
Abstract
Antimicrobial resistance (AMR) poses a serious threat to the clinical management of typhoid fever. AMR in Salmonella Typhi (S. Typhi) is commonly associated with the H58 lineage, a lineage that arose comparatively recently before becoming globally disseminated. To better understand when and how H58 emerged and became dominant, we performed detailed phylogenetic analyses on contemporary genome sequences from S. Typhi isolated in the period spanning the emergence. Our dataset, which contains the earliest described H58 S. Typhi organism, indicates that ancestral H58 organisms were already multi-drug resistant (MDR). These organisms emerged spontaneously in India in 1987 and became radially distributed throughout South Asia and then globally in the ensuing years. These early organisms were associated with a single long branch, possessing mutations associated with increased bile tolerance, suggesting that the first H58 organism was generated during chronic carriage. The subsequent use of fluoroquinolones led to several independent mutations in gyrA. The ability of H58 to acquire and maintain AMR genes continues to pose a threat, as extensively drug-resistant (XDR; MDR plus resistance to ciprofloxacin and third generation cephalosporins) variants, have emerged recently in this lineage. Understanding where and how H58 S. Typhi originated and became successful is key to understand how AMR drives successful lineages of bacterial pathogens. Additionally, these data can inform optimal targeting of typhoid conjugate vaccines (TCVs) for reducing the potential for emergence and the impact of new drug-resistant variants. Emphasis should also be placed upon the prospective identification and treatment of chronic carriers to prevent the emergence of new drug resistant variants with the ability to spread efficiently.
Similar content being viewed by others

Introduction
Salmonella enterica serovar Typhi (S. Typhi) is the etiologic agent of typhoid fever, a disease associated with an estimated 10.9 million new infections and 116,800 deaths annually1. The disease classically presents as a non-differentiated fever and can progress to more severe manifestations or even death2. Typhoid fever necessitates antimicrobial therapy, as the associated mortality rate in the pre-antimicrobial era ranged from 10–30%3,4; presently, typhoid has a case fatality rate (CFR) of <1% when treated with effective antimicrobials3. S. Typhi is spread via the faecal-oral route, typically through the ingestion of contaminated food or water2. Therefore, high prevalence rates of typhoid fever were historically associated with urban slums in South Asia with poor sanitation5. Recent multicentre surveillance studies have demonstrated that typhoid fever is also a major problem in both urban and rural areas in sub-Saharan Africa6,7,8.
Given the importance of antimicrobials for the management and control of typhoid, antimicrobial resistance (AMR) in S. Typhi is a major public health issue. Indeed, the problem of AMR in S. Typhi first appeared in the 1950s with the emergence of resistance against the most widely used drug, chloramphenicol3,9. Multi-drug resistant typhoid (MDR; resistance to first-line antimicrobials chloramphenicol, trimethoprim-sulfamethoxazole, and ampicillin) was first identified in the 1970s and became common in the early 1990s10,11. MDR in S. Typhi is frequently conferred by self-transmissible IncH1 plasmids carrying a suite of resistance genes, including resistance determinants for chloramphenicol (catA1 or cmlA), ampicillin (blaTEM-1D, blaOXA-7), and co-trimoxazole (at least one dfrA gene and at least one sul gene)12. Lower efficacy of first-line antimicrobials led to the increased use of fluoroquinolones, but decreased fluoroquinolone susceptibility became apparent in the mid-1990s, was widespread in South and Southeast Asia in the early 2000s13,14, and has now become ubiquitous globally15. Inevitably, as treatment options have become limited, third generation cephalosporins and azithromycin have been used more widely for effective treatment of typhoid fever16,17,18. However, newly circulating extensively-drug resistant (XDR; MDR plus resistance to fluoroquinolones and third generation cephalosporins) variants of S. Typhi have left azithromycin as the only feasible oral antimicrobial for the treatment of typhoid fever across South Asia19. We are at a potential tipping point, as azithromycin-resistant S. Typhi has since been reported in Bangladesh, Pakistan, Nepal, and India, thereby threatening efficacy of common oral antimicrobials for effective typhoid treatment20,21,22,23. If an XDR organism were to acquire azithromycin resistance (induced by a single base pair mutation), this would lead to what Hooda and colleagues have referred to as pan-oral drug-resistant (PoDR) S. Typhi, which would likely require inpatient management24. This would come at substantial additional cost to patients and their families, and place additional strain on already overburdened health systems25. A recently published case report of a clinical relapse caused by XDR S. Typhi that was also resistant to both carbapenems and azithromycin in Pakistan signals the potential emergence of “untreatable typhoid”26; however; it is unclear what circumstances would permit the emergence of a successful clone exhibiting resistance to all of these antimicrobials.
In contrast to many other Gram-negative bacteria, S. Typhi is human restricted with limited genetic diversity that can be described by a monophyletic phylogenetic structure27. Therefore, the phylogeny and evolution of S. Typhi provide a model for how AMR emerges, spreads, and becomes maintained in a human pathogen. AMR phenotypes in S. Typhi are typically dominated by a single lineage; H58 (genotype 4.3.1 and consequent sublineages), which was the 58th S. Typhi haplotype to be described in the original genome-wide typing system28. This highly successful lineage is commonly associated with MDR phenotypes and decreased fluoroquinolone susceptibility14. Previous phylogeographic analysis suggested that H58 emerged initially in Asia between 1985 and 1992 and then disseminated rapidly to become the dominant clade in Asia and subsequently in East Africa14. H58 is currently subdivided into three distinct lineages—lineage I (4.3.1.1) and lineage II (4.3.1.2), which were first identified in a paediatric study conducted in Kathmandu29, and lineage III (4.3.1.3), which was identified in Dhaka, Bangladesh30. A recent study of acute typhoid fever patients and asymptomatic carriers in Kenya demonstrated the co-circulation of genotypes 4.3.1.1 and 4.3.1.2 in this setting, and closer analysis showed that these East African sequences had distinct AMR profiles and were the result of several introduction events31,32. These events led to the designation of three additional genotypes: H58 lineage I sublineage East Africa I (4.3.1.1.EA1), H58 lineage II sublineage East Africa II (4.3.1.2.EA2), and H58 lineage II sublineage East Africa III (4.3.1.2.EA3)32. In addition, the XDR S. Typhi clone, which was caused by a monophyletic outbreak of genotype 4.3.1.1 organisms, was designated genotype 4.3.1.1.P1 to facilitate monitoring of its spread.
