differences between the control and CytoD-treated oocytes (Fig. 6e, f).
Depolymerization of actin filaments, which inhibits the stirring movements of the cytoplasm and fluctuations of the nucleus shape, also substantially reduces the motility of the nucleolus. The space-time trajectories of nucleolus centroid and corresponding mean squared displacement80 (MSD) for the control and CytoD-treated oocytes are plotted in Fig. 6d, e, respectively. MSD describes average squared displacement for time delay \(\tau ={nt}\), where \(t\) is the time between consecutive points in the trajectory, that passed from the start of the observation. In the experiment, we monitor a single cross-section in time, thus, measured trajectory is a 2D projection of a 3D motion, which underestimates the actual nucleolus displacement. Nonetheless, the MSD is substantially higher in the control oocytes than in the CytoD-treated ones for all time delays \(\tau\) indicating higher mobility of nucleolus. The MSD can be described by the generalized diffusion model which provides additional insight into the physical nature of the motion. The essential parameter in the diffusion model is the slope α of MSD in log–log plot. The linear relationship between MSD and time (i.e., the slope α of MSD in log–log plot \(\alpha =1\)) indicates random walk motion characteristic for classical free diffusion process (Brownian motion). In the subdiffusive processes, the free diffusion is inhibited resulting in slope values \(\alpha < 1\). On the contrary, superdiffusive processes with enhanced diffusion are characterized by \(1 < \alpha < 2\)80,81. For control oocytes, the motion of nucleolus in short time scales (i.e., time delays below 20 s) is substantially enhanced by external forces (\(\alpha =1.65\)). However, for longer time scales this motion is significantly inhibited transiting from super- to subdiffusive process (\(\alpha =0.48\)). Possible reason for this difference is the interaction of nucleolus with obstacles like chromatin or nuclear envelope that prevent large amplitude free diffusion. For CytoD-treated oocytes, the movement of nucleolus is close to standard diffusion in time scales below 10 s, but, similarly to control samples, becomes inhibited at longer time scales. Interestingly the transition from super to subdiffusive characteristics occurs at much longer delays for control samples than CytoD-treated ones.
The presented analysis indicates that nucleolus motility is forced by the deformation of the nuclear envelope in response to actin-filament-driven movement of the cytoplasm. Notably, our analysis of nuclear dynamics in GV oocytes agrees with the data presented in the literature, obtained with confocal fluorescence imaging51,74. As nuclear envelope fluctuations reflect the functionality of actin cytoskeleton and regulate chromatin remodeling and gene expression51,74, they may be another potential marker of oocyte quality that can be assessed by FF-OCM.
To conclude, our study demonstrates a novel FF-OCM tool for label-free imaging of living mouse oocytes and embryos. Our system achieves high resolution, optical sectioning and high contrast that enables imaging intracellular structures, which are not visible in standard BF or DIC microscopy, and measuring dynamics of cytoplasmic stirring motion and nuclear envelope fluctuations. Nonetheless, the system lacks automatic 3D scanning functionality that would greatly enhance its imaging capabilities. 3D scanning poses several technical difficulties, such as (1) the need for motorized z axis in the microscope or z-scanning stage, (2) the need for automatic correction of reference arm length due to focus defect82, and (3) compromise between the need for signal averaging in a single cross-section and the time of the 3D scan. We believe that with further development the system can be fully motorized and the FF-OCM sensitivity can be significantly increased by using immersion objectives, which would decrease the overall measurement time. Nonetheless, our best-quality images from single cross-section are acquired in 10 s, which is comparable to the scanning time in confocal fluorescence microscopy. In fact, confocal fluorescence microscopy and FF-OCM can be regarded as complementary techniques for basic biological research. Both techniques possess optical sectioning capabilities with FF-OCM providing a non-specific overview of the sample, whilst confocal fluorescence microscopy provides specific visualization of a targeted molecule. Importantly, the modular design of our FF-OCM system allows for such imaging capabilities at a fraction of the cost of a confocal microscope.
