

Abstract
Generating large omics datasets has become routine for gaining insights into cellular processes, yet deciphering these datasets to determine metabolic states remains challenging. Kinetic models can help integrate omics data by explicitly linking metabolite concentrations, metabolic fluxes and enzyme levels. Nevertheless, determining the kinetic parameters that underlie cellular physiology poses notable obstacles to the widespread use of these mathematical representations of metabolism. Here we present RENAISSANCE, a generative machine learning framework for efficiently parameterizing large-scale kinetic models with dynamic properties matching experimental observations. Through seamless integration of diverse omics data and other relevant information, including extracellular medium composition, physicochemical data and expertise of domain specialists, RENAISSANCE accurately characterizes intracellular metabolic states in Escherichia coli. It also estimates missing kinetic parameters and reconciles them with sparse experimental data, substantially reducing parameter uncertainty and improving accuracy. This framework will be valuable for researchers studying metabolic variations involving changes in metabolite and enzyme levels and enzyme activity in health and biotechnology.

Similar content being viewed by others
Main
Advancement in biotechnology and health sciences hinges heavily on our capability to integrate different varieties of data produced by high-throughput techniques and obtain coherent insights into cellular processes1,2,3. Considerable effort has been invested in using genome-scale models, mathematical representations of metabolic information about living organisms, to reconcile and make sense of such constantly growing disparate datasets4,5. Genome-scale models integrate omics data by considering constraints imposed by genetics and physicochemical laws6,7,8,9,10. For instance, researchers use inequality constraints stemming from the second law of thermodynamics to relate metabolic fluxes (fluxome) to metabolite profiles (metabolome)11,12,13,14. However, data integration using these inequality constraints results in considerable uncertainty about intracellular metabolic states15. Consequently, despite the availability of large omics datasets, determining the exact intracellular levels of metabolite profiles and metabolic reaction rates with these constraint-based models remains elusive.
Kinetic models of metabolism can address these issues by consolidating several types of omics data, such as metabolomics, fluxomics, transcriptomics and proteomics, within a common and coherent mathematical framework16. Indeed, these models contain information about enzyme kinetics and metabolic regulation, allowing them to explicitly couple metabolite concentrations, metabolic reaction rates and enzyme levels through mechanistic relations. Additionally, unlike constraint-based models, kinetic models capture time-dependent responses of cellular metabolism. Taken altogether, these models show great promise for addressing complex phenomena in biomedical sciences and biotechnology, such as metabolic reprogramming in the tumour microenvironment and disease17,18,19, relationships between cancer, metabolism and circadian rhythms20, dynamics of drug absorption and drug metabolism21, and engineering and modulating cell phenotypes22,23,24.
Despite the capacity of kinetic models to reconcile data and identify metabolic features associated with phenotype, the application of these models is somewhat limited16,25,26,27,28,29,30. The major challenge in developing kinetic models is the lack of knowledge about the characteristic kinetic parameter values that govern the cellular physiology of the studied organism in vivo. Overcoming this requires employing intricate computational procedures and the extensive expertise of researchers. It is often impractical to build and use these models for studying multiple physiological conditions and large cohorts31. Therefore, there is a need for accelerated approaches for parameterizing kinetic models that would allow the broader research community access to these models.
Recent efforts employing new tailor-made parameterization28 and machine learning32,33,34 improved the efficiency of constructing near-genome-scale kinetic models. Nevertheless, challenges remain regarding extensive computational time28 and the need for training data from traditional kinetic modelling approaches32,33,34. Here, we present RENAISSANCE (REconstruction of dyNAmIc models through Stratified Sampling using Artificial Neural networks and Concepts of Evolution strategies), a machine learning framework that efficiently parameterizes biologically relevant kinetic models of metabolism without requiring training data. The behaviour of parameterized kinetic models is highly nonlinear yet deterministic and depends on the intracellular state, defined by network topology and integrated data. To capture this nonlinear behaviour, we use feed-forward neural networks of comparable complexity and optimize them with natural evolution strategies (NES)35,36 to obtain kinetic models with desired properties (Fig. 1a). This dramatically reduces the extensive computation time required by traditional kinetic modelling methods, thus allowing its broad utilization for high-throughput dynamical studies of metabolism. We showcase RENAISSANCE through three studies: generating a population of large-scale dynamic models of Escherichia coli metabolism, characterizing intracellular metabolic states in the E. coli metabolic network accurately, and integrating and reconciling available experimental kinetic data.

a, A NES algorithm iteratively generates candidate solutions for an optimization problem based on their assigned fitness scores until satisfactory solutions are obtained. b, Context-specific structural properties of the metabolic networks are established and incorporated into the model. c, Once the model structure is fixed, available omics data are integrated into the model. d, Generators for parameterizing biologically relevant (valid) kinetic models are optimized iteratively in four steps to meet the design objective: a population of generators is randomly initialized (step I); generators produce parameters needed to parameterize kinetic models (step II); the fitness of the kinetic models (circles and bars in shades of red) is assessed on the basis of the dominant time constants of the model responses (Methods), and the generator is assigned a score based on this performance (step III); the rewards for each generator are fed back to NES to find the best-performing generator (step IV); the best-performing generator is then perturbed to obtain the next generation of generators (step I). e, A few applications of RENAISSANCE-generated models presented in this paper.
Results
Parameterizing biologically relevant kinetic models
We developed RENAISSANCE, a machine-learning framework for parameterizing biologically relevant kinetic models. These models are consistent with experimentally observed steady states and produce dynamic metabolic responses with timescales37 that match experimental observations in cellular organisms. The input to RENAISSANCE is a steady-state profile of metabolite concentrations and metabolic fluxes computed by integrating structural properties of the metabolic network (stoichiometry, regulatory structure and rate laws) and available data (metabolomics, fluxomics, thermodynamics, proteomics and transcriptomics) into the model (Fig. 1b,c and Methods).
RENAISSANCE uses feed-forward neural networks (generators) to parameterize kinetic models, with the size of generator networks dictated by the complexity of the kinetic model. Using NES, it optimizes the weights of generators in four iterative steps until they produce biologically relevant models (Fig. 1d and Methods). The iterative process starts by initializing a population of generators with random weights (step I). The use of multiple generators facilitates a more thorough and more efficient exploration of parametric space. Each generator takes multivariate Gaussian noise as input and produces a batch of kinetic parameters consistent with the network structure and integrated data. These parameter sets are then used to parameterize the kinetic model (step II). Next, we evaluate the dynamics of each parameterized model by computing the eigenvalues of its Jacobian and the corresponding dominant time constants (Methods). These quantities allow us to assess if the generated kinetic models have dynamic responses corresponding to experimental observations (valid models) or not (invalid models). Based on this evaluation, we assign a reward to the generator (step III). NES repeats steps II and III for every generator in the population, followed by normalizing all rewards. The weights of the parent generator for the next generation are then obtained by using the weights of all the members of the previous generation, weighted by their normalized rewards. Although high-performing generators have a greater impact on the weight of the parent generator in the next generation, lower-performing individuals also contribute. NES subsequently mutates this parent generator by injecting a pre-defined noise level into its weights, thus recreating a population of generators (step I). We iterate steps I–IV until we obtain a generator that meets the user-defined design objective, such as maximizing the incidence of biologically relevant kinetic models (Methods).