It is apparent from investigating the phylogeny of S. Typhi that H58 is atypical in comparison to other lineages. This lineage became dominant in less than a decade, and first appeared on a long basal branch length, indicative of a larger number of single base pair mutations separating it from its nearest neighbour (Fig. 1a). These observations suggest that there is something ‘unique’ about the evolution of this lineage, but we have limited understanding of how H58 emerged, what enabled its rapid spread, and when it initially appeared. Here, by collating new genome sequences of S. Typhi that were associated with travel to South Asia in the late 1980s and early 1990s and comparing them to a global population over the same period, we explore an expanded early phylogenetic dataset to resolve the origins and rapid success of this important and successful AMR clone.

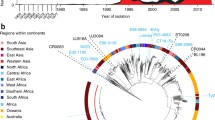
a A phylogenetic overview of historical (1980–1995) S. Typhi from the UKHSA. Maximum likelihood outgroup rooted phylogenetic tree depicting the genomic sequences of 463 S. Typhi isolated from returning travellers to the United Kingdom between 1980 and 1995. Branch colour indicates genotype, and rings outside of the tree indicate country of origin, MDR, and presence of IncH1 plasmid, coloured as per the inset legend. Individual mutations in the quinolone resistance determining region (QRDR) indicated by stars outside the rings. b A phylogenetic overview of historical H58 (4.3.1) S. Typhi isolates. Zoomed-in view of H58 S. Typhi isolates from historical collection. Genotype, country of origin, presence of MDR, AMR mutations, and presence of IncH1 plasmids are indicated by bars to the right of the tree and coloured as per the inset legend. Individual mutations in the quinolone resistance determining region (QRDR) indicated by stars to the right of the bars. c A bar graph showing distribution of samples by year (1980–1995) and countries of origin are indicated by colour as per inset legend.
Results and discussion
Sampling
The main questions that we aimed to address with this study were: (i) when and where did H58 S. Typhi first emerge; (ii) can we better resolve the evolutionary events that lead to the long branch length observed for H58 S. Typhi; and (iii) how quickly did this lineage spread and why? Therefore, to investigate the origins of S. Typhi H58, data from United Kingdom Health Security Agency (UKHSA, formerly Public Health England) containing information on archived S. Typhi organisms isolated between 1980 and 1995 from travellers returning to the UK from overseas and receiving a blood culture were analysed.
The database was queried and organisms were selected from the following three categories: (i) 126 S. Typhi with the E1 Phage type, which was thought to be associated with H5812, originating from South Asia (India, Nepal, Pakistan, and Bangladesh), (ii) 159 S. Typhi organisms with a variety of non-E1 phage types originating from South Asia, and (iii) 184 S. Typhi organisms with a variety of phage types (both E1 and non-E1) originating from locations outside of South Asia. A total of 470 S. Typhi organisms (out of 3751 total S. Typhi isolated from returning travellers by UKHSA between 1980 and 1995) meeting these criteria were randomly selected, revived, subjected to DNA extraction and whole genome sequenced. Ultimately, our dataset was composed of 463 novel sequences generated as a component of this study and 305 existing sequences known to belong to the H58 lineage and its nearest neighbours30,33,34, yielding a total of 768 whole genome sequences on which to structure subsequent analysis.
Population structure, genotype distribution, and antimicrobial resistance profiles of historical S. Typhi
We inferred a maximum likelihood phylogenetic tree from the sequencing data to examine the population structure of this historical (1980–1995) collection of global S. Typhi sequences (Fig. 1). Notably, unlike the extant global population of S. Typhi, which is largely dominated by a single lineage34, this historic population exhibited considerable genetic diversity, with 37 genotypes represented (Fig. 1a; Supplementary Table 1). Most isolates belonged to primary clade 2 (194/463), of which clade 2.0 was most common (36%, 69/194) followed by subclades 2.3.3 (14%, 28/194) and 2.2.2 (13%, 25/194). An additional 23% of isolates belonged to primary clade 3 (108/463) and 3% of isolates were classified as major lineage 1 (12/463). Ultimately, 29% of isolates belonged to primary clade 4 (134/463), of which 63% (84/134) were H58. Among these, H58 lineage I (genotype 4.3.1.1) was most common (67%, 56/84), followed by genotype 4.3.1 (H58 not differentiated into any sublineage; 24%, 20/84). The earliest H58 isolates in our dataset are illustrated in higher resolution in Fig. 1b. The temporal distribution and country of origin of the sequences are shown in Fig. 1c.
Although we enriched our historical dataset for samples isolated from travellers returning from South Asia, our final dataset still provided substantial geographic coverage, with 39 different countries represented over the study period (1980–1995). Therefore, these data are likely representative of the circulating S. Typhi during this period of investigation. Notably, the earliest H58 organism in our dataset that was classified as H58 based on a single marker SNP according to the GenoTyphi scheme was isolated in 1983 from an individual entering the UK from India, followed by two additional Indian isolates (1988) that were also classified as H58. These three organisms differed from the larger cluster of H58 organisms by 61–64 single nucleotide polymorphisms (SNPs) (Fig. 1a); therefore, we considered them to be distinct from the larger cluster and not truly H58. All H58 S. Typhi in this dataset were isolated from travellers returning from South Asia, with the majority (51/84; 61%) originating from Pakistan and the remainder from India and Bangladesh; 31/84 (37%) and 2/84 (2%), respectively.
Given that this dataset included isolates from the early MDR era and then following the emergence of reduced fluoroquinolone susceptibility, we analysed the data for genes associated with MDR and mutations in the DNA gyrase gene, gyrA. Overall, 7% (34/463) of the organisms in this historical dataset were genetically defined as being MDR; importantly, all were H58 (genotypes 4.3.1, 4.3.1.1, 4.3.1.2, and 4.3.1.3) and isolated between 1991 and 1995, and 97% (33/34) possessed an IncH1 plasmid (Fig. 1b). Thirteen H58 organisms contained an IncH1 plasmid carrying AMR genes but did not possess the genes conferring resistance to all three first-line antimicrobials, and thus were not genetically defined as MDR (Fig. 1b, Supplementary Table 1). The first mutation in the quinolone resistance determining region (QRDR) in our dataset was a gyrA-D87Y substitution identified in a genotype 2.5 organism originating in South Africa in 1986 (Fig. 1a). Based on our data, this was a spontaneous mutation that evolved de novo that did not become fixed in the population. Similarly, two single QRDR mutations (gyrA-S83F and gyrA-S83Y) occurred independently in H58 organisms (genotype 4.3.1) in India in 1995 (Fig. 1b) and were not observed in descendant populations. No mutations in gyrB or parC were observed in this dataset.