In this paper, we demonstrate that state-of-the-art research on intracellular dynamics can be performed with no need for fluorescent labeling and confocal fluorescence microscopy. Additionally, all characteristic features of oocytes and embryos recognized in BF microscopy as indicators of cell quality are visualized by FF-OCM with much higher contrast. Furthermore, a number of new static and dynamic parameters can be extracted from FF-OCM images to characterize oocytes and embryos that present great potential for facilitating evaluation of oocyte and embryo quality, i.e., chromatin conformation in GV oocytes, shape and internal structure of metaphase spindle, intensity and distribution of the cortical layer signal, presence of cytoplasmic protrusions, zona pellucida texture, dynamics of cytoplasm motion, and nuclear envelope fluctuations. Therefore, FF-OCM is a powerful tool that holds promise not only for high-resolution and high-contrast imaging in basic research, but also for efficient label-free assessment of oocyte developmental potential and preimplantation embryo quality in assisted reproduction.
Methods
FF-OCM optical setup
Our FF-OCM system is based on wide-field spatially incoherent illumination interferometry and operates in the time domain. This configuration is advantageous for reduced cross-talk83, reduced optical aberrations84, and high NA illumination and detection. This results in 0.5 µm lateral and 1.2 µm axial resolution governed by the NA of the objective.
The FF-OCM module is fully integrated with a commercial inverted wide-field fluorescence microscope (Nikon Eclipse Ti2-U). Its compact design (total dimensions 180 × 250 × 80 mm) allows for the module to fit in the space of a standard filter wheel in the upper level of the two-level microscope. This enables sequential imaging of a specimen by BF, wide-field fluorescence and FF-OCM techniques. The FF-OCM, disascopic (i.e., BF), and epi-fluorescence light paths in the microscope are schematically shown in Fig. 7a. The system uses the same LED illumination source (CoolLED pE-300 ultra) as epifluorescence. All imaging methods use the same camera with sCMOS sensor (Hamamatsu Orca Flash 4.0 v3).

a Schematic of light paths in the microscope allowing for sequential bright-field, wide-field fluorescence and FF-OCM imaging. b Schematic optical layout of the FF-OCM imaging setup. The components marked by the blue box constitute the FF-OCM module, all other optical components are a part of a commercial microscope. PZT piezoelectric transducer, BS beamsplitter, ND neutral density filter, BP bandpass filter, TL tube lens. c Schematic of FF-OCM signal acquisition and processing.
The optical layout of our FF-OCM system is schematically depicted in Fig. 7b. The configuration of FF-OCM is based on a Linnik interferometer (i.e., identical microscope objectives in both interferometer arms) comprising Nikon Plan Fluor 40×/0.75 objectives. The wide-field illumination of the specimen is controlled by the aperture and field diaphragms (Nikon LAPP system). The illumination wavelength and bandwidth are controlled by a bandpass (BP) filter. Here, we use green light illumination at central wavelength of 525 nm and bandwidth of 45 nm (Semrock 525/45). A custom-made neutral density (ND) reflective filter is used to control light intensity in the reference arm. The reference mirror is mounted on a piezoelectric actuator (ThorLabs PA3CKW) providing phase modulation of the reference field. The mirror and objective in the reference arm are mounted on a motorized translation stage to control the delay between the object and reference fields. The microscope was placed on a 5 cm thick breadboard (Standa) and isolated from the laboratory desk by rubber dumping feet (ThorLabs AV5/M).
FF-OCM data processing
A schematic of the data acquisition and processing procedures of our FF-OCM system is shown in Fig. 7c. The camera acquisition is synchronized with the phase modulation of the reference field. The phase is modulated in a saw tooth pattern and the amplitude is adjusted such that n frames are acquired during a 2π phase shift. Processing involves fast Fourier transform (FFT) calculated for each pixel in time direction for Nc modulation periods (\({n\times N}_{c}\) raw frames). The OCM signal is taken as the FFT magnitude at frequency bin \({N}_{c}\). This signal represents the Doppler frequency shift in the interference between the sample and reference fields caused by the constant velocity of the phase ramp. Increasing the number \({N}_{c}\) of frames taken for FFT allows for pronounced filtering of high-frequency noise. However, taking too many frames would also filter out the fluctuations of the OCM signal caused by the natural motion of the cell cytoplasm. Next, \({N}_{i}\) FF-COM images are averaged incoherently to improve image quality by increasing signal-to-noise ratio and reduce speckle contrast. Speckle reduction takes the advantage of natural motion of the cytoplasm, which results in multiple realizations of speckle field. The quality of final image depends on the total number of raw frames, \({N}_{T}\), taken for processing, and values of parameters \(n\), \({N}_{c}\) and \({N}_{i}\), where:
This equation defines the trade-off between imaging frame rate, high-frequency noise filtering set by parameter \({N}_{c}\), and reduction of speckle set by parameter\({N}_{i}\). For the data presented in this paper, we use a camera frame rate of 200 Hz. Four frames were acquired per modulation period (\(n=4\)) and five periods were taken for FFT (\({N}_{c}=5\)) resulting in FF-OCM imaging frame rate of 10 Hz. The maximum imaging speed is limited by the camera frame rate. The FF-OCM images presented in this paper result from incoherent averaging of \({N}_{c}=100\) frames.