The generated kinetic models are versatile and applicable to a broad range of metabolism studies (Fig. 1e).
Generating large-scale kinetic models of E. coli metabolism
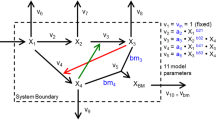
We studied the anthranilate-producing E. coli strain W3110 trpD9923 to test and validate RENAISSANCE. The kinetic model structure for this strain, adopted from Narayanan et al.38, consisted of 113 nonlinear ordinary differential equations parameterized by 502 kinetic parameters, including 384 Michaelis constants, KMs (Methods and Supplementary Fig. 4). It encompasses 123 reactions and describes core metabolic pathways, including glycolysis, the pentose phosphate pathway (PPP), the tricarboxylic cycle (TCA), anaplerotic reactions, the shikimate pathway, glutamine synthesis and a lumped reaction for growth (Methods and Supplementary Fig. 5). The objective was to find kinetic parameters resulting in dynamic models consistent with an experimentally observed doubling time of 134 min for the studied E. coli strain39. A valid kinetic model satisfying this requirement should produce metabolic responses with the dominant time constant of 24 min, corresponding to having the largest eigenvalue λmax < −2.5 (Methods).
We used thermodynamics-based flux balance analysis13,40 to integrate experimental data39 and compute 5,000 steady-state profiles of metabolite concentrations and fluxes (Methods). We selected one of these profiles as input for RENAISSANCE (Methods) and identified a set of hyperparameters yielding the best framework performance with a three-layer generator neural network (Methods and Supplementary Notes 2–4). RENAISSANCE was then executed for 50 evolution generations using the optimized settings. We repeated the optimization process ten times with a randomly initialized generator population to obtain statistical replicates. For each generation, we generated 100 kinetic parameter sets for every generator in the population and computed the maximum eigenvalue, λmax, for each parameter set. To evaluate and rank the generators, we used the incidence of valid models, defined as the proportion of the generated models that are valid (with λmax < −2.5; Methods). We observed that the incidence of valid models steadily increases with the number of generations, with the mean incidence converging around 92% after 50 generations (Fig. 2a, thick black line and Supplementary Figs. 14 and 16). For some repeats, we could achieve incidence up to 100% (Fig. 2a, green-shaded region).

a, The incidence of models exhibiting the desired dynamic properties increases with the number of generations, as indicated by the mean incidence (black line) and the maximum and minimum incidence (green-shaded region) observed over ten statistical repeats for every generation. The dashed line indicates the incidence of a repeat with fast convergence. The black diamonds indicate the generators selected for subsequent analysis from that repeat. b, The distribution of the maximum eigenvalues (λmax) for the generated models over generations. The vertical dashed lines indicate λmax = −2.5 (left) and λmax = 0 (right). c, Robustness analysis. The time evolution of the normalized perturbed biomass, \(v(t)/{v}_{{{\mathrm{ref}}}}\), and concentrations, \(\left(X\left(t\right)-{X}_{\mathrm{ref}}\right)/{X}_{\mathrm{ref}}\), of nicotinamide adenine dinucleotide reduced (NADH), adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide phosphate reduced (NADPH). Shown are the mean response (dashed black line), the 25th–75th percentile (dark-orange region) and the 5th–95th percentile (light-orange region) of the ensemble of responses. The vertical dashed line corresponds to t = 24 min. d, Bioreactor simulations. The time evolution of biomass, glucose concentration and anthranilate concentration in the bioreactor runs of the 13 models closely fitting the experimental data39. The black dots and error bars represent the mean and standard deviation from triplicate experiments, respectively.
For further analysis of the generated models, we selected a statistical repeat with fast convergence (Fig. 2a, dashed line) and chose ten generators from that repeat with monotonically increasing incidence over generations (Fig. 2a, black diamonds). For each of the ten chosen generators, we generated 500 kinetic parameter sets and examined the distribution of the resulting maximum eigenvalues (Fig. 2b). Remarkably, the generated models gradually shifted over the optimization process from having slow dynamics (λmax > −2.5) to having fast dynamics, with the metabolic processes settling before the subsequent cell division, indicating that RENAISSANCE-generated models could capture the experimentally observed dynamics.
Since cellular organisms maintain phenotypic stability when faced with perturbations41, the generated models that describe cellular metabolism should possess the same property. To test the robustness of the models, we perturbed the steady-state metabolite concentrations up to ±50% and verified if the perturbed system returned to the steady state. For this purpose, we generated 1,000 relevant kinetic models using the final of 10 selected generators (Fig. 2a, generation 45), chosen for yielding the highest incidence of valid models. Inspection of the time evolution of the normalized biomass showed that the biomass returned to the reference steady state (v(t)/vref = 1) within 24 min for 100% of the perturbed models (Fig. 2c). Similarly, the perturbed time responses of a few critical metabolites, namely NADH, ATP and NADPH, returned to their steady-state values within 24 min for 99.9%, 99.9% and 100% of the 1,000 generated kinetic models, respectively (Fig. 2c). Examining every cytosolic metabolite collectively revealed that 75.4% of the models returned to the steady state within 24 min and 93.1% returned within 34 min, demonstrating that the generated kinetic models are robust and obey imposed context-specific observable biophysical timescale constraints.
Next, we tested the generated models in nonlinear dynamic bioreactor simulations closely mimicking real-world experimental conditions38,39. The temporal evolution of biomass production showed similar trends as typical experimental observations with clear exponential and stationary phases of E. coli growth (Fig. 2d, Supplementary Note 6 and Supplementary Fig. 6). Similarly, glucose uptake and anthranilate production also reproduce trends observed in experiments with glucose consumption halted and anthranilate production saturating at around 20 h. This study indicates that the RENAISSANCE models can accurately reproduce the physiologically observable and emergent properties of cellular metabolism, even without implicit training to reproduce fermentation experiments.
Characterizing the intracellular metabolic states of E. coli
Accurately determining the intracellular levels of metabolite profiles and metabolic reaction rates is crucial for associating metabolic signatures with phenotype. Yet, our capabilities to establish the intracellular metabolic state are limited. Even with the ever-increasing availability of physiological and omics data, a considerable amount of uncertainty in the intracellular states remains. We propose using kinetic models to reduce this uncertainty because of their explicit coupling of enzyme levels, metabolite concentrations and metabolic fluxes. Moreover, kinetic models allow us to consider dynamic constraints in addition to steady-state data, thus allowing us further uncertainty reduction.
After integrating available physiology and omics data39,42,43,44 using the constraint-based thermodynamics-based flux balance analysis40, substantial uncertainty was present in the intracellular metabolic state as indicated by the wide ranges of metabolite concentrations and metabolic fluxes. We sampled 5,000 steady-state profiles of metabolite concentrations and metabolic fluxes from this uncertain space and deployed RENAISSANCE to find the fastest possible dynamics (maximum negative eigenvalues, λmax) for each steady state (Methods and Supplementary Fig. 7). We visualized the steady-state profiles by performing dimension reduction with principal component analysis (PCA)45 and t-distributed stochastic neighbour embedding (t-SNE)46 (Methods) and coloured each steady-state profile according to the obtained λmax (Fig. 3a). We observed a high variation in the dynamics (λmax) of the studied steady-state profiles (Fig. 3c, blue distribution). Of 5,000 steady-state profiles, 918 (18.4%) had λmax larger than −2.5, meaning these intracellular metabolic states could not correspond to the experimental observations. Indeed, the dynamic responses corresponding to these states have a time constant superior to 24 min, that is, slower than the experimental observations.