In order to contextualise these isolates to understand the evolutionary events leading to this clone, we selected H58 and nearest neighbours (from genotypes 4.1 and 4.2) sequences (n = 305) that were available in the public domain from previous studies30,33,34 (Supplementary Table 2) and generated a phylogenetic tree combining these isolates with early H58 and nearest neighbour isolates from our contemporary dataset (n = 117). In our H58 and nearest neighbour dataset (n = 422, Supplementary Fig. 1), which included both published data as well as our data, 17 countries were represented30,33,34. Of the non-H58 isolates (nearest neighbours), 42% were genotype 4.1 (32/76), 13% (10/76) were genotype 4.2, 28% (21/76) were 4.2.1, and 16% (12/76) were 4.2.3. These non-H58 nearest neighbour organisms were isolated between 1981 and 2000. None of them were MDR, and none carried an IncH1 plasmid. Of our H58 isolates (defined as having informative SNPs indicative of lineage 4.3.1), the earliest organism was isolated in 1983 in India, followed by two additional Indian isolates (1988) that were also classified as H58 based on a single marker SNP within the GenoTyphi typing scheme. However, we observed that there were no more recent isolates from this founder group (Supplementary Fig. 1), implying that this lineage became extinct; these isolates did not contain incH1 plasmids and were non-MDR. In addition, these isolates were relatively genetically distinct from the other early H58 isolates and were therefore considered to not truly belong to the same lineage. Within the H58 lineage, most of the organisms belonged to H58 sublineage I (4.3.1.1; 84%, 290/346), followed by sublineage II (4.3.1.2; 7%, 25/346), genotype 4.3.1 (6%, 21/346), and sublineage III (4.3.1.3; 3%, 10/346). Overall, 63% (219/346) of these H58 organisms were MDR, and 87% (191/219) of these MDR H58 organisms carried an IncH1 plasmid. All the MDR H58 organisms lacking an IncH1 plasmid were genotype 4.3.1.1, the earliest of which was isolated in India in 1991. Within this group, the first single point mutation in the QRDR occurred comparatively early in an organism isolated in India in 199133.
Evolutionary history of H58 S. Typhi
Using BEAST35, we determined that the median substitution rate of H58 was 2.79 × 10−7 substitutions base−1 year−1 [95% highest posterior density (HPD): 2.40 × 10−7–3.24 × 10−7], which is comparable to that observed in previous studies36. We found that the most recent common ancestor (MRCA) of the first H58 was estimated to have emerged in late 1987 (95% HPD: 1986–1988). Two H58 sublineages (4.3.1.1 and 4.3.1.2) then emerged almost simultaneously in India in 1987 and 1988 (Fig. 2). A time-inferred phylogeny presented a clonal expansion of H58 that originated from South Asia, specifically in India, and then disseminated globally. As noted above, 63% (219/346) of isolates were MDR, and 87% (191/219) of those isolates contained an IncH1 plasmid known to carry AMR genes. Detailed genetic analysis of the IncH1 plasmids observed in most of these MDR isolates revealed high genetic similarity, with an average of ~1 single nucleotide polymorphism (SNPs) difference between them (Supplementary Fig. 2). These data strongly suggest that the ancestral H58 organism that was the basis for the major clonal expansion was already MDR before undergoing clonal expansion and subsequent global dissemination; some ensuing H58 organisms then lost the MDR plasmid in certain locations, presumably because of decreased antimicrobial selection pressure owing to less frequent use of first-line antimicrobials, with a corresponding impact on fitness. Recent large-scale genomic analyses have shown that the MDR cassette carried on these early IncH1 plasmids has become chromosomally integrated in some settings and within some lineages (e.g., Nigeria, Malawi) where the MDR phenotype has been maintained at high levels of prevalence, and in other places where chromosomal integration has not occurred, plasmid loss (and associated loss of MDR phenotype) has been observed15. The fact that the three early precursor organisms that were not MDR appear to have become extinct supports our hypothesis that the presence of an MDR phenotype was a pivotal selective event.

BEAST-generated dated phylogeny of H58 S. Typhi isolates from UKHSA collection and published literature (n = 345). Tip colours indicate presence of specific mutation(s) in the quinolone resistance determining region (QRDR) as per inset legend. Branch colour and the first column to the right of the tree indicate genotype, the second column indicates country of origin, the third represents presence of MDR, and the final column indicates presence of an incH1 plasmid. Our analysis suggests that the Most Recent Common Ancestor (MCRA) of H58 appeared in 1987, and that two sublineages (I and II) emerged almost simultaneously in India in 1987 and 1988. The first single point mutation in the quinolone resistance determining region (QRDR) was observed in 1990, and the first “triple mutant” was observed in Bangladesh in 1999.
A stark observation is that QRDR mutations appeared in H58 organisms quickly and frequently. Within this H58 dataset, organisms with one or more QRDR mutations appeared within six independent lineages of H58, as illustrated in Fig. 2 and Supplementary Fig. 1. The earliest H58 lineage (lineage II) to develop QRDR mutations was observed in 1991 in India, with a single S83Y mutation in the gyrA gene, and the first isolate containing two QRDR mutations (gyrAS83Y; parC-S80R) appeared in a genotype 4.3.1.3 organism in Bangladesh in 1999 (Fig. 2, Supplementary Fig. 1). The first “triple mutant” (mutations in gyrA-D87G, gyrA-S83F, and parC-E84K) was found in a 4.3.1.1 Bangladeshi isolate in 1999 (Fig. 2, Supplementary Fig. 1).
Genetic variation associated with H58 S. Typhi
Our data support the general hypothesis that H58 S. Typhi was successful specifically because of the acquisition and maintenance of an MDR plasmid. This selection meant that later gyrA mutations were more likely to occur in this lineage given its dominance (and therefore, higher rates of replication leading to additional opportunities for mutations to occur), as well as assumed frequent fluoroquinolone exposure, given its existing MDR phenotype. Based on the results of our mapping, we undertook further genetic analysis to identify non-synonymous SNPs unique to the early H58 isolates, as well as SNPs that were unique to early H58 isolates that were MDR. The motivation was to explain the origins of the long branch length illustrated in Fig. 1a, in an attempt to infer why this lineage was so globally successful and identify genetic markers that may stabilize an MDR IncH1 plasmid.