To control the acquisition and provide a live preview we created a dedicated LabView software, version 2021 SP1. The synchronization and modulation signals are generated using DAQ card (NI USB-6363). The live preview is enabled by GPU accelerated FFT processing (GeForce RTX 3090, NVIDIA).
Preparation of oocytes and embryos
Mice were maintained in the animal facility of the Faculty of Biology, University of Warsaw at 14:10 h light/darkness cycle and provided with food and water ad libitum. Animals were sacrificed by cervical dislocation. All experiments were performed in compliance with the ARRIVE guidelines and national regulations.
GV oocytes were obtained from 2-month-old unprimed F1 (C57Bl6/Tar × CBA/Tar) mice. Fully grown GV oocytes were released from ovarian antral follicles by puncturing with a needle into M2 medium (M16 medium buffered with HEPES)85 with 3′,5′-cyclic AMP (dbcAMP, 150 μg/mL, Merck). Oocytes were separated from the cumulus cells by gentle pipetting.
MII oocytes were obtained by in vitro maturation of GV oocytes. In vitro maturation was performed for 15–16 h in M16 medium (Merck) at 37.5 °C and in 5% CO2 in the air. Aging of MII oocytes involved an additional 24- or 48-h long culture in M16 medium.
Zygotes were obtained by spontaneous mating of 2-month-old unprimed F1 (C57Bl6/Tar × CBA/Tar) females with 6–8-month-old F1 (C57Bl6/Tar × CBA/Tar) males. Zygotes were released from the oviducts into M2 medium in the morning of the 1st day after mating. Two-cell embryos, morulae and blastocysts were obtained by in vitro culture of the zygotes in EmbryoMax Advanced KSOM Embryo Medium (Merck) for 24, 48 and 72 h, respectively.
In some experiments, oocytes were stained for 30 min with Hoechst 33342 (100 ng/mL; Merck) or incubated with CytoD (2 μg/mL; Merck).
FF-OCM imaging protocols of oocytes and early embryos
Oocytes/embryos were placed in droplets of M2 medium on glass-bottom dish (no. 1.5 coverslip 0.16–0.19 mm, MatTek Corporation). Droplets were covered with a layer of mineral oil (Irvine Scientific, FujiFilm) to prevent evaporation. For imaging of GV oocytes, M2 medium was supplemented with dbcAMP (150 μg/mL); additionally, in the experiments with disrupted actin cytoskeleton, CytoD (2 μg/mL) was added as well. As our FF-OCM system was not equipped with an incubator, oocytes/embryos were imaged at room temperature and air concentration of CO2. For each sample, an optical cross-section of interest was selected in live preview mode and then imaged instantaneously for 100 s which allowed us to capture stirring motion of the cytoplasm. Images presented throughout the paper are an average of 100 FF-OCM frames (10 s acquisition) unless stated otherwise.
The illumination power was 68 µW for a circular imaging area of ~120 µm diameter defined by the field diaphragm. This gives illumination power density of 120 mW/mm2, which is comparable to wide-field fluorescence. Acquired raw images were processed after the imaging session. By default, FF-OCM is presented on a linear scale. Images presented on a logarithmic scale are marked throughout the paper.