a, The two-dimensional representation of the fastest linearized dynamic modes (corresponding to the maximum eigenvalue λmax) of 5,000 intracellular steady states (reaction fluxes and metabolite concentrations) obtained with PCA and t-SNE46 (Methods). Each point represents a steady state, with its colour indicating the corresponding λmax value computed by RENAISSANCE. b, Magnified view of 22 neighbouring steady states with fast dynamics (−3.8 ≤ λmax ≤ −8.5). The colour scheme is the same as in a. c, Distributions of the fastest linearized dynamic (λmax) for all 5,000 steady states (blue) and the 22 steady states shown in b (green). d, The linearized dynamics landscape of the 22 fast steady states in the reduced space (Methods). The triangles represent the location of the steady states in the landscape. e, The landscape in d is enhanced by sampling 90 additional steady states in the neighbourhood of the initial 22 steady states. The circles represent the location of the newly sampled steady states in the same landscape as in d. f, Distributions of the fastest linearized dynamic (λmax) for all steady states (blue) and 90 steady states sampled in e (pink). g, The concentration of 3-phosphoglyceric acid (3pg) (mM) versus the fastest linearized dynamic in every steady state. The colour scheme is the same as that in a. The horizontal black line indicates the cut-off for valid models (λmax = −2.5). The peach-shaded region shows the range of 3-phosphoglyceric acid concentration that does not allow fast dynamics. h, The dynamic landscape of 40 steady states sampled by constraining the metabolite concentrations of 30 metabolites to ranges that do not support fast dynamics. The diamonds represent the location of the steady states. i, Distributions of the fastest linearized dynamic (λmax) for the 40 steady states sampled in g (peach), compared with all steady states (blue) and those sampled in d and e (pink).
As t-SNE optimizes the preservation of local distances between points when projecting them from a high-dimensional space to a lower-dimensional one46,47, we hypothesized that sampling from a region containing closely positioned steady-state profiles associated with fast dynamics (Fig. 3a, blue dots) would yield steady-state profiles that satisfy dynamic requirements. Conversely, sampling from a region around adjacent profiles corresponding to slow dynamics (Fig. 3a, yellow dots) would probably result in profiles not meeting dynamic requirements.
To test this hypothesis, we selected one of these local regions (Fig. 3b), which contained 22 steady states with fast dynamics with −3.8 ≤ λmax ≤ −8.5 (Fig. 3c, green distribution), and analysed its neighbourhood (Fig. 3d). We sampled 90 additional steady states within this neighbourhood from the Gaussian distribution with a mean and standard deviation estimated on the initial 22 steady states. The sampled steady states allowed us to improve the resolution of the initial dynamic landscape (Fig. 3e, circles). Crucially, the sampled steady states had linearized dynamics in the same range as the initial 22 states (Fig. 3d–f), confirming our hypothesis. Therefore, RENAISSANCE allows us to select subsets of intracellular states consistent with experimentally observed dynamics and generate additional ones with the same characteristics. Moreover, it allows us to discard subregions with experimentally inconsistent states, thus reducing uncertainty. Indeed, sampling from a region containing closely positioned steady-state profiles associated with slow dynamics yielded steady states corresponding to similarly slow dynamics (Supplementary Fig. 13).
We next examined individual metabolite concentrations of the 5,000 steady-state profiles to identify patterns corresponding to the experimentally observed phenotype. We observed a clear bias in the dynamics depending on the concentrations for some of the metabolites (Fig. 3g and Supplementary Fig. 7). For example, in the case of 3-phosphoglyceric acid, we obtain models with relevant dynamics only when the concentration of this metabolite is less than ∼0.002 mM. In contrast, steady-state profiles with 3-phosphoglyceric acid concentrations between 0.002 and 0.003 mM do not have relevant dynamics (Fig. 3g). To investigate this further, we identified 30 cytosolic metabolites that showed such concentration biases by visual inspection (Supplementary Fig. 8) and sampled 40 new steady states from the same Gaussian distribution as before (Fig. 3d) but constrained the selected 30 metabolites to concentration ranges that do not support relevant dynamics (for example, the peach-shaded region in Fig. 3g). As expected, almost all of these new intracellular states did not yield models with relevant dynamics (Fig. 3h,i). This result demonstrates that information stemming from the dynamic responses can be used to constrain values of intracellular metabolites to specific ranges.
Overall, the dynamic characterization of a broad range of intracellular states allows us to reduce uncertainty at the level of steady-state profiles, as well as individual metabolite concentrations and metabolic fluxes.
Integration and reconciliation of experimental information
Experimentally measured Michaelis constants, KMs, are curated in comprehensive databases containing functional and molecular information of enzymes such as BRENDA48. However, as we transition to large genome-scale kinetic models, a vast majority of the associated kinetic parameters remain unknown. Integrating experimental results from in vivo and in vitro studies, despite the disparities in their parameter values, can help further constrain uncertainty and lead to a more accurate description of intracellular metabolic states. To this end, we retrieved experimentally measured values for 108 out of 384 KMs in our model from BRENDA (Methods).
To investigate how the integrated kinetic data constrain unknown kinetic parameters, we started by integrating four KM values of aconitase (ACONTa,b) from TCA (Fig. 4a and Methods), obtained generators with a high incidence of valid models (>99%) and generated 500 valid kinetic models (Supplementary Fig. 9). To quantify the effect of integrating one experimental KM value on the generated values of the other kinetic parameters, we compared the estimates of the other KMs and maximum velocities, vmax, with ones obtained when no kinetic parameters were integrated. Integration of KM values of aconitase at a reaction level restricted the estimates of \({v}_{\max }^{{{\mathrm{ACONTa}}}}\) (Fig. 4b). Due to the correlation in the vmax values throughout the network, restricting \({v}_{\max }^{{{\mathrm{ACONTa}}}}\) estimates through KM integration constrained the estimated ranges of other maximal velocities, such as \({v}_{\max }^{{{\mathrm{ICDHyr}}}}\) (Fig. 4b). This restriction further affected downstream KM values in the network, such as \({K}_{{\mathrm{M}},{{\mathrm{akg}}}}^{{\,{\mathrm{ICDHyr}}}}\) and \({K}_{{\mathrm{M}},{{\mathrm{succoa}}}}^{{{\mathrm{AKGDH}}}}\) (Fig. 4b). These results suggest that integrating only a small amount of experimental data, localized to one enzyme (ACONTa,b), propagates throughout the metabolic network and alters the rest of the kinetic parameters.