We identified 16 unique non-synonymous SNPs that were exclusive to the early H58 isolates as compared to precursor 4.1 and 4.2 organisms, the majority of which were present in genes associated with central metabolism and outer membrane structures; one of which was associated with pathogenicity (Table 1). Within the early H58 isolates that were also MDR, we identified an additional 23 unique non-synonymous SNPs, most of which were found in genes encoding proteins predicted to regulate metabolism, degrade small molecules, membrane/surface structures, as well as regulators, pathogenicity adaptation, and information transfer (Table 2). We additionally identified mutations in a gene (t2518/STY0376) encoding a hypothetical protein with an EAL (diguanylate phosphodiesterase) domain with a non-synonymous mutation, which has previously been identified as being associated with H58 organisms37. The homologous gene (STM0343) in Salmonella Typhimurium has been described as regulating motility and invasion, which suggests that SNPs in this gene might also contribute to virulence38,39. In addition, we identified SNPs in genes that have previously been associated with tolerance to bile in S. Typhi (sirA, recB, wecF, dsdA, and yjjV) among the early MDR H58 isolates40,41.
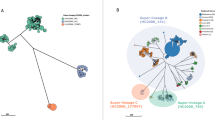
We additionally sought to compare the genetic diversity of the IncH1 plasmids carried by most early H58 S. Typhi organisms to further investigate our hypothesis that the original H58 organism harboured an MDR IncH1 plasmid. We postulated that this plasmid co-evolved with H58 S. Typhi before the MDR locus within it became chromosomally integrated or the plasmid became lost in subsequent years, as compared to multiple independent acquisitions of an MDR plasmid within the H58 lineage. As described above, these plasmids appeared to be nearly identical. We generated a recombination-filtered alignment of our plasmid sequences (n = 185), identified unique plasmid haplotypes (unique plasmid allele combinations), and aligned these to a core-genome chromosomal tree (available via Microreact: https://microreact.org/project/uaEn32y5NKEPGVvbxh8HLn-old-h58-inchi1). We also generated a minimum spanning tree to further examine the relatedness of these plasmids (Fig. 3). We observed eight unique plasmid haplotypes, separated by a maximum of 4 SNPs. Most plasmids shared an identical plasmid backbone (n = 175; large central node in Fig. 3, red colour on Microreact), with occasional evidence of microevolution (one to two unique SNPs; represented by smaller nodes in Fig. 3). Moreover, singleton plasmid haplotypes were randomly distributed throughout the chromosomal phylogeny and plasmid haplotypes with more than one member sequence were clustered by their host sequences. This is highly indicative that all early H58 S. Typhi organisms carried the same plasmid variant, which underwent subsequent microevolution, rather than several distinct plasmid acquisition events.

Nodes indicate unique IncH1 haplotypes observed among n = 185 plasmid sequences. Nodes are represented as pie charts, where node size indicates frequency of haplotype, and are coloured by the genotype(s) of the host S. Typhi sequences carrying each plasmid haplotype.
Our analyses suggest that there was a cascade of events that corresponded with the origins of the first H58 S. Typhi organism. Cumulative mutations signified by the observed long branch length is uncommon in S. Typhi and has two feasible explanations. The first is that the progenitor organism was a hyper mutator, and that a key mutation in a gene such as mutS was responsible for generating a large amount of genetic diversity in a short time frame42. However, no such informative SNPs were observed in the early H58 isolates, although any such mutations may have reverted. The second, and more likely explanation, is that the organism was in an environment that created an atypical selective pressure to induce mutations that facilitated its ability become exposed to, and then accept, an MDR plasmid. Our previous data on S. Typhi carriage in the gallbladder determined that this environment creates an atypical selective pressure and stimulates mutations in metabolism and outer membrane structures31,43. This genetic variation was associated with organisms being located on signature long branches; our observations here are comparable. We suggest that H58 S. Typhi became successful due to its early ability to accept and stabilise a large MDR plasmid, which may have occurred whilst in the gallbladder; this one-off event and onward transmission then created a successful lineage. Therefore, we speculate that gallbladder carriage acts as a niche for generation of new variants with both modest (single SNPs) and large (plasmid acquisition) events capable of generating new lineages of S. Typhi with a selective advantage.
Implications
Our study of the origins of H58 S. Typhi has implications for how we understand the emergence and spread of new drug-resistant variants and can help inform optimal use of typhoid conjugate vaccines (TCVs). Our data suggest that a rare series of events associated with specific selective pressures can trigger MDR organisms to arise and spread rapidly. This observation comes at a critical time in global typhoid control. There TCVs have been prequalified by the World Health Organization, with additional candidates in late-stage clinical development, and promising clinical efficacy and effectiveness data44,45. The continued spread of drug resistant H58 S. Typhi is a major argument for the use of TCV, particularly as resistance to all antimicrobials used to treat typhoid fever has been reported in S. Typhi in South Asia. We continue to observe a pattern in which drug-resistant variants emerge in South Asia and spread radially, with H58 being the only major genotype to do so to date14,46,47,48,49,50. H58 S. Typhi was first isolated in Kenya shortly after its estimated emergence in 1988, further illustrating the potential for rapid spread of this lineage49. This suggests that prioritization of widespread use of TCVs in South Asia can prevent substantial morbidity and mortality in the region. Additionally, introduction of TCVs should limit the continued emergence and spread of drug-resistant organisms elsewhere by preventing infections caused by drug-resistant S. Typhi and by lowering selection pressure both through reduction of incidence of disease and associated antimicrobial use. This approach may extend the useful lifespan of existing therapeutic options in some parts of the world until TCVs become more widely available. Given that resistance frequently emerges and spread from South Asia46, it makes sense to prioritise vaccine interventions in this region (while supply is constrained) to have the greatest potential impact on disease incidence and AMR.
Our data strongly suggest that H58 S. Typhi, which is highly associated with being MDR and decreased fluoroquinolone susceptibility, is adept at acquiring and maintaining drug resistance determinants, which is likely facilitated by unique mutations that occurred in the earliest H58 organism. The rapid international dissemination of the H58 lineage, starting in South Asia and spreading throughout Southeast Asia14,33, into Africa14,31,48,49,51,52,53 and more recently, Latin America54, suggest that expanded genomic surveillance is warranted to monitor its continued global spread. Such information has also helped to inform the development of transmission dynamics models that predict the spread of newer drug-resistant variants, like XDR, which can also inform TCV introduction decision-making55.