Dynamic FF-OCM
Our analysis of cytoplasm motion dynamics is based on the PSD of FF-OCM signal. The analysis was done by a custom routine written in LabView, version 2021 SP1. It consists in the following steps: (1) FF-OCM images are recorded for 100 s at 10 fps, (2) a mask is constructed by to eliminate pixels with white noise (TSA Whiteness Test VI, confidence level 95%), (3) PSD is calculated for each pixel (Power Spectrum VI), (4) region of cytoplasm or zona pellucida (ellipsoidal masks) is manually selected for each oocyte, (5) PSD is averaged over the region, (6) PSD is fitted with inverse power law: \(y=a{x}^{-b}\) over the frequency range 0.01–3 Hz, and the exponent \(b\) is extracted.
Nuclear envelope analysis
The nuclear envelope fluctuations were analyzed using Fiji image processing software86. The FF-OCM images acquired at 10 fps, are averaged using a moving window of 10 s and a step of 1 s. The envelope was extracted based on the contrast difference between the nucleus and cytoplasm. The procedure includes following steps: (1) use median filter (radius = 4), (2) manually set threshold to segment nucleus (enable option ‘stack histogram’), (3) manually select region of interest (ROI) containing nucleus, (4) use ‘analyze particles’ function to extract nucleolus (parameters: size range 1000-infinity pixels, circularity 0.0–1.0, show ‘Bare Outlines’, enable option ‘include holes’). Described procedure results in a time stack of nuclear envelopes. Time average plots presented in Fig. 6b are obtained by taking average intensity projection of the time stack centered about the nucleus centroid.
The procedure of calculating average nuclear envelope shape (Fiji macro) includes following steps: (1) get profile of nuclear envelope image along a line starting at nucleus centroid at a defined angle, (2) calculate the distance from nucleus centroid to the envelope along the line, (3) repeat for 360° angle and all slices in the time stack, (4) calculate average distance and standard deviation for each angle, (5) plot average shape in polar coordinates. The described procedure is valid for convex envelope shapes. For concave (i.e., wrinkled) shapes the procedure would largely overestimate the envelope fluctuations.
The procedure of calculating nuclear envelope stability parameter includes the following steps: (1) take a time stack of nuclear envelope shapes of length L, (2) calculate average intensity projection for the first \(L-k\) slices, for \(k\) ranging from 0 to \(L-1\), (3) calculate number of non-zero pixels in each average intensity projection and divide the result by envelope perimeter, (4) calculate time fraction as \(k/L\). The nuclear envelope stability shows exponential decay with increasing measurement time fraction tf.
Nucleolus trajectory analysis
The nucleolus trajectories were analyzed using Fiji image processing software86. The FF-OCM images acquired at 10 fps, are averaged using a moving window of 10 s and a step of 1 s. The nucleolus was segmented based on the contrast difference between the nucleolus and chromatin condensed around the nucleolus in an SN-type oocyte. The procedure includes following steps: (1) use median filter (radius = 4), (2) take logarithm of OCM intensity, (3) manually set threshold to segment nucleolus (enable option ‘stack histogram’), (4) manually select ROI that containing nucleolus, (5) calculate centroid position using analyze particles function (parameters: size range 1000-infinity pixels, circularity 0.7–1.0, show ‘Bare Outlines’). MSD is calculated from nucleolus centroid trajectory as an average squared displacement for each delay ranging from 1 to 100 s using a custom-made Matlab script. MSD is computed for time delays \(\tau ={nt}\), where \(t\) is sampling time between consecutive points in the trajectory, of using equation80:
for a trajectory \(r\left(t\right)\) of total length N.
Statistics and reproducibility
A number of independent experiments are specified in figure legends. All statistical analyses were performed using Matlab. Statistical significance was determined using a two-tailed paired sample t-test. Differences with a p value of 0.05 or less were considered statistically significant; *p < 0.05, **p < 0.01, ***p < 0.001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data for the plots shown in all figures are available as Supplementary Data. Any additional data that support the findings of this study are available from the corresponding author upon reasonable request.
Code availability
Codes used for data analysis are available from the corresponding author upon reasonable request.
References
Ghosh, B. & Agarwal, K. Viewing life without labels under optical microscopes. Commun. Biol. 6, 559 (2023).
Chan, C. J., Bevilacqua, C. & Prevedel, R. Mechanical mapping of mammalian follicle development using Brillouin microscopy. Commun. Biol. 4, 1133 (2021).