a, RENAISSANCE allows direct integration of KM values from literature and reconciling them with unknown parameters that collectively lead to valid kinetic models. b, Propagation of the integrated KM experimental data around ACONTa,b through the metabolic network. Comparison of RENAISSANCE-generated values for \({K}_{{\mathrm{M}}{{,}}{{\text{acon-C}}}}^{{{\mathrm{ACONTa}}}}\) versus \({v}_{\max }^{{{\mathrm{ACONTa}}}}\), \({v}_{\max }^{{{\mathrm{ICDHyr}}}}\) versus \({v}_{\max }^{{{\mathrm{ACONTa}}}}\), \({v}_{\max }^{{{\mathrm{ICDHyr}}}}\) versus \({K}_{{\mathrm{M}},{{\mathrm{akg}}}}^{{\,{\mathrm{ICDHyr}}}}\), and \({K}_{{\mathrm{M}},{{\mathrm{succoa}}}}^{{{\mathrm{AKGDH}}}}\) versus \({K}_{{\mathrm{M}},{{\mathrm{akg}}}}^{{\,{\mathrm{ICDHyr}}}}\) when (i) no kinetic parameters are integrated (grey circles) and when (ii) four parameters are integrated (orange circles; Methods). ICDHyr, isocitrate dehydrogenase; AKGDH, 2-oxoglutarate dehydrogenase; SUCDi, succinate dehydrogenase (irreversible); SUCOAS, succinyl-CoA synthetase (ADP-forming); CS, citrate synthase; MDH, malate dehydrogenase; MALS, malate synthase; FUM, fumarase; q8h2_c, ubiquinol-8; q8_c, ubiquinone-8; succ_c, succinate; coa_c, coenzyme A; pi_c, phosphate; succoa, succinyl-CoA; nadh_c, nicotinamide adenine dinucleotide-reduced; nad_c, nicotinamide adenine dinucleotide; nadph_c, nicotinamide adenine dinucleotide phosphate-reduced; nadph, nicotinamide adenine dinucleotide phosphate; icit_c, isocitrate; cit_c, citrate; accoa_c, acetyl-CoA; oaa_c, oxaloacetate; mal-L_c, L-Malate; fum_c, fumarate; glx_c, glyoxylate; acon-C, cis-aconitate; akg, 2-oxoglutarate; succoa, succinyl coenzyme-A.
We next enquired if RENAISSANCE improves its KM estimates as the number of integrated experimental KM values increases. We also examined how the localization of integrated KM values, such as the integration of KM values from TCA, affects the estimation of KM values in other subsystems of the metabolic network. Specifically, we integrated 10 random combinations of half (9) of the 17 available experimentally measured KM values associated with the TCA of E. coli, one combination at a time. For each of the 10 combinations, we obtained generators with a high incidence of valid models (>90%) and generated 2,000 of these models. In total, we generated 20,000 models containing 10 distinct combinations of the remaining 8 Michaelis constants to be estimated. This process ensured that each of the 17 Michaelis constants was integrated at least once and estimated at least once within the 10 combinations.
The comparison between the experimentally observed and the RENAISSANCE estimated range of TCA KM values, quantified through the overlap score (OS) between these two ranges (Fig. 5g), showed that integrating KMs improves the estimates of the non-integrated individual KMs within the same subsystem (Fig. 5a, red bars and Supplementary Fig. 9), compared with when no KM values are integrated (Supplementary Fig. 15, black diamonds). Indeed, noteworthy improvement was observed in the predictions of 16 out of the 17 KM values in TCA when experimental values of KM were integrated. The average prediction accuracy for the entire subsystem also increased (Fig. 5b, red bars) compared with the case with no integration of experimental KMs (blue bars). A similar analysis was conducted for other subsystems, PPP, glycolysis, anaplerotic reactions, shikimate pathway and pyruvate metabolism, and consistently, estimates of KM values within the same subsystem improved upon the integration of experimental information for all the cases (Fig. 5c,d and Supplementary Fig. 10). These findings indicate that integrating experimental information may improve prediction accuracy beyond the subsystem level.

a, The OS (defined in d) of RENAISSANCE estimates for 17 experimentally available Michaelis constants, KMs, from TCA (red). The estimation process involved integrating experimental values for 9 out of the 17 parameters and estimating other model parameters including the remaining 8 parameters from TCA. The procedure was repeated ten times with a different random combination of nine parameters. The black diamonds represent the OS of the estimates without integrating experimental data. The error bars indicate the standard error in the OS over ten combinations of integrated parameters. b, The mean OS for the estimated KMs within metabolic subsystems before (blue) and after (red) integrating experimentally measured KMs. The estimation process for each subsystem was conducted in a similar manner as in a. c, The OS of RENAISSANCE estimates for 91 experimentally available KMs not belonging to TCA when 10 different combinations of 9 (out of 17) KMs from TCA were integrated. The black diamonds represent the OS without integration. The error bars represent the standard error in OS. d, The mean OS of the estimated KMs from all metabolic subsystems except for the subsystem labelled on the x axis before (blue) and after (red) integrating KMs from the labelled subsystem. e, The metabolic subsystems of the top 15 KMs with the highest increase in OS when Michaelis constants from the labelled subsystem were integrated. The donut plots indicate the count of integrated experimental KM values from each labelled subsystem. f, The two-component PCA48 representation of the 276 (out of 384) experimentally unverified kinetic parameters with and without integrating experimentally measured Michaelis constants. g, The OS of an estimated KM is calculated as the ratio between (x) the overlap of the RENAISSANCE predicted (teal) and the experimentally observed range (violet), and (y) the RENAISSANCE predicted range. anpl, anaplerotic reactions; glu, glutamate metabolism; gly, glycolysis/gluconeogenesis; glse, glycine/serine metabolism; hist, histidine metabolism; nsp, nucleotide salvage pathway; oxp, oxidative phosphorylation; pyr, pyruvate metabolism; shkk, shikimate pathway; oth, other reactions.
Inspecting the distributions of the generated KMs that were not part of the TCA subsystem revealed that the predictions for a vast majority of these KMs (85 out 91) improved upon the integration of TCA KMs (Fig. 5c, coloured bars) compared with the case where no KMs were integrated (black diamonds). Similarly, the mean OS of the entire set increased (Fig. 5d). We then examined the top 15 KMs that exhibited the most marked improvement in their estimates and determined the metabolic subsystem in which they are located. The integration of experimental KM values from TCA yielded the most notable improvement in the estimates of the shikimate pathway (6 in the top 15), followed by glycolysis (3 out of 15) and anaplerotic reactions (2 out of 15) (Fig. 5e, leftmost donut plot). Interestingly, a similar analysis conducted by integrating KMs from other subsystems showed that the estimates from these three subsystems (shikimate pathway, glycolysis and anaplerotic reactions) consistently yielded the most notable improvement (Fig. 5e and Supplementary Fig. 11). These results provide evidence that RENAISSANCE effectively incorporates experimental kinetic data from a specific subsystem of the metabolic network, resulting in improved parameter estimates across the entire network.
We further examined the impact of integrating experimental kinetic data on parameters that lack verifiable experimental measurements, which accounted for 276 out of 384 KMs. To obtain a qualitative assessment of the effects of integration, we employed PCA45 to visualize the RENAISSANCE predictions for these unknown KMs (Fig. 5f). The analysis revealed notable shifts in the estimates of these KMs when experimental data were integrated compared with the case where no data were integrated (Fig. 5f, blue cluster). Additionally, the estimates for the cases with integrated experimental data exhibited greater similarity than those without integration. We provide the generator, trained to incorporate all 108 KMs available from BRENDA (Supplementary Fig. 12), in Supplementary Information.