Ultimately, we postulate that H58 S. Typhi likely emerged from a chronic carrier, which is supported by the indicative long branch length between early H58 organisms and its nearest non-H58 neighbours. This deduction is consistent with observations from previous studies conducted in Nepal and Kenya, in which higher mean branch lengths were observed in carriage isolates as compared to isolates from symptomatic patients31,43. This phenomenon is to be expected, assuming that chronic carriers will have had a longer time from acquisition of infection to shedding and sampling. Notably, considering the structure of the phylogenetic tree and the loss of the early non-MDR H58 organisms from the population, it is likely that the MDR phenotype was the main catalysing factor for the success of this lineage. Additionally, we observed non-synonymous mutations in genes associated with outer membrane structures, metabolism and virulence, which we have been observed previously in organisms isolated directly from the gallbladder31,43. Our analysis also supports previous transcriptomic analysis showing that H58 S. Typhi has higher bile tolerance relative to other laboratory strains (Ty2 and CT18) and exhibits increased virulence in the presence of bile, thereby increasing the potential of H58 organisms to colonize and persist in the gallbladder. These observations suggest that gallbladder is the ideal location for the generation of variants and highlights the potential for chronic carriage to lead to the emergence of S. Typhi (and other invasive Salmonella) that are genetically predisposed to express new phenotypes, which may include drug resistance. We suggest that more emphasis should be placed upon the prospective identification and treatment of chronic carriers to prevent the emergence of new variants with the ability to spread. Contemporary data from returning travellers to the UK suggest that 1.4% of those infected with S. Typhi are chronic carriers, and 0.7% are carrying MDR S. Typhi56. A comparable frequency of carriage (1.1%) has been observed among children aged 16 years and younger in Mukuru, an informal settlement close to Nairobi, Kenya31. This prevalence rate is likely to be higher in older age groups57,58,59, and among people living in settings where typhoid is hyperendemic, and thus, may present a more substantial risk in terms of sustained transmission of drug-resistant S. Typhi and the potential emergence of additional drug-resistant variants. Scalable, low-cost assays to detect carriers will become vital if we aim to eliminate typhoid and prevent future resurgence.
Our study has several limitations. Our dataset was enriched for samples of E1 phage type, which was thought to be associated with H58, and samples isolated from travellers returning from South Asia, so was not fully globally representative. This approach was taken to provide the highest possibility of identifying early H58 isolates. However, subsequent work conducted by others has suggested that S. Typhi WGS data isolated from returning travellers is representative of the circulating S. Typhi populations in the countries of origin53 and that routine surveillance of S. Typhi isolated from returning travellers could be used as informal sentinel surveillance for countries of travel. We acknowledge potential bias associated with data obtained from returning travellers including differences in health-seeking behaviours, which may be associated with increased disease severity and AMR, differences in patterns of travel, and a potential overrepresentation of samples from countries that have common travel links with the UK. Additionally, we were unable to conduct experimental validation of our hypotheses about the roles of specific SNPs in maintenance of an MDR plasmid and likelihood of acquisition of QRDR mutations; we intend to explore these questions experimentally in the future.
Our data suggest that H58 S. Typhi likely emerged from a chronic carrier in India in 1987. The prototype organism of the successful clonal expansion was already MDR and became highly successful across South Asia in over a period of <10 years. Ultimately, sustained use of, and exposure to, fluoroquinolones led to selective mutations in gyrA on many independent occasions. The dominance of this lineage and its ability to maintain AMR genes has latterly meant it has become resistant to additional antimicrobials. Our work represents a blueprint of how such organisms can arise and become dominant, but also provides the justification and evidence for the introduction of new interventions for disease control; if we reduce disease burden by vaccination, we will additionally reduce the likelihood of comparable events occurring in other S. Typhi organisms and other pathogens. Widespread vaccine deployment, as well as screen and treat programmes, may not only impact AMR directly through the prevention of drug-resistant infections, but also indirectly, as reduced transmission leads to decreased selection pressure on account of lower bacterial replication rates and decreased antimicrobial use. While these efficacious and cost-effective interventions should be deployed widely in all typhoid-endemic settings, it makes sense to prioritise vaccine use in the region from which drug-resistance most often emerges and spreads for maximal potential impact on AMR.
Methods
DNA extraction and Whole Genome sequencing
Genomic DNA from S. Typhi isolates was extracted using the Wizard Genomic DNA Extraction Kit (Promega, Wisconsin, USA), following standardized manufacturer’s protocol. Two ng of genomic DNA from each organism was fragmented and tagged for multiplexing with Nextera DNA Sample Preparation Kits, followed by paired-end sequencing on an Illumina HiSeq2000 Platform to produce 101 bp paired-end reads (Illumina, Cambridge, UK). Raw reads were deposited in the European Nucleotide Archive (ENA) under study accession number PRJEB15284 (Supplementary Table 1).
Read alignment and SNP analysis
FastQC and FASTX-Toolkit bioinformatics pipelines were used to check the quality of raw reads60,61. Six samples were excluded from the analysis, one was determined to not be Salmonella, one appeared to be comprised of multiple genotypes, and four samples were on a long branch length and were determined to be contaminated. Paired end reads for the remaining 464 samples were mapped to the S. Typhi CT18 reference genome (accession number: AL513382)62 using the RedDog mapping pipeline (v1beta.10b, available at http://githib.com/katholt/reddog). RedDog uses Bowtie2 v2.2.963 to map all raw reads to the CT18 reference genome and then uses SAMtools v1.3.164 to identify high quality SNP calls. SNPs that did not meet predefined criteria (a minimal phred quality score of 30 and depth coverage of 5 were filtered out)65. A failed mapping sequence was defined as when <50% of total reads mapped to the reference genome. 2 isolates were excluded from additional analysis after mapping failed, due to depth coverage of less than 10 (as per the RedDog pipeline default). A concatenation of core SNPs that were present in >95% of all genomes was generated and filtered to exclude all SNPs from phage regions or repetitive sequences in the genome reference CT18 as defined previously (Supplementary Table 2)62. Briefly, SNPs were filtered from excluded regions totalling 346,834 bases from an alignment of 43,100 SNPs using the Python script embedded in the RedDog pipeline. Gubbins (v2.3.2)66 was used to filter out additional SNPs in recombinant regions. Finally, the alignment of 16,324 SNPs from mapping of the remaining 462 isolates was utilized for phylogenetic analysis of the UKHSA dataset (Fig. 1) and an alignment of 2118 SNPs was used to construct phylogenetic analysis for H58 and nearest neighbours (Fig. 2). Resultant BAM files for all isolates from RedDog mapping were used to determine previously defined genotypes according to an extended genotyping framework using the GenoTyphi pipeline32 (available: https://github.com/katholt/genotyphi).