Bols, P. E. J., Jorssen, E. P. A., Goovaerts, I. G. F., Langbeen, A. & Leroy, J. L. M. R. High throughput non-invasive oocyte quality assessment: the search continues. Anim. Reprod. Sci. 9, 420–425 (2012).
Barberet, J. et al. Can novel early non-invasive biomarkers of embryo quality be identified with time-lapse imaging to predict live birth? Hum. Reprod. 34, 1439–1449 (2019).
Pandit, S. & Sharma, R. Non invasive assessment of human oocytes and embryos in assisted reproduction: review on present practices and future trends. Med. J. Armed Forces India 78, 7–16 (2022).
Ajduk, A. et al. Rhythmic actomyosin-driven contractions induced by sperm entry predict mammalian embryo viability. Nat. Commun. 2, 417 (2011).
Rubessa, M. & Wheeler, M. B. Label-free microscopy: a non-invasive new tool to assess gametes and embryo quality. Theriogenology 150, 241–246 (2020).
Dardikman-Yoffe, G., Mirsky, S. K., Barnea, I. & Shaked, N. T. High-resolution 4-D acquisition of freely swimming human sperm cells without staining. Sci. Adv. 6, eaay7619 (2020).
Iyer, S., Mukherjee, S. & Kumar, M. Watching the embryo: evolution of the microscope for the study of embryogenesis. BioEssays 43, 1–17 (2021).
Jonkman, J., Brown, C. M., Wright, G. D., Anderson, K. I. & North, A. J. Tutorial: guidance for quantitative confocal microscopy. Nat. Protoc. 15, 1585–1611 (2020).
Ajduk, A. & Szkulmowski, M. Light microscopy of mammalian gametes and embryos: methods and applications. Int. J. Dev. Biol. 63, 235–244 (2019).
Magidson, V. & Khodjakov, A. Circumventing photodamage in live-cell microscopy. in Methods in Cell Biology Vol. 114, 545–560 (Elsevier Inc., 2013).
Nikiforov, D., Grøndahl, M. L., Hreinsson, J. & Andersen, C. Y. Human oocyte morphology and outcomes of infertility treatment: a systematic review. Reprod. Sci. 29, 2768–2785 (2022).
Ozturk, S. Selection of competent oocytes by morphological criteria for assisted reproductive technologies. Mol. Reprod. Dev. 87, 1021–1036 (2020).
Gardner, D. K. & Balaban, B. Assessment of human embryo development using morphological criteria in an era of time-lapse, algorithms and ‘OMICS’: Is looking good still important? Mol. Hum. Reprod. 22, 704–718 (2016).
Montag, M., Köster, M., van der Ven, K. & van der Ven, H. Gamete competence assessment by polarizing optics in assisted reproduction. Hum. Reprod. Update 17, 654–666 (2011).
Bormann, C. L. et al. Consistency and objectivity of automated embryo assessments using deep neural networks. Fertil. Steril. 113, 781–787.e1 (2020).
Nguyen, T. H., Kandel, M. E., Rubessa, M., Wheeler, M. B. & Popescu, G. Gradient light interference microscopy for 3D imaging of unlabeled specimens. Nat. Commun. 8, 210 (2017).
An, R., Wang, C., Turek, J., Machaty, Z. & Nolte, D. D. Biodynamic imaging of live porcine oocytes, zygotes and blastocysts for viability assessment in assisted reproductive technologies. Biomed. Opt. Express 6, 963 (2015).
Bouma, B. E. et al. Optical coherence tomography. Nat. Rev. Methods Prim. 2, 79 (2022).
Podoleanu, A. G. Optical coherence tomography. J. Microsc. 247, 209–219 (2012).
Singh, M. et al. Applicability, usability, and limitations of murine embryonic imaging with optical coherence tomography and optical projection tomography. Biomed. Opt. Express 7, 2295 (2016).
Wang, S., Larina, I. V., Wang, S. & Larina, I. V. In vivo dynamic 3D imaging of oocytes and embryos in the mouse oviduct. Cell Rep. 36, 109382 (2021).
