
Abstract
Glioblastoma (GBM) is the most aggressive glial tumor of the adult brain, associated with invariably fatal outcome, and a deeper understanding of the underlying malignant mechanisms is necessary to address the current therapeutic failure. We previously demonstrated the role of the CXCL12/CXCR4 axis in GBM cell migration and resistance to ionizing radiation. The atypical chemokine receptor ACKR3, responsible for CXCL12 scavenging, was previously suggested as additional important player in the context of GBM. Following validation of the detection tools, we observed that ACKR3 is expressed within GBM patient tumor tissue, distributed in diverse cell types. In contrast to CXCR4, ACKR3 expression in patient-derived stem-like cells (GSCs) remains however low, while ACKR3 gene expression by tumor cells appears to be modulated by the in-vivo environment. Using overexpression models, we also showed that in vitro ACKR3 had no significant direct effect on cell proliferation or invasion. Altogether, these results suggest that in vitro ACKR3 plays a minor role in malignant GBM cell biology and that its expression is possibly regulated by in-vivo influences. The subtle and multifaceted functions ACKR3 could exert in GBM should therefore only be tackled within a comprehensive tumor microenvironment considering tumoral but also non-tumoral cells.
Similar content being viewed by others


Introduction
Gliomas are glial primary tumors of the central nervous system (CNS) which are classified by the World Health Organization (WHO) based on their specific histological characteristics and molecular features1. According to this classification, glioblastoma (GBM) is identified as the most common and aggressive grade 4 glioma and is distinguished by a set of genetic alterations1,2. These include the absence of the isocitrate dehydrogenase (IDH) mutation, designating GBM as an IDH wild-type (IDH WT) tumor, the presence of mutations in the telomerase reverse transcriptase (TERT) promoter and copy number alterations in chromosome 7 and 10 (+ 7/-10), most often associated with epithelial growth factor receptor (EGFR) amplification1,2. The standard-of-care therapy associating maximal safe surgery and concomitant radio-chemotherapy using temozolomide (TMZ) allows a median survival of about 16 months from diagnosis3. Moreover, the progression of the disease is characterized by a systematic recurrence that relies on (1) GBM cell infiltration through the brain tissue, hindering total resection of the tumor4, and (2) an extreme heterogeneity5 of GBM cells that transit through diverse functional states6,7,8, which overall lead to therapeutic resistance.
Chemokines constitute a subgroup of chemotactic cytokines secreted by various cell types in different tissues and playing an important role in inducing and guiding cell migration9. They are important regulators of various processes such as development, immune responses and tissue repair9. Chemokine receptors, which are G protein-coupled receptors, have been widely studied for their role in cancer development and metastasis10,11. Besides malignant cells, chemokines and their receptors are also expressed by a wide range of cell subtypes found within the tumor bed including tumor-associated macrophages, tumor infiltrating-lymphocytes, vascular cells, non-malignant glial cells and neurons, and play diverse roles in GBM growth, angiogenesis and resistance to treatment12.
We have previously demonstrated the function of CXCR4, the receptor for the CXCL12 chemokine, in the migration of GBM cells towards the subventricular zone (SVZ) in orthotopic xenografts models, as well as in the CXCL12-mediated protection from radiation therapy13,14. ACKR3 (formerly named CXCR7), a second receptor for CXCL12, expressed in diverse cell types, including leukocytes, neurons or endothelial cells was identified15,16. ACKR3 was subsequently demonstrated to play a crucial role in regulating CXCL12-dependent processes including cardiovascular and neuronal development as well as in the migration and homing of hematopoietic stem/progenitor cells17,18,19,20. Unlike CXCR4 which signals via G protein pathways to induce cell migration and proliferation, ACKR3 activity relies mainly on β-arrestin recruitment and its ability to signal through G protein-independent pathways is still controversial. ACKR3 is proposed to act as a scavenger or “sink” receptor for CXCL12, shaping its gradient and regulating its availability, thereby controlling the directional migration and homing of CXCR4-expressing cells21,22,23,24,25. So far, the exact role and expression of ACKR3 in GBM and its crosstalk with CXCR4 through their shared ligand, CXCL12, remain to be elucidated.