These results suggest that integrating experimental kinetic information reduces quantitative uncertainties in the intracellular metabolic state of the cell, allowing RENAISSANCE to make more informed predictions on the dynamic properties of the entire metabolic network. We anticipate that the inclusion of new experimental data and their subsequent integration will enhance the predictive capabilities of RENAISSANCE even further.
Discussion
Metabolism plays a defining role in shaping the overall health of living organisms. A reprogrammed or altered metabolism is not only associated with the most common causes of death in humans—cancer, stroke, diabetes, heart disease and others—but is also related to many congenital diseases49. Thus, a better understanding of metabolic processes is crucial to accelerate the development of new drugs, personalized therapies and nutrition. Biotechnological advances such as the bioproduction of industrially essential compounds and environmental bioremediation also hinge on our ability to describe cellular metabolism accurately.
Kinetic models provide the most thorough mathematical representation of metabolism. The efficient construction of these models will open new possibilities for various biomedical and biotechnological applications. However, acquiring the parameters of these models with traditional kinetic modelling approaches is computationally expensive and arduous15,34. Several machine learning methods were recently proposed for more efficient kinetic model generation, including iSCHRUNK32,33,50 and REKINDLE34. REKINDLE, in particular, has demonstrated remarkable gains in model generation efficiency by using generative adversarial networks (GANs)51. Nevertheless, existing kinetic modelling approaches were required to create the data needed for the GAN training. The proposed RENAISSANCE framework retains the model generation efficiency of REKINDLE without the need for training data because it employs the NES, requiring only a scoring function to train generators.
In its conception, RENAISSANCE can parameterize kinetic models to satisfy a broad range of biochemical properties or physiological conditions. For example, it can parameterize models reproducing experimentally observed fermentation curves or drug adsorption patterns. Herein, we use RENAISSANCE to parameterize kinetic models to be consistent with an experimentally observed steady state. This approach to model construction was introduced within the ORACLE conceptual framework50,52,53,54,55, which parameterizes kinetic models by unbiased sampling. In contrast, in RENAISSANCE, we leverage machine learning to perform stratified sampling biased towards kinetic models producing metabolic responses over time with timescales37 matching experimental observations in studied organisms. Due to its capability to bias parameter sampling towards desired model properties, the proposed framework substantially improves model construction efficiency, enabling comprehensive studies of multiple physiological conditions.
RENAISSANCE can train model generators on a standard workstation in 3–20 min (Supplementary Note 5). Once trained, the generators generate ~1 million models in 15–20 s, making this framework several orders of magnitude more efficient than traditional sampling-based kinetic approaches. RENAISSANCE also does not require specialized hardware to execute. The proof-of-concept applications shown here demonstrate RENAISSANCE’s applicability to a broad range of studies. In this work, we deployed RENAISSANCE to parameterize valid models of metabolism consistent with an experimentally observed steady state, with validity being characterized by the biological relevance of their timescales. However, conceptually, any other requirement can be imposed or data used, such as consistency with knockout studies or time series from drug absorption trials.
As RENAISSANCE is agnostic to the nature, range and number of the parameters it needs to generate, it is straightforward to adjust the framework to meet the specific demands the models need to satisfy. The parameters this framework can handle are not restricted to Michaelis constants only and can include other kinetic parameters, such as enzyme saturations52 and enzyme states56, and other unknown quantities in the studied system, such as metabolite concentrations.
Crucially, given proteomic data, RENAISSANCE can predict unknown enzyme turnover number, kcat, values and consolidate them with experimentally measured kcat values from databases such as BRENDA and SABIO-RK57. As such, it represents a valuable complement to current machine learning methods that estimate kcat values directly58,59,60.
In summary, we provide a fast and efficient framework that leverages machine learning to generate biologically relevant kinetic models. The open-access code of RENAISSANCE will facilitate experimentalists and modellers to apply this framework to their metabolic system of choice and integrate a broad range of available data.
Methods
E. coli model structure and data integration
The kinetic model structure is based on a previous study by Narayanan et al.38. The reduced stoichiometry was obtained using redGEM61 and lumpGEM62, and it includes core carbon pathways such as glycolysis, PPP, TCA, anaplerotic reactions, the shikimate pathway, glutamine synthesis and a lumped reaction for growth. The anthranilate phosphoribosyltransferase was removed to tailor the general E. coli model to strain W3110 trpD9923. The resulting model structure had 113 mass balances, including one for biomass accumulation, involving 123 reactions parametrized with 507 kinetic parameters including 384 KMs and 123 vmaxs (Supplementary Fig. 4). Further details on the kinetic model structure can be found in Supplementary Data 1 of Narayanan et al.38.
A context-specific model of W3110 trpD9923 was created by integrating metabolomics and fluxomics data from previous experimental studies. The lower bounds of the growth rate (0.26 h−1) and anthranilate secretion rate (0.14 mmol gDW−1 h−1 (gram per weight per hour)) were set to the reported values from Balderas-Hernandez et al.39, and the glucose uptake rate was adjusted to be consistent with the secretion and growth rates. Extracellular metabolites neither found in the media nor listed as secreted were assigned upper bounds on their secretion rates of 0.01 µM gDW−1 h−1 and concentrations of 1 μM, whenever possible. Intracellular metabolite concentrations reported in Park et al.44 were constrained to be within twofold of the reported values. Next, constraints were imposed on thermodynamic variables, calculated using the group contribution method42,43, to ensure that the sampled flux directionalities and metabolite concentrations were consistent with the second law of thermodynamics.
Then, 5,000 sets of steady-state profiles consistent with the integrated data were sampled from this context-specific model using thermodynamics-based flux balance analysis implemented in the pyTFA tool40. Each steady-state profile comprises metabolite concentrations, metabolic fluxes and thermodynamic variables. Once these profiles are available, we can generate kinetic models around each of these steady states27,53,55 using the RENAISSANCE framework. We used the profile with index 1712 as input for RENAISSANCE in all studies, except for the study detailed in ‘Characterizing the intracellular metabolic states of E. coli’ section where all 5,000 steady states were employed (Fig. 3).
At present, RENAISSANCE allows for integrating transcriptomics and proteomics data at the steady-state level. When computing the steady-state profiles around which the kinetic models are built, REMI63 and TEX-FBA64 tools allow simultaneous integration of transcriptomics, metabolomics and fluxomics data. Other tools for integrating transcriptomics are provided elsewhere65. As discussed in Sanchez and Zhang et al.9, proteomics data can be integrated by imposing the upper bounds on the fluxes through the expression \({v}_{j}\le {k}_{{{\mathrm{cat}}}}^{i,\,j}{E}_{i}\), where kcat is the turnover number and E denotes enzyme concentration. The transcriptomics and proteomics data were not available for the present study.
Determining the validity of kinetic models
The Jacobian matrix and its eigenvalues are used in the control66 and nonlinear dynamics theory67 to analyse the stability and behaviour of a nonlinear dynamical system in the vicinity of an equilibrium point by performing linearization. The Jacobian matrix is derived by taking the partial derivatives of a set of differential equations with respect to the state variables. The sign of the Jacobian eigenvalues provides information on the local stability of the generated models, where a model is locally stable if the real parts of all eigenvalues are negative52. The inverse of the real part of the largest eigenvalue of the Jacobian defines the dominant time constant of the linearized system.