Karnowski, K. et al. Optical coherence microscopy as a novel, non-invasive method for the 4D live imaging of early mammalian embryos. Sci. Rep. 7, 4165 (2017).
Caujolle, S. et al. Speckle variance OCT for depth resolved assessment of the viability of bovine embryos. Biomed. Opt. Express 8, 5139 (2017).
Masuda, Y. et al. Three-dimensional live imaging of bovine embryos by optical coherence tomography. J. Reprod. Dev. 67, 149–154 (2021).
Fluks, M., Tamborski, S., Szkulmowski, M. & Ajduk, A. Optical coherence microscopy allows for quality assessment of immature mouse oocytes. Reproduction 164, 83–95 (2022).
Fluks, M., Milewski, R., Tamborski, S., Szkulmowski, M. & Ajduk, A. Spindle shape and volume differ in high- and low-quality metaphase II oocytes. Reproduction 167, e230281 (2024).
Sobkowiak, A. et al. The number of nuclei in compacted embryos, assessed by optical coherence microscopy, is a non-invasive and robust marker of mouse embryo quality. Mol. Hum. Reprod. 30, gaae012 (2024).
Wang, L., Fu, R., Xu, C. & Xu, M. Methods and applications of full-filed optical coherence tomography: a review. J. Biomed. Opt. 27, 1–26 (2022).
Dubois, A., Vabre, L., Boccara, A.-C. & Beaurepaire, E. High-resolution full-field optical coherence tomography with a Linnik microscope. Appl. Opt. 41, 805 (2002).
Zheng, J. et al. Label-free subcellular 3D live imaging of preimplantation mouse embryos with full-field optical coherence tomography. J. Biomed. Opt. 17, 1 (2012).
Zheng, J. et al. Noninvasive three-dimensional live imaging methodology for the spindles at meiosis and mitosis. J. Biomed. Opt. 18, 050505 (2013).
Scholler, J. et al. Dynamic full-field optical coherence tomography: 3D live-imaging of retinal organoids. Light Sci. Appl. 9, 140 (2020).
Durand, T. et al. Visualizing enteric nervous system activity through dye-free dynamic full-field optical coherence tomography. Commun. Biol. 6, 1–10 (2023).
Monfort, T. et al. Dynamic full-field optical coherence tomography module adapted to commercial microscopes allows longitudinal in vitro cell culture study. Commun. Biol. 6, 1–17 (2023).
Groux, K. et al. Dynamic full-field optical coherence tomography allows live imaging of retinal pigment epithelium stress model. Commun. Biol. 5, 1–11 (2022).
Moore, E. L., Wang, S. & Larina, I. V. Staging mouse preimplantation development in vivo using optical coherence microscopy. J. Biophotonics 12, 1–6 (2019).
Chen, S. et al. Contrast of nuclei in stratified squamous epithelium in optical coherence tomography images at 800 nm. J. Biophotonics 12, 1–10 (2019).
Joo, K. et al. The importance of cytoplasmic strings during early human embryonic development. Front. Cell Dev. Biol. 11, 1–8 (2023).
Ma, B. X., Yang, L., Tian, Y., Jin, L. & Huang, B. Cytoplasmic strings between ICM and mTE are a positive predictor of clinical pregnancy and live birth outcomes: a time-lapse study. Front. Med. 9, 934327 (2022).
Chapman, M., Cooke, S., Eastick, J. & Venetis, C. Detailed analysis of cytoplasmic strings in human blastocysts: new insights. Zygote 31, 78–84 (2023).
Eastick, J., Venetis, C., Cooke, S. & Chapman, M. The presence of cytoplasmic strings in human blastocysts is associated with the probability of clinical pregnancy with fetal heart. J. Assist. Reprod. Genet. 38, 2139–2149 (2021).
Coticchio, G., Lagalla, C., Sturmey, R., Pennetta, F. & Borini, A. The enigmatic morula: mechanisms of development, cell fate determination, self-correction and implications for ART. Hum. Reprod. Update 25, 422–438 (2019).
Bouniol-Baly, C. et al. Differential transcriptional activity associated with chromatin configuration in fully grown mouse germinal vesicle oocytes. Biol. Reprod. 60, 580–587 (1999).