ACKR3 can also heterodimerize with CXCR4 and modify its signaling properties26. Several years ago, different studies reported high expression of ACKR3 in brain tumor cell lines and tissue samples, which correlated with bad prognosis and increased aggressiveness in preclinical models23. ACKR3 was also shown to be expressed on cells delimiting the SVZ in the mouse brain27,28, which suggests an important role ACKR3 could play in glioma29. On that basis, we decided to investigate the precise function ACKR3 could exert in GBM cells, in a potential interplay with CXCL12 and CXCR4, using patient-derived models. Recently, based on large-scale patient-based transcriptomic data, we have shown that ACKR3 is one of the most abundant chemokine receptor-encoding genes in glioma tissue12. However, how these results are correlated to detection of the corresponding protein ex vivo and in various patient-derived cell cultures remain elusive.
In this study, we assessed the expression of ACKR3 protein expression in patient-derived GBM tissue and GBM stem-like cells, in different in vitro and ex vivo settings, with the ultimate aim to better understand its function and impact on GBM and to guide novel ACKR3-targeting therapeutic strategies.
Results
Validation of monoclonal antibodies for the detection of ACKR3 at the cell membrane
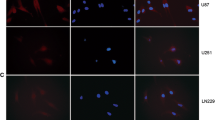
Detecting ACKR3 at the protein level is a widely acknowledged challenge in the field. While genetic models (e.g. reporter genes) can be employed for this purpose12,21, the crucial requirement remains the recognition under native conditions, especially in order to elucidate ACKR3 role and relevance in cancer. In this study, we first aimed to assess the efficacy of two ACKR3-specific monoclonal antibodies: the 8F11-M16 antibody and the 11G8 antibody, commonly employed in our experiments for ACKR3 detection using flow cytometry and immunofluorescence, respectively. Results indicate that both antibodies yield a strong signal in U87 stably expressing ACKR3 (Fig. 1A). No signal was observed in U87 and U87 CXCR4 cells, showing a good specificity of both antibodies for ACKR3 (Fig. 1A). Additionally, we provide experimental evidence that, among several antibodies that fail to specifically detect the receptor, the 11G8 antibody reliably identifies ACKR3 in immunostaining and immunoblotting experiments using overexpression GBM cell models, as well as in MCF-7 breast cancer cells, which were previously described to endogenously express ACKR323,24. (Fig. 1B-C). In contrast, diverse other commercially available antibodies provided signals that appeared similar in both U87 and U87 ACKR3. Based on these results, we consider that both and 8F11-M16 and 11G8 antibodies are reliable tools for ACKR3 detection, using flow cytometry and immunostaining experiments.

ACKR3 tool validation using flow cytometry. (A) Cell surface ACKR3 expression in U87, U87 ACKR3 and U87 CXCR4 cell lines by using 8F11-M16 and 11G8 antibodies. (B) Immunofluorescent staining of ACKR3 (green) in U87 and U87 ACKR3 cells using 11G8 MAB42273 antibody against ACKR3. DAPI (blue) was used to counterstain nuclei. Scale bar = 50 μm. (C) Western Blot (WB) analysis of ACKR3 in protein extracts from U87, U87 ACKR3, T033-mRFP, T033-hACKR3 and MCF-7 cells, using different commercially available antibodies (11G8/MAB42273, ab72100, ACR-037 and 7TM0080N). Original blots/gels are presented in Supplementary Figure S3.