The time constant defines the time required for the system response to decay to \(\frac{1}{{\mathrm{e}}}\approx 36.8 \%\) of its initial value. The dominant time constants allow us to characterize the model dynamics; small time constants characterize fast metabolic processes such as glycolysis and electron transport chain. In contrast, polymerization processes involving the synthesis of DNA, RNA and proteins typically occur at slower timescales.
In this context, we consider a kinetic model valid (biologically relevant) if all time constants of the model response are consistent with the experimental observations of the studied organism.
To ensure that a perturbation of the metabolic processes settles within 1% of the steady state before cell division, the dominant time constants of the model response should be five times faster than the cell’s doubling time38. The biochemical response should also have a characteristic time slower than the timescale of proton diffusion within the cell34. With these properties, models can reliably describe the experimentally measured metabolic responses.
The doubling time of the E. coli strain used in this study is tdoubling = 134 min, which corresponds to a growth rate of \({\mathrm{ln}}2\times \frac{60}{{t}_{\mathrm{doubling}}}=0.31{{\rm{h}^{-1}}}\). Therefore, the dominant time constant of the model’s responses should be smaller than one-fifth of the doubling time (26.8 min). Here, we imposed a stricter dominant time constant of 24 min, corresponding to an upper limit of Re(λi) <−2.5 (or −60/24), on the real parts of the eigenvalues, λi, of the Jacobian. All kinetic parameter sets resulting in the model obeying this constraint are labelled valid and the rest invalid.
Assigning rewards to determine fitness in RENAISSANCE
In RENAISSANCE, we employ deep neural networks known as generators to produce kinetic parameter sets for a given metabolic mode l structure. Technically and structurally, these neural networks are similar to those in GANs51 or other deep generative algorithms such as variational autoencoders68. The key distinction lies in the training methodology for this neural network. GANs or variational autoencoders rely on explicit training data. For instance, in a prior study34, we used kinetic parameter sets derived from traditional kinetic modelling methods for training. Unlike traditional gradient-based deep learning methods, which rely on training data to train a neural network, RENAISSANCE employs the NES (Supplementary Note 1), which only requires a scoring function.
To optimize the weights of the generator network, the NES algorithm produces a population of candidate solutions to an optimization problem and assigns a fitness score to each candidate solution (Fig. 1a). The algorithm uses the fitness scores of the current solutions to generate the next generation of candidate solutions, which are likely to have better fitness scores than the current generation. The iterative procedure stops as soon as the obtained solutions are satisfactory. This method is particularly advantageous in scenarios where the fitness landscape is complex, non-differentiable or unknown, and it avoids the need for backpropagation or direct gradient computation.
The NES algorithm includes several steps:
Step I: NES collects the rewards of all the generators (neural networks) in a generation. In our case, the reward is the percentage of relevant generated models (thus, the reward for each neural network or generator in a generation is between 0 and 1).
Step II: The rewards for each generation member are normalized by subtracting the mean and dividing by the standard deviation of all the rewards in the generation. The normalization ensures that the update direction depends on how each member’s performance compares with the average rather than on absolute reward values.
Step III: For each weight in the generator neural network, the algorithm computes an update proportional to the dot product between the population’s perturbations (the added noise for generating new individuals) and the normalized rewards. If a perturbation consistently results in higher rewards, it will exert a more important influence on the direction of the weight update. Thus, the reward determines the selection of the ‘best’ generator.
Step IV: The update to each weight is scaled by the learning rate and inversely scaled by the population size and the noise level (σ). Scaling prevents drastic changes that might destabilize the learning process (Supplementary Note 2).
The design objective of the conducted studies is to maximize the occurrence of biologically relevant kinetic models. In case all generators of the current generation fail to generate valid models, the generator producing models closest to the cut-off eigenvalue (−2.5) is rewarded higher, thus having a more important impact on the weight of the parent generator in the next generation. The implementation of this concept is as follows.
To calculate the local gradient estimate, NES requires an objective function, F, to evaluate the fitness of each generator network, G. In our study, we use the incidence of the generator, I(G), as the objective function, which is defined as the fraction of the generated models that are relevant (0 ≤ I(G) ≤ 1). Thus, generator networks with a higher incidence of relevant models are ‘fitter’ than those with low incidence and have a higher weight in determining the parameters of the seed generator network for the next generation. In many cases, we observed that initially the generator neural networks do not generate any relevant models, and thus the optimization does not proceed as the fitness is always 0. To mitigate this, we added a sigmoidal term defined as
where λfastest corresponds to the smallest maximal eigenvalue of the generated models and λpartition is the maximal eigenvalue partition that determines the relevancy of the kinetic model. In this study, λpartition = −2.5 (‘Determining the validity of kinetic models’). This term rewards generators that generate models with dynamics closer to the relevant range more than those that generate models with slower, irrelevant or unstable dynamics. This effectively pushes the optimization process towards finding generators that generate relevant models. So, the overall reward, R, for a generator, G, can be summarized as
For the large-scale analysis of intracellular states (Fig. 3), the fitness for NES was no longer the incidence of the generators but the fastest possible dynamic for the models generated by a given generator. Thus, the reward was changed suitably as
where λmean is the mean of the 10 fastest maximum eigenvalues (Supplementary Fig. 6) generated by a generator (out of 100 for this case study). This reward function ensured that the generators that generated models with more negative maximum eigenvalues (faster linearized dynamics, λmax) were rewarded more than the others.
Hyperparameter tuning of RENAISSANCE
The hyperparameters of the NES algorithm used in RENAISSANCE are (1) the population size, n, determining the number of generator networks initiated/created and evaluated in each RENAISSANCE generation; (2) search radius, σ, representing the level of noise injected into the weights of the parent generator in each generation; (3) learning rate, α, determining the step size taken by the optimizer in the gradient space, that is, it determines the magnitude of the updates to the generator neural network weights during each iteration of the optimization process; and (4) learning rate decay, d, representing the rate at which the learning rate decreases at each generation, helping the optimization process to converge more effectively. In this study, we tuned the hyperparameters of RENAISSANCE to maximize the incidence of valid models (Supplementary Notes 3 and 4).
The optimal set of hyperparameters found after grid search is as follows: the population size of the generator networks, n = 20; the noise level in generating the agent population from the mean optimal weights in each generation, σ = 10−2; the learning rate of the gradient step, α = 10−3; and the decay rate of learning, d = 5%. In addition, the generated KMs were constrained strictly in the range [1.3 × 10−11, 20] to accurately represent experimentally measured KM values as curated in the BRENDA database48.
Generator neural networks
The generator neural networks were composed of three layers with 1,076,352 parameters: layer 1, dense, with 256 units, dropout (0.5); layer 2, dense, with 512 units, dropout (0.5); and layer 3, dense, with 1,024 units, dropout (0.5). All software programs were implemented in Python (v3.6). Neural networks were implemented using the TensorFlow library69 (v2.3.0).
Dimension reduction and visualization of steady states
For generating Fig. 3a,d,f (left), the following steps were followed: (1) the steady-state matrix (consisting of 1,127 features) was subjected to PCA45; (2) the components of PCA that contributed to over 99% of the total expected variance were reduced to two dimensions using t-SNE46; and (3) the t-SNE components {xq, xp} were then subjected to polar coordinate transformation as
{x1, x2} were then plotted to generate the figures.