Zuccotti, M. et al. The analysis of chromatin organisation allows selection of mouse antral oocytes competent for development to blastocyst. Zygote 10, 73–78 (2002).
Debey, P. et al. Competent mouse oocytes isolated from antral follicles exhibit different chromatin organization and follow different maturation dynamics. Mol. Reprod. Dev. 36, 59–74 (1993).
Fluks, M., Szczepanska, K., Ishikawa, T. & Ajduk, A. Transcriptional status of mouse oocytes corresponds with their ability to generate Ca2+ release. Reproduction 157, 465–474 (2019).
Tan, J. H. et al. Chromatin configurations in the germinal vesicle of mammalian oocytes. Mol. Hum. Reprod. 15, 1–9 (2009).
Scheffler, K., Giannini, F., Lemonnier, T. & Mogessie, B. The prophase oocyte nucleus is a homeostatic G-actin buffer. J. Cell Sci. 135, jcs259807 (2022).
Almonacid, M. et al. Active fluctuations of the nuclear envelope shape the transcriptional dynamics in oocytes. Dev. Cell 51, 145–157 (2019).
Tilia, L., Chapman, M., Kilani, S., Cooke, S. & Venetis, C. Oocyte meiotic spindle morphology is a predictive marker of blastocyst ploidy—a prospective cohort study. Fertil. Steril. 113, 105–113 (2020).
Wang, Q. & Sun, Q.-Y. Evaluation of oocyte quality: morphological, cellular and molecular predictors. Reprod. Fertil. Dev. 19, 1 (2007).
Kilani, S., Cooke, S., Kan, A. & Chapman, M. Are there non-invasive markers in human oocytes that can predict pregnancy outcome? Reprod. Biomed. Online 18, 674–680 (2009).
Rama Raju, G. A., Prakash, G. J., Krishna, K. M. & Madan, K. Meiotic spindle and zona pellucida characteristics as predictors of embryonic development: a preliminary study using PolScope imaging. Reprod. Biomed. 14, 166–174 (2007).
Di Nisio, V., Antonouli, S., Damdimopoulou, P., Salumets, A. & Cecconi, S. In vivo and in vitro postovulatory aging: when time works against oocyte quality? J. Assist. Reprod. Genet. 39, 905–918 (2022).
Takahashi, T., Igarashi, H., Amita, M., Hara, S. & Kurachi, H. Cellular and molecular mechanisms of various types of oocyte aging. Reprod. Med. Biol. 10, 239–249 (2011).
Li, L., Lu, X. & Dean, J. The maternal to zygotic transition in mammals. Mol. Asp. Med. 34, 919–938 (2013).
Fu, X., Zhang, C. & Zhang, Y. Epigenetic regulation of mouse preimplantation embryo development. Curr. Opin. Genet. Dev. 64, 13–20 (2020).
Otsuki, J. et al. Noninvasive embryo selection: kinetic analysis of female and male pronuclear development to predict embryo quality and potential to produce live birth. Fertil. Steril. 112, 874–881 (2019).
Araki, E. et al. Correlation between the pronucleus size and the potential for human single pronucleus zygotes to develop into blastocysts. J. Assist. Reprod. Genet. 35, 817–823 (2018).
Royen, E. V. Multinucleation in cleavage stage embryos. Hum. Reprod. 18, 1062–1069 (2003).
Clarke, H. J. Transzonal projections: Essential structures mediating intercellular communication in the mammalian ovarian follicle. Mol. Reprod. Dev. 89, 509–525 (2022).
Wassarman, P. M. & Litscher, E. S. Female fertility and the zona pellucida. Elife 11, 1–11 (2022).
Baena, V. & Terasaki, M. Three-dimensional organization of transzonal projections and other cytoplasmic extensions in the mouse ovarian follicle. Sci. Rep. 14, 1–13 (2019).
McGinnis, L. K., Rodrigues, P. & Limback, D. Structural aspects of oocyte maturation. in Encyclopedia of Reproduction 2nd edn (ed. Skinner, M. K. B. T.-E. of R.) 176–182 (Elsevier, 2018). https://doi.org/10.1016/B978-0-12-801238-3.64445-8.
Lim, H. Y. G. & Plachta, N. Cytoskeletal control of early mammalian development. Nat. Rev. Mol. Cell Biol. 22, 548–562 (2021).