ACKR3 exhibits diverse expression patterns in GBM tissue
Over the last years, different studies reported ACKR3 expression in numerous cancer cell lines and tissue23. Based on data from The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) databases (via the GEPIA platform), ACKR3 expression appears higher in low-grade gliomas (LGGs) and GBM samples compared to normal brain (Fig. 2A), which speaks for a potential role of ACKR3 in malignant cells. In line with this, we performed immunofluorescence staining to detect ACKR3 at the protein level in patient-derived FFPE tissues. We observed that ACKR3 is expressed in both glioma tissue and non-tumoral brain tissue with high inter- and intra-tumoral heterogeneity. We identified three different expression patterns of ACKR3 expression (named here after type 1, 2 and 3). In most GBM samples, ACKR3 appeared present around cell nuclei (“type 1”). Similar pattern was found in non-GBM brain samples, such as the hippocampus (HIPPO) or the subventricular zone (SVZ). In “type 2”, ACKR3 positive regions displayed blood vessel-like shapes and were found in GBM tissue, as well as in the non-tumoral SVZ, in line with previous reports showing the expression of ACKR3 on tumor-associated vasculature30. Finally, in several samples, ACKR3 was detectable in the whole tissue, without apparent specificity to any cell type or structure (“type 3”). No ACKR3 expression was found in non-tumoral cortical (CORTEX) tissue samples (Fig. 2B-C).

ACKR3 is expressed in different patterns in human glioma tissue and non-tumor brain tissue. (A) In blue, ACKR3 expression in GBM, Low Grade Glioma (LGG), Colon Adenocarcinoma (COAD), Acute Myeloid Leukemia (LAML), Skin Cutaneous Melanoma (SKCM) and Lung adenocarcinoma (LUAD) (TCGA data) vs. in grey, non-tumoral samples (GTEx data, using GEPIA tool in TCGA database). (B) Immunofluorescent staining of ACKR3 (green) in glioma patient FFPE tissue. White arrow indicates ACKR3 expression patterns. (C) ACKR3 immunofluorescent staining (green) in various regions of non-tumoral FFRE brain tissue: subventricular zone (SVZ), hippocampus (HIPPO), and cortex. of non-tumoral FFPE brain tissue. DAPI (blue) was used to counterstain nuclei. Scale bar = 10 μm.
Different co-staining experiments have been performed to analyze the cell types that appeared positive for the receptor. ACKR3 was expressed in SOX2 + cells (“stem-like”) and in GFAP + cells (astrocytes), with “type 1” features. ACKR3 did not colocalize with PDGFRβ (pericytes) or Iba1 (microglia) markers. Importantly, EGFR + cells in tumor tissue were rarely ACKR3+ (Fig. 3A-B). Blood vessel-like (“type 2”) CXCR4 + cells also appeared ACKR3+ (Fig. 3B). Altogether, these results show that ACKR3 expression within GBM tissue is rather heterogeneous, and it can be distributed among different cell types, which mostly correspond to SOX2, GFAP, and CXCR4-expressing cells. This is in line with single-cell RNAseq data showing that ACKR3 is expressed, although at a low level, in cells assigned to various categories (Fig. 3C).

Characterization of ACKR3-positive cells in glioblastoma tissue. (A–B) Immunofluorescent staining of ACKR3 (green) and cell type specific markers, including SOX2, GFAP, Iba1, EGFR, PDGFRb and CXCR4 (red). DAPI (blue) was used to counterstain nuclei. Scale bar = 10 μm. (C) ACKR3 expression in various cell types within patient glioma samples. Single cell RNAseq data from Neftel 2019 dataset8 show ACKR3 expression in different cell types. The “% expressing” value indicates the proportion of cells in the signature that are positive for a given transcript, and the “scaled mean expression” is relative to each gene expression level (logTPM) across all cells within the signature (using Single Cell Portal online platform).