Integrating known kinetic parameters from BRENDA
If there were multiple experimentally measured values for a single KM in BRENDA, we took the geometric mean, \({K}_{{\mathrm{M}},\exp }\), of the different values and added an experimental error rate of ±20% to \({K}_{{\mathrm{M}},\exp }\). The same error rate was applied if there was only one recorded experimental value, \({K}_{{\mathrm{M}},\exp }\). Then, the value of an integrated KM was sampled uniformly from the range \({K}_{{\mathrm{M}},\exp }\) ± 20% when integrated into RENAISSANCE for the training process and generation.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting this study’s findings are publicly available in the Zenodo repository at https://doi.org/10.5281/zenodo.7628650 (ref. 70) and the links therein.
Code availability
A Python implementation of the RENAISSANCE workflow is publicly available via GitHub at https://github.com/EPFL-LCSB/renaissance and https://gitlab.com/EPFL-LCSB/renaissance. The ORACLE framework is implemented in the SKimPy (Symbolic Kinetic models in Python)62 toolbox, available via GitHub at https://github.com/EPFL-LCSB/skimpy.
References
Bui, A. A. T., Van Horn, J. D., NIH BD2K Centers consortium. Envisioning the future of ‘big data’ biomedicine. J. Biomed. Inform. 69, 115–117 (2017).
Monk, J. M. et al. iML1515, a knowledgebase that computes Escherichia coli traits. Nat. Biotechnol. 35, 904–908 (2017).
Brunk, E. et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 36, 272–281 (2018).
O’Brien, E. J., Monk, J. M. & Palsson, B. O. Using genome-scale models to predict biological capabilities. Cell 161, 971–987 (2015).
Fang, X., Lloyd, C. J. & Palsson, B. O. Reconstructing organisms in silico: genome-scale models and their emerging applications. Nat. Rev. Microbiol. 18, 731–743 (2020).
Lewis, N. E., Nagarajan, H. & Palsson, B. O. Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 10, 291–305 (2012).
Lerman, J. A. et al. In silico method for modelling metabolism and gene product expression at genome scale. Nat. Commun. 3, 929 (2012).
Bordbar, A., Monk, J. M., King, Z. A. & Palsson, B. O. Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 15, 107–120 (2014).
Sánchez, B. J. et al. Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints. Mol. Syst. Biol. 13, 935 (2017).
Salvy, P. & Hatzimanikatis, V. The ETFL formulation allows multi-omics integration in thermodynamics-compliant metabolism and expression models. Nat. Commun. 11, 30 (2020).
Beard, D. A., Liang, S. & Qian, H. Energy balance for analysis of complex metabolic networks. Biophys. J. 83, 79–86 (2002).
Kümmel, A., Panke, S. & Heinemann, M. Putative regulatory sites unraveled by network-embedded thermodynamic analysis of metabolome data. Mol. Syst. Biol. 2, 2006.0034 (2006).
Henry, C. S., Broadbelt, L. J. & Hatzimanikatis, V. Thermodynamics-based metabolic flux analysis. Biophys. J. 92, 1792–1805 (2007).
Oftadeh, O. et al. A genome-scale metabolic model of saccharomyces cerevisiae that integrates expression constraints and reaction thermodynamics. Nat. Commun. 12, 4790 (2021).
Miskovic, L., Tokic, M., Fengos, G. & Hatzimanikatis, V. Rites of passage: requirements and standards for building kinetic models of metabolic phenotypes. Curr. Opin. Biotechnol. 36, 146–153 (2015).
Saa, P. A. & Nielsen, L. K. Formulation, construction and analysis of kinetic models of metabolism: a review of modelling frameworks. Biotechnol. Adv. 35, 981–1003 (2017).
Munger, J. et al. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 26, 1179–1186 (2008).
DeBerardinis, R. J. & Chandel, N. S. Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200 (2016).
DeBerardinis, R. J. & Keshari, K. R. Metabolic analysis as a driver for discovery, diagnosis, and therapy. Cell 185, 2678–2689 (2022).
Masri, S. & Sassone-Corsi, P. The emerging link between cancer, metabolism, and circadian rhythms. Nat. Med. 24, 1795–1803 (2018).
Cascante, M. et al. Metabolic control analysis in drug discovery and disease. Nat. Biotechnol. 20, 243–249 (2002).
Na, D. et al. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat. Biotechnol. 31, 170–174 (2013).
Gupta, A., Reizman, I. M. B., Reisch, C. R. & Prather, K. L. J. Dynamic regulation of metabolic flux in engineered bacteria using a pathway-independent quorum-sensing circuit. Nat. Biotechnol. 35, 273–279 (2017).
Guijas, C., Montenegro-Burke, J. R., Warth, B., Spilker, M. E. & Siuzdak, G. Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat. Biotechnol. 36, 316–320 (2018).
Khodayari, A. & Maranas, C. D. A genome-scale Escherichia coli kinetic metabolic model k-Ecoli457 satisfying flux data for multiple mutant strains. Nat. Commun. 7, 13806 (2016).
Foster, C. J., Gopalakrishnan, S., Antoniewicz, M. R. & Maranas, C. D. From Escherichia coli mutant 13C labeling data to a core kinetic model: a kinetic model parameterization pipeline. PLoS Comput. Biol. 15, e1007319 (2019).
Hameri, T., Fengos, G., Ataman, M., Miskovic, L. & Hatzimanikatis, V. Kinetic models of metabolism that consider alternative steady-state solutions of intracellular fluxes and concentrations. Metab. Eng. 52, 29–41 (2019).
Gopalakrishnan, S., Dash, S. & Maranas, C. K-FIT: an accelerated kinetic parameterization algorithm using steady-state fluxomic data. Metab. Eng. 61, 197–205 (2020).
St John, P. C., Strutz, J., Broadbelt, L. J., Tyo, K. E. J. & Bomble, Y. J. Bayesian inference of metabolic kinetics from genome-scale multiomics data. PLoS Comput. Biol. 15, e1007424 (2019).
Haiman, Z. B., Zielinski, D. C., Koike, Y., Yurkovich, J. T. & Palsson, B. O. MASSpy: building, simulating, and visualizing dynamic biological models in python using mass action kinetics. PLoS Comput. Biol. 17, e1008208 (2021).
Bordbar, A. et al. Personalized whole-cell kinetic models of metabolism for discovery in genomics and pharmacodynamics. Cell Syst. 1, 283–292 (2015).
Andreozzi, S., Miskovic, L. & Hatzimanikatis, V. iSCHRUNK—in silico approach to characterization and reduction of uncertainty in the kinetic models of genome-scale metabolic networks. Metab. Eng. 33, 158–168 (2016).
Miskovic, L., Beal, J., Moret, M. & Hatzimanikatis, V. Uncertainty reduction in biochemical kinetic models: enforcing desired model properties. PLoS Comput. Biol. 15, e1007242 (2019).
Choudhury, S. et al. Reconstructing kinetic models for dynamical studies of metabolism using generative adversarial networks. Nat. Mach. Intell. 4, 710–719 (2022).
Koutník, J., Cuccu, G., Schmidhuber, J. & Gomez, F. Evolving large-scale neural networks for vision-based reinforcement learning. In Proc. of the 15th annual conference on Genetic and evolutionary computation 1061–1068 (ACM, 2013).