Phylogenetic analysis
RAxML (v8.2.9)67 was used to infer maximum likelihood (ML) phylogenetic trees from the final chromosomal SNP alignment, with a generalized time-reversible model, a gamma distribution to model site-specific rate variation (the GTR+ Γ substitution model; GTRGAMMA in RAxML), and 100 bootstrap pseudo-replicates to assess branch support. Salmonella Paratyphi A AKU1_12601 (accession no: FM200053)68 was used as an outgroup. The resultant trees were visualized using Interactive Tree of Life (iTOL)69 and the ggtree package in R70. An interactive visualisation of this phylogeny and associated metadata can be found in Microreact (https://microreact.org/project/hzELvWqY3UCvsyAw892fnd-origins-of-h58-s-typhi)71.
Characterisation of AMR associated genes and mobile elements
SRST2 (v0.2.0)72 was used to detect AMR genes and plasmid replicons using the ARGannot73 and PlasmidFinder74 databases, respectively. Mutations in the gyrA and parC genes, as well as the R717Q mutation in acrB, were detected using Mykrobe v0.10.075.
Raw read data for all S. Typhi sequences included in the chromosomal SNP analysis described above were mapped to the reference sequence of IncHI1 plasmid pAKU_1 (accession number AM412236) using RedDog (as described above). Those plasmid sequences where a read depth of at least 10-fold and coverage across the reference sequence of at least 75% were observed were included in SNP analysis, with repetitive regions excluded with Gubbins (v.2.3.2)66. An alignment of 15 SNPs was used as input for plasmid haplotype assignment, which was carried out manually in R (v4.1.2) using the package ape (v5.7.1). SNP distances were determined using snp-dists (v0.7.0). Minimum spanning trees were inferred and visualised using the MSTree method within GrapeTree (v. 1.5.0)76.
Bayesian phylogenetic analysis of H58 and nearest neighbours
Our estimation of the temporal signal of our H58 and nearest neighbour data exhibited a strong correlation between the sampling dates and the root-to-tip distances, with a positive value for the slope and an R2 value of 0.4743 (Supplementary Fig. 3). Additionally, the randomly reassigned sampling time of sequences 20 times to generate the mean rates indicated that there was no overlap between the 95% credible intervals of the mean rate of the real data set and that of the date randomization data (Supplementary Fig. 4). To infer where and when the first H58 (genotype 4.3.1) organism emerged, we conducted Bayesian phylogenetic analyses on a subset (n = 345) of H58 (genotype 4.3.1) from our dataset and from published literature isolated between 1980 and 200014,30,33. This analysis of 345 S. Typhi isolates was conducted in BEAST v1.8.435. The temporal signal of the data was checked initially. The maximum likelihood tree, constructed using the GTR+ Γ substitution model and GTRGAMMA, was subjected to TempEst v1.5 to test the best fit of linear regression between sampling dates and their root-to-tip genetic distances, using default TempEst parameters77. To further test temporal signal, the TipDatingBeast R package was used to randomly reassign the sampling dates of sequences 20 times to create date-randomized data sets. BEAST analyses were conducted for these randomized data sets and the mean rates were compared between runs. The data had sufficient temporal signal if the 95% credible interval of mean rates of the date-randomized datasets did not overlap with that of the original sampling dataset78,79.
An automatic model selection programme (ModelFinder)80 was implemented through IQ-TREE81 and run on the non-recombinant SNP alignment (724 variable sites) to select the best-fit sequence evolution model for BEAST analysis. ModelFinder showed that GTR had the lowest Bayesian Information Criteria (BIC) score and thus it was chosen as the best-fit substitution model.
As part of the BEAST analysis, six different model combinations were run for six combinations, and the final analysis was conducted using the best fitting model. The path sampling and stepping-stone sampling approaches were applied to compare the log marginal likelihoods of the different runs36,82. The GTR+Γ4 with strict clock and Bayesian skyline was identified as the best-fit model for running BEAST. Finally, BEAST was run three independent times using the best-fit model, using a Bayesian Markov Chain Monte Carlo (MCMC) parameter-fitting approach (generated 107 chains and sampled every 1000 iterations). The log files after three runs were combined using LogCombiner v1.8.383 with a burn-in rate of 10%. The effective sample size (ESS) of all parameters was assessed by Tracer v1.8.384. If the ESS of any parameters was less than 200, we increased the MCMC chain length by 50% and reduced the sampling frequency accordingly36. The trees were combined and summarized using LogCombiner v1.8.3 and TreeAnnotator v1.8.335.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
References
GBD 2017 Typhoid and Paratyphoid Collaborators. The global burden of typhoid and paratyphoid fevers: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Infect. Dis. 19, 369–381 (2019).
Crump, J. A. et al. Epidemiology, clinical presentation, laboratory diagnosis, antimicrobial resistance, and antimicrobial management of invasive salmonella infections. Clin. Microbiol. Rev. 28, 901–937 (2015).
Andrews, J. R. et al. Extensively drug-resistant typhoid—are conjugate vaccines arriving just in time? N. Engl. J. Med. 379, 1493–1495 (2018).
Stuart, B. M. & Pullen, R. L. Typhoid; clinical analysis of 360 cases. Arch. Intern Med. (Chic.) 78, 629–661 (1946).
Ochiai, R. L. et al. A study of typhoid fever in five Asian countries: disease burden and implications for controls. Bull. World Health Organ 86, 260–268 (2008).
Carey, M. E. & Steele, A. D. The severe typhoid fever in Africa program highlights the need for broad deployment of typhoid conjugate vaccines. Clin. Infect. Dis. 69, S413–s416 (2019).
Marks, F. et al. Incidence of invasive salmonella disease in sub-Saharan Africa: a multicentre population-based surveillance study. Lancet. Glob. Health 5, e310–e323 (2017).
Baker, S., Hombach, J. & Marks, F. What have we learned from the typhoid fever surveillance in Africa program? Clin. Infect. Dis. 62, S1–S3 (2016).
Anderson, E. S. The problem and implications of chloramphenicol resistance in the typhoid bacillus. J. Hyg. (Lond.) 74, 289–299 (1975).