Patient-derived GBM stem-like cell cultures express low levels of ACKR3 at the membrane
Fresh tumor tissue obtained after surgical resection was processed in order to obtain patient-derived GBM stem-like cells (GSCs) cultured as floating, 3D tumorospheres (Fig. 4A). The expression of ACKR3 was assessed at mRNA level by qRT-PCR as well as at the cell membrane by flow cytometry. ACKR3 mRNA was detected in patient-derived GSCs (Fig. 4B), although weakly expressed, e.g. compared to the SOX2 gene (Figure S1A). Using flow cytometry, we aimed at detecting ACKR3 at the cell surface of GSCs. The signal intensity for ACKR3 appeared rather close to the negative control values (Fig. 4C). The percentage of ACKR3-positive cells in each GSC culture was as follows: T08 = 1.22±0.76%; T013 = 3.91±1.78%; T018 = 2.90±0.66%; T033 = 0.78±0.18% (vs. U87 ACKR3 = 81.40±2.31%) (Fig. 4D). We selected T018 and T033 GSCs to assess whether ACKR3 expression at the cell membrane could be upregulated upon stimulation with the ACKR3-binding chemokines CXCL12 and CXCL11. T018 and T033 cells were incubated for 24–48 h with different concentrations of chemokines and flow cytometry analysis was performed to detect ACKR3 as well as CXCR4, which also binds CXCL12 but not CXCL11. When T018 cells were stimulated for 24 h with 10 nM of CXCL12, we discovered a statistically significant decrease of the percentage of CXCR4-positive cells (82.6±4.8%), compared to untreated cells (92.4±2.2%), which suggested CXCL12-mediated CXCR4 downregulation (N = 4; p = 0.03) (Fig. 4E). No differences of CXCR4 expression were observed when T018 cells were stimulated with a lower concentration of CXCL12 (2.5 nM) or with CXCL11 (10 nM) (Fig. 4E). After 48 h of stimulation, we observed again a reduced expression of the CXCR4 receptor at the membrane (Fig. 4F). Importantly, no difference was observed for ACKR3 expression upon stimulation with CXCL12 or CXCL11, for 24–48 h (Fig. 4E–F). Whereas reduction in CXCR4-positive cells was suggested in T033 cells as well after 24 h of stimulation by CXCL12, no significant differences were recorded. Again, the expression of ACKR3 remained very low across all conditions (Figure S1B–C). We observed a strong signal for ACKR3 detection in U87 genetically modified for expressing stably ACKR3 (positive control), but also in MCF-7 breast cancer cells, considered as positive control for endogenous ACKR3 expression23,24. Then, we tested a 5-minute incubation of CXCL12, which revealed a significant decrease in ACKR3 signal, revealing receptor internalization in the MCF-7 cells endogenously expressing ACKR3 (N = 4 or 5; p = 0.014), but the signal in GSCs remained close to zero due to scarce surface expression of ACKR3 (Figure S1D). In conclusion, these results indicate that ACKR3 is not detected at the membrane of patient-derived GSCs, irrespective of chemokine presence in the culture medium. To make sure ACKR3 is not localized exclusively inside the cells and therefore not detected by surface staining, we used immunofluorescence and high-magnification microscopy. This revealed a very mild ACKR3 staining, without specific colocalization with Rab5, ruling out a restricted localization of ACKR3 in endosomes in GSCs (Figure S1E).

ACKR3 surface expression in patient-derived GSCs is low and remains unchanged upon CXCL11 or CXCL12 stimulation. (A) Phase-contrast images of patient-derived GSCs (T08, T013, T018 and T033) cultured as 3D tumorospheres (scale bar = 100 μm). (B) ACKR3 mRNA expression relative to GAPDH mRNA expression in patient-derived GSCs and U87 cells (U87 overexpressing ACKR3 were used as positive control) (n = 4). (C) Fluorescence intensity of ACKR3 membrane staining in patient-derived GSCs and U87 cells using flow cytometry (U87 overexpressing ACKR3 were used as positive control) (n = 4). (D) Percentage of ACKR3 positive cells in patient-derived GSCs using flow cytometry. (E–F) ACKR3 and CXCR4 fluorescence intensity (and % of positive T018 GSCs) after CXCL11 or CXCL12 stimulation for 24–48 h (n = 4). Statistical significance was determined by one-way ANOVA (∗ p < 0.05).