Dumortier, J. G. et al. Hydraulic fracturing and active coarsening position the lumen of the mouse blastocyst. Science 365, 465–468 (2019).
Kruppa, A. J. & Buss, F. Motor proteins at the mitochondria–cytoskeleton interface. J. Cell Sci. 134, jcs226084 (2021).
Zhang, D. Interplay between endoplasmic reticulum membrane contacts and actomyosin cytoskeleton. Cytoskeleton 77, 241–248 (2020).
Venkatesh, K., Mathew, A. & Koushika, S. P. Role of actin in organelle trafficking in neurons. Cytoskeleton 77, 97–109 (2020).
Kreitzer, G. & Myat, M. M. Microtubule motors in establishment of epithelial cell polarity. Cold Spring Harb. Perspect. Biol. 10, a027896 (2018).
Sun, Q. & Schatten, H. Regulation of dynamic events by microfilaments during oocyte maturation and fertilization. Reproduction 131, 193–205 (2006).
Al Jord, A. et al. Cytoplasmic forces functionally reorganize nuclear condensates in oocytes. Nat. Commun. 13, 5070 (2022).
Almonacid, M., Terret, M.-E. & Verlhac, M.-H. Control of nucleus positioning in mouse oocytes. Semin. Cell Dev. Biol. 82, 34–40 (2018).
Nolte, D. D. Coherent light scattering from cellular dynamics in living tissues. Rep. Prog. Phys. 87, 036601 (2024).
Oldenburg, A. L. et al. Inverse-power-law behavior of cellular motility reveals stromal–epithelial cell interactions in 3D co-culture by OCT fluctuation spectroscopy. Optica 2, 877 (2015).
Tseng, Q. et al. Spatial organization of the extracellular matrix regulates cell-cell junction positioning. Proc. Natl. Acad. Sci. USA 109, 1506–1511 (2012).
Almonacid, M. et al. Active diffusion positions the nucleus in mouse oocytes. Nat. Cell Biol. 17, 470–479 (2015).
Kettmayer, C., Gratton, E. & Estrada, L. C. Comparison of MSD analysis from single particle tracking with MSD from images. Getting the best of both worlds. Methods Appl. Fluoresc. 12, 15001 (2024).
Bruno, L., Levi, V., Brunstein, M. & Despósito, M. A. Transition to superdiffusive behavior in intracellular actin-based transport mediated by molecular motors. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 80, 1–7 (2009).
Dubois, A. Focus defect and dispersion mismatch in full-field optical coherence microscopy.Appl. Opt. 56, D142 (2017).
Stremplewski, P. et al. In vivo volumetric imaging by crosstalk-free full-field OCT. Optica 6, 608 (2019).
Xiao, P., Fink, M. & Boccara, A. C. Full-field spatially incoherent illumination interferometry: a spatial resolution almost insensitive to aberrations. Opt. Lett. 41, 3920 (2016).
Fulton, B. P. & Whittingham, D. G. Activation of mammalian oocytes by intracellular injection of calcium. Nature 273, 149–151 (1978).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Acknowledgements
Measurements were supported by the infrastructure of the National Laboratory for Photonics and Quantum Technologies (NLPQT). A.A. acknowledges support from the National Science Centre (Poland) by the OPUS Grant (2017/27/B/NZ5/00405). B.F.K. acknowledges funding from the NAWA Chair programme (BPN/PRO/2022/1/00003/U/00001).
Author information
Authors and Affiliations
Contributions
S.M. developed experimental setup, performed imaging, data processing and data analysis, A.A. prepared oocytes and embryos, P.S. contributed to system design, S.M., A.A. and B.K. prepared manuscript, A.A, B.K. and M.S. supervised the project. All authors discussed the results and agreed on the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Kirill Larin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Dr. Chao Zhou and Dr. Ophelia Bu.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Morawiec, S., Ajduk, A., Stremplewski, P. et al. Full-field optical coherence microscopy enables high-resolution label-free imaging of the dynamics of live mouse oocytes and early embryos. Commun Biol 7, 1057 (2024). https://doi.org/10.1038/s42003-024-06745-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06745-x
- Springer Nature Limited