Olarte, J. & Galindo, E. Salmonella typhi resistant to chloramphenicol, ampicillin, and other antimicrobial agents: strains isolated during an extensive typhoid fever epidemic in Mexico. Antimicrob. Agents Chemother. 4, 597–601 (1973).
Mirza, S. H., Beeching, N. J. & Hart, C. A. Multi-drug resistant typhoid: a global problem. J. Med. Microbiol. 44, 317–319 (1996).
Holt, K. E. et al. Emergence of a globally dominant IncHI1 plasmid type associated with multiple drug resistant typhoid. PLoS Negl. Trop. Dis. 5, e1245 (2011).
Britto, C. D. et al. A systematic review of antimicrobial resistance in Salmonella enterica serovar Typhi, the etiological agent of typhoid. PLoS Negl. Trop. Dis. 12, e0006779 (2018).
Wong, V. K. et al. Phylogeographical analysis of the dominant multidrug-resistant H58 clade of Salmonella Typhi identifies inter- and intracontinental transmission events. Nat. Genet 47, 632–639 (2015).
Carey, M. E. et al. Global diversity and antimicrobial resistance of typhoid fever pathogens: Insights from a meta-analysis of 13,000 Salmonella Typhi genomes. Elife 12, e85867 (2023).
Balasegaram, S. et al. Guidelines for the public health management of typhoid and paratyphoid in England: practice guidelines from the National Typhoid and Paratyphoid Reference Group. J. Infect. 65, 197–213 (2012).
Rai, S. et al. Rationale of azithromycin prescribing practices for enteric fever in India. Indian J. Med. Microbiol. 30, 30–33 (2012).
Basnyat, B. et al. Enteric fever. Br. Med. J. 372, n437 (2021).
Klemm, E. J. et al. Emergence of an extensively drug-resistant salmonella enterica serovar typhi clone harboring a promiscuous plasmid encoding resistance to fluoroquinolones and third-generation cephalosporins. mBio 9, e00105–e00118 (2018).
Hooda, Y. et al. Molecular mechanism of azithromycin resistance among typhoidal Salmonella strains in Bangladesh identified through passive pediatric surveillance. PLoS Negl. Trop. Dis. 13, e0007868 (2019).
Duy, P. T. et al. The emergence of azithromycin-resistant Salmonella Typhi in Nepal. JAC Antimicrob. Resist 2, dlaa109 (2020).
Carey, M. E. et al. Spontaneous emergence of azithromycin resistance in independent lineages of Salmonella Typhi in Northern India. Clin. Infect. Dis. 72, e120–e127 (2021).
Iqbal, J. et al. A race against time: reduced azithromycin susceptibility in Salmonella enterica serovar typhi in Pakistan. mSphere 5, e00215–e00220 (2020).
Hooda, Y. et al. Mass azithromycin administration: considerations in an increasingly resistant world. BMJ Glob. Health 5, e002446 (2020).
Mejia, N. et al. Typhoid and paratyphoid cost of illness in Pakistan: patient and health facility costs from the surveillance for enteric fever in Asia project II. Clin. Infect. Dis. 71, S319–s335 (2020).
Nizamuddin, S. et al. Case of carbapenem-resistant salmonella typhi infection, Pakistan, 2022. Emerg. Infect. Dis. 29, 2395–2397 (2023).
Achtman, M. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu. Rev. Microbiol. 62, 53–70 (2008).
Roumagnac, P. et al. Evolutionary history of Salmonella typhi. Science 314, 1301–1304 (2006).
Holt, K. E. et al. High-throughput bacterial SNP typing identifies distinct clusters of Salmonella Typhi causing typhoid in Nepalese children. BMC Infect. Dis. 10, 144 (2010).
Tanmoy, A. M. et al. Salmonella enterica Serovar Typhi in Bangladesh: exploration of genomic diversity and antimicrobial resistance. mBio 9, e02112–e02118 (2018).
Kariuki, S. et al. Multiple introductions of multidrug-resistant typhoid associated with acute infection and asymptomatic carriage, Kenya. Elife 10, e67852 (2021).
Dyson, Z. A. & Holt, K. E. Five years of GenoTyphi: updates to the global salmonella typhi genotyping framework. J. Infect. Dis. 224, S775–s780 (2021).
Pragasam, A. K. et al. Phylogenetic analysis indicates a longer term presence of the globally distributed H58 haplotype of salmonella typhi in Southern India. Clin. Infect. Dis. 71, 1856–1863 (2020).
Wong, V. K. et al. An extended genotyping framework for Salmonella enterica serovar Typhi, the cause of human typhoid. Nat. Commun. 7, 12827 (2016).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018).
Duchêne, S. et al. Genome-scale rates of evolutionary change in bacteria. Micro. Genom. 2, e000094 (2016).
Johnson, R. et al. Comparison of Salmonella enterica Serovars Typhi and Typhimurium Reveals Typhoidal Serovar-Specific Responses to Bile. Infect Immun. 86, e00490-17 (2018).
Ahmad, I. et al. Complex c-di-GMP signaling networks mediate transition between virulence properties and biofilm formation in Salmonella enterica serovar Typhimurium. PLoS One 6, e28351 (2011).
Bogomolnaya, L. M. et al. Identification of novel factors involved in modulating motility of Salmonella enterica serotype typhimurium. PLoS One 9, e111513 (2014).
Langridge, G. C. et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res 19, 2308–2316 (2009).
Prieto, A. I., Ramos-Morales, F. & Casadesús, J. Repair of DNA damage induced by bile salts in Salmonella enterica. Genetics 174, 575–584 (2006).
Zahrt, T. C., Buchmeier, N. & Maloy, S. Effect of mutS and recD mutations on Salmonella virulence. Infect. Immun. 67, 6168–6172 (1999).
Thanh Duy, P. et al. Gallbladder carriage generates genetic variation and genome degradation in Salmonella Typhi. PLoS Pathog. 16, e1008998 (2020).
Carey, M. E., McCann, N. S. & Gibani, M. M. Typhoid fever control in the 21st century: where are we now? Curr. Opin. Infect. Dis. 35, 424–430 (2022).
Yousafzai, M.T. et al., Effectiveness of typhoid conjugate vaccine against culture-confirmed Salmonella enterica serotype Typhi in an extensively drug-resistant outbreak setting of Hyderabad, Pakistan: a cohort study. Lancet Glob Health 9, e1154–e1162 (2021).
da Silva, K. E. et al. The international and intercontinental spread and expansion of antimicrobial-resistant Salmonella Typhi: a genomic epidemiology study. Lancet Microbe 3, e567–e577 (2022).