ACKR3 overexpression does not directly influence GBM cell proliferation and invasion
To investigate the role ACKR3 could play in GBM cells that initially express low levels of this receptor, we took advantages of U87 cells and U87 ACKR3 cells and carried out various assays to determine whether the expression of ACKR3 in GBM cells influenced cell proliferation (Fig. 5A). Cell counting revealed no changes in proliferation between U87 ACKR3 and U87 parental cells. CFSE labeling experiments confirmed these results (Fig. 5B–C). We further modified patient-derived GSC T033 via lentiviral transduction to overexpress the ACKR3 receptor (Fig. 5D). Again, no difference in cell proliferation was observed in T033 hACKR3 cells compared to T033 mRFP in the presence or absence of CXCL12 (Fig. 5E–F). Next, we aimed at comparing T033 mRFP and T033 hACKR3 tumorigenicity in vivo. Cells were engrafted in the right striatum of immunodeficient nude mice. After 10 weeks, we observed that both cell types developed large, highly invasive tumors that spread all over the mouse brain. No obvious difference could be observed in T033 hACKR3 compared to mRFP, in terms of growth or invasiveness (Fig. 5G). We also checked in vitro whether ACKR3 expression modified the invasive properties of U87 cells using Boyden chamber assays. The invasive capacities of U87 ACKR3 cells were not modified compared to U87 wild-type cells, even upon stimulation with CXCL12 or CXCL11. Conversely, the presence of CXCL12 increased the invasiveness of U87 CXCR4 cells, which was not observed upon CXCL11 stimulation (p = 0,010) (Fig. 5G).

ACKR3 expression in U87 and T033 cells does not modify cell proliferation or invasion (A) ACKR3 mRNA expression relative to GAPDH in U87 vs. U87 ACKR3 cells (n = 4). (B) Proliferation assay on U87 vs. U87 ACKR3 cells. Cells were counted at day 0, 4 and 7. (n = 5) (C) CFSE assay (Cell Trace proliferation assay) on U87, U87 ACKR3 and U87 CXCR4 cells after 5 days with or without CXCL12 (10 nM) (n = 3) (D) ACKR3 mRNA expression relative to GAPDH in T033 mRFP vs. T033 hACKR3 cells. (E) Proliferation assay on T033 mRFP and T033 hACKR3 cells. Cells were counted at day 0, 4 and 7. (F) CellTrace Violet assay on T033 GSCs after 4 days with or without CXCL12 (10 nM). (G) T033 mRFP and T033 hACKR3 cells were engrafted in the right striatum of nude mice, sacrificed 10 weeks post engraftment, and detected using immunohistochemistry staining against human vimentin. (H) Transwell invasion assay on U87, U87 ACKR3 and U87 CXCR4 cells for 24 h with CXCL11 or CXCL12 (n = 3) (*p < 0.05; **p < 0.01).
ACKR3 protein expression is not detected in orthotopic tumors, but gene expression is upregulated ex vivo
In vivo, T033 GSCs induce the formation of a highly infiltrative tumors within a few weeks (Figure S2A). At 7 weeks post-graft, immunohistochemistry against human vimentin shows that T033 cells had invaded the whole right hemisphere and reached the contralateral hemisphere via white matter tracts (Figure S2B). We verified that the tumor core was enriched in CXCR4-positive cells, but ACKR3 expression was not detected (Figure S2C). In a second experiment, in vivo engrafted T033 GSCs expressing the RFP and luciferase genes (T033-RFP-Luc) were harvested from different brain regions at 6 weeks post-implantation (before clinical endpoint), dissociated and put back in culture. We were particularly interested in the impact of the subventricular zone on GBM cell behavior31. Interestingly, RFP-positive T033 cells were detected in the tissue harvested from the right SVZ (“SVZ in”) as well as in the right temporal cortex, away from the SVZ (“SVZ out”). We let RFP-positive T033 cells “SVZ in” and “SVZ out” form new tumorospheres in serum-free culture for seven days, then collected them to analyze their ACKR3 gene expression. Of note, only few RFP-positive cells could be harvested from the contralateral SVZ and cortex which did not regrow. Surprisingly, quantitative RT-PCR analysis showed a significant increase of ACKR3 expression of T033 GSCs ex vivo, especially from “SVZ in”, compared to T033 in vitro (Fig. 6D–E). Given that, endothelial cells present on the lateral wall of ventricle is described as a major source of CXCL1232, we stimulated T033 GSC with CXCL12 in vitro. However, no differences were observed in terms of ACKR3 gene expression when GSCs