Salimans, T., Ho, J., Chen, X., Sidor, S. & Sutskever, I. Evolution strategies as a scalable alternative to reinforcement learning. Preprint at http://arxiv.org/abs/1703.03864 (2017).
Shamir, M., Bar-On, Y., Phillips, R. & Milo, R. SnapShot: timescales in cell biology. Cell 164, 1302.e1 (2016).
Narayanan, B., Weilandt, D., Masid, M., Miskovic, L. & Hatzimanikatis, V. Rational strain design with minimal phenotype perturbation. Nat. Commun. 15, 723 (2024).
Balderas-Hernández, V. E. et al. Metabolic engineering for improving anthranilate synthesis from glucose in Escherichia coli. Microb. Cell Fact. 8, 19 (2009).
Salvy, P. et al. pyTFA and matTFA: a Python package and a Matlab toolbox for thermodynamics-based flux analysis. Bioinformatics 35, 167–169 (2019).
Stelling, J., Sauer, U., Szallasi, Z., Doyle, F. J. & Doyle, J. Robustness of cellular functions. Cell 118, 675–685 (2004).
Mavrovouniotis, M. L. Group contributions for estimating standard gibbs energies of formation of biochemical compounds in aqueous solution. Biotechnol. Bioeng. 36, 1070–1082 (1990).
Jankowski, M. D., Henry, C. S., Broadbelt, L. J. & Hatzimanikatis, V. Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys. J. 95, 1487–1499 (2008).
Park, J. O. et al. Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat. Chem. Biol. 12, 482–489 (2016).
Dunteman, G. Principal Components Analysis (Sage, 1989).
van der Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
van der Maaten, L. Learning a parametric embedding by preserving local structure. In Proc. Twelth International Conference on Artificial Intelligence and Statistics 384–391 (PMLR, 2009).
Chang, A. et al. BRENDA, the ELIXIR core data resource in 2021: new developments and updates. Nucleic Acids Res. 49, D498–D508 (2021).
Ezgu, F. Inborn errors of metabolism. Adv. Clin. Chem. 73, 195–250 (2016).
Tokic, M., Hatzimanikatis, V. & Miskovic, L. Large-scale kinetic metabolic models of Pseudomonas putida KT2440 for consistent design of metabolic engineering strategies. Biotechnol. Biofuels 13, 33 (2020).
Goodfellow, I. et al. Generative adversarial networks. Commun. ACM 63, 139–144 (2020).
Wang, L., Birol, I. & Hatzimanikatis, V. Metabolic control analysis under uncertainty: framework development and case studies. Biophys. J. 87, 3750–3763 (2004).
Miskovic, L. & Hatzimanikatis, V. Production of biofuels and biochemicals: in need of an ORACLE. Trends Biotechnol. 28, 391–397 (2010).
Andreozzi, S. et al. Identification of metabolic engineering targets for the enhancement of 1,4-butanediol production in recombinant E. coli using large-scale kinetic models. Metab. Eng. 35, 148–159 (2016).
Miskovic, L. et al. A design–build–test cycle using modeling and experiments reveals interdependencies between upper glycolysis and xylose uptake in recombinant S. cerevisiae and improves predictive capabilities of large-scale kinetic models. Biotechnol. Biofuels 10, 166 (2017).
Mišković, L. & Hatzimanikatis, V. Modeling of uncertainties in biochemical reactions. Biotechnol. Bioeng. 108, 413–423 (2011).
Wittig, U., Rey, M., Weidemann, A., Kania, R. & Müller, W. SABIO-RK: an updated resource for manually curated biochemical reaction kinetics. Nucleic Acids Res. 46, D656–D660 (2018).
Li, F. et al. Deep learning-based kcat prediction enables improved enzyme-constrained model reconstruction. Nat. Catal. 5, 662–672 (2022).
Heckmann, D. et al. Machine learning applied to enzyme turnover numbers reveals protein structural correlates and improves metabolic models. Nat. Commun. 9, 5252 (2018).
Boorla, V. S., Upadhyay, V. & Maranas, C. D. ML helps predict enzyme turnover rates. Nat. Catal. 5, 655–657 (2022).
Ataman, M., Hernandez Gardiol, D. F., Fengos, G. & Hatzimanikatis, V. redGEM: systematic reduction and analysis of genome-scale metabolic reconstructions for development of consistent core metabolic models. PLoS Comput. Biol. 13, e1005444 (2017).
Ataman, M. & Hatzimanikatis, V. lumpGEM: systematic generation of subnetworks and elementally balanced lumped reactions for the biosynthesis of target metabolites. PLoS Comput. Biol. 13, e1005513 (2017).
Pandey, V., Hadadi, N. & Hatzimanikatis, V. Enhanced flux prediction by integrating relative expression and relative metabolite abundance into thermodynamically consistent metabolic models. PLoS Comput. Biol. 15, e1007036 (2019).
Pandey, V.; Gardiol, D. H.; Chiappino-Pepe, A.; Hatzimanikatis, V. TEX-FBA: A constraint-based method for integrating gene expression, thermodynamics, and metabolomics data into genome-scale metabolic models. Preprint at bioRxiv https://doi.org/10.1101/536235 (2019).
Machado, D. & Herrgård, M. Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of metabolism. PLoS Comput. Biol. 10, e1003580 (2014).
Astrom, K. J. & Murray, R. M. Feedback Systems: An Introduction for Scientists and Engineers (Princeton Univ. Press, 2008).
Strogatz, S. Nonlinear Dynamics and Chaos: With Applications to Physics, Biology, Chemistry, and Engineering 2nd Edn (Westview Press, 2015).
Kingma, D. P. & Welling, M. An introduction to variational autoencoders. Found. Trends Mach. Learn. 12, 307–392 (2019).
Abadi, M. TensorFlow: learning functions at scale. ACM https://doi.org/10.1145/2951913.2976746 (2016).
Choudhury, S. et al. Supporting data for generative machine learning produces kinetic models that accurately characterize intracellular metabolic states. Zenodo https://doi.org/10.5281/zenodo.7628650 (2023).
Acknowledgements
This work was supported by funding from the Swiss National Science Foundation grant 200021_188623 (S.C., V.H. and L.M.), the European Union’s Horizon 2020 research and innovation programme under grant agreement 814408 (S.C. and B.N.), Swedish Research Council Vetenskapsradet grant 2016-06160 (S.C. and B.N.) and the Ecole Polytechnique Fédérale de Lausanne (EPFL) (M.M., V.H. and L.M.).
Author information
Authors and Affiliations
Contributions
S.C., M.M. and L.M. designed the overall method and approach. V.H. and L.M. supervised the research. S.C. and L.M. developed the RENAISSANCE method. S.C., B.N. and M.M. designed the code. S.C., B.N. and L.M. analysed the data. S.C. and L.M. wrote the manuscript. All authors read and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Catalysis thanks Xiaozhou Luo, Ruibo Wu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Notes 1–6, Figs. 1–16, Tables 1–3 and References.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Choudhury, S., Narayanan, B., Moret, M. et al. Generative machine learning produces kinetic models that accurately characterize intracellular metabolic states. Nat Catal (2024). https://doi.org/10.1038/s41929-024-01220-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41929-024-01220-6
- Springer Nature Limited