Britto, C. D. et al. Persistent circulation of a fluoroquinolone-resistant Salmonella enterica Typhi clone in the Indian subcontinent. J. Antimicrob. Chemother. 75, 337–341 (2020).
Feasey, N. A. et al. Rapid emergence of multidrug resistant, H58-lineage Salmonella typhi in Blantyre, Malawi. PLoS Negl. Trop. Dis. 9, e0003748 (2015).
Kariuki, S. et al. Typhoid in Kenya is associated with a dominant multidrug-resistant Salmonella enterica serovar Typhi haplotype that is also widespread in Southeast Asia. J. Clin. Microbiol 48, 2171–2176 (2010).
Chau, T. T. et al. Antimicrobial drug resistance of Salmonella enterica serovar typhi in Asia and molecular mechanism of reduced susceptibility to the fluoroquinolones. Antimicrob. Agents Chemother. 51, 4315–4323 (2007).
Mashe, T. et al. Salmonella enterica serovar Typhi H58 clone has been endemic in Zimbabwe from 2012 to 2019. J. Antimicrob. Chemother. 76, 1160–1167 (2021).
Park, S. E. et al. The phylogeography and incidence of multi-drug resistant typhoid fever in sub-Saharan Africa. Nat. Commun. 9, 5094 (2018).
Ingle, D. J. et al. Informal genomic surveillance of regional distribution of Salmonella Typhi genotypes and antimicrobial resistance via returning travellers. PLoS Negl. Trop. Dis. 13, e0007620 (2019).
Maes, M. et al. Multiple introductions of Salmonella enterica serovar Typhi H58 with reduced fluoroquinolone susceptibility into Chile. Emerg. Infect. Dis. 26, 2736–2740 (2020).
Walker, J. et al. Assessing the global risk of typhoid outbreaks caused by extensively drug resistant Salmonella Typhi. Nat. Commun. 14, 6502 (2023).
Chattaway, M. A. et al. Phylogenomics and antimicrobial resistance of Salmonella Typhi and Paratyphi A, B and C in England, 2016-2019. Micro. Genom. 7, 000633 (2021).
Gunn, J. S. et al. Salmonella chronic carriage: epidemiology, diagnosis, and gallbladder persistence. Trends Microbiol. 22, 648–655 (2014).
Senthilkumar, B., Senbagam, D. & Rajasekarapandian, M. An epidemiological surveillance of asymptomatic typhoid carriers associated in respect to socioeconomic status in India. J. Public Health 22, 297–301 (2014).
Levine, M. M., Black, R. E. & Lanata, C. Precise estimation of the numbers of chronic carriers of Salmonella typhi in Santiago, Chile, an endemic area. J. Infect. Dis. 146, 724–726 (1982).
Hannon, G.J. FASTX-Toolkit. 2010; Available from: http://hannonlab.cshl.edu/fastx_toolkit.
Andrews, S. FASTQC. A quality control tool for high throughput sequence data. 2010; Available from: https://github.com/s-andrews/FastQC/.
Holt, K. E. et al. High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nat. Genet 40, 987–993 (2008).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Rahman, S. I. A. et al. Population structure and antimicrobial resistance patterns of Salmonella Typhi isolates in urban Dhaka, Bangladesh from 2004 to 2016. PLoS Negl. Trop. Dis. 14, e0008036 (2020).
Croucher, N. J. et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43, e15 (2015).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Holt, K. E. et al. Pseudogene accumulation in the evolutionary histories of Salmonella enterica serovars Paratyphi A and Typhi. BMC Genom. 10, 36 (2009).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–w296 (2021).
Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinforma. 69, e96 (2020).
Argimón, S. et al. Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Micro. Genom. 2, e000093 (2016).
Inouye, M. et al. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 6, 90 (2014).
Gupta, S. K. et al. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58, 212–220 (2014).
Carattoli, A. et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903 (2014).
Bradley, P. et al. Ultrafast search of all deposited bacterial and viral genomic data. Nat. Biotechnol. 37, 152–159 (2019).
Zhou, Z. et al. GrapeTree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 28, 1395–1404 (2018).
Rambaut, A. et al. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2, vew007 (2016).
Firth, C. et al. Using time-structured data to estimate evolutionary rates of double-stranded DNA viruses. Mol. Biol. Evol. 27, 2038–2051 (2010).
Duchêne, S. et al. The performance of the date-randomization test in phylogenetic analyses of time-structured virus data. Mol. Biol. Evol. 32, 1895–1906 (2015).
Kalyaanamoorthy, S. et al. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Nguyen, L. T. et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Xie, W. et al. Improving marginal likelihood estimation for Bayesian phylogenetic model selection. Syst. Biol. 60, 150–160 (2011).
Hill, V. & Baele, G. Bayesian estimation of past population dynamics in BEAST 1.10 using the skygrid coalescent model. Mol. Biol. Evol. 36, 2620–2628 (2019).
Rambaut, A. et al. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Acknowledgements
This work was supported by a Wellcome senior research fellowship to Stephen Baker (215515/Z/19/Z). The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. M.A.C. is affiliated to the National Institute for Health Research Health Protection Research Unit (NIHR HPRU) in Genomics and Enabling Data at University of Warwick in partnership with the UK Health Security Agency (UKHSA), in collaboration with University or Cambridge and Oxford. M.A.C. is based at UKHSA. The views expressed are those of the author(s) and not necessarily those of the NIHR, the Department of Health and Social Care or the UK Health Security Agency.
Author information
Authors and Affiliations
Contributions
Conceptualization, funding acquisition, supervision: S.B.; data curation: M.E.C., T.N.T.N., T.D.H.N., S.N., M.C.; formal analysis: M.E.C., T.N.T.N., Z.A.D., P.T.D.; investigation: M.E.C., T.N.T.N., T.D.H.N., Z.A.D., P.T.D., E.M., J.A.K.; resources: S.N. and M.C., project administration: S.B., M.C.; writing—original draft preparation: M.E.C., S.B., writing—review & editing: M.E.C., T.N.T.N., T.D.H.N., Z.A.D., P.T.D., E.M., S.N., M.C., S.B., and J.A.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Joy Scaria, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: George Inglis and Tobias Goris.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Carey, M.E., Thi Nguyen, T.N., Tran, D.H.N. et al. The origins of haplotype 58 (H58) Salmonella enterica serovar Typhi. Commun Biol 7, 775 (2024). https://doi.org/10.1038/s42003-024-06451-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06451-8
- Springer Nature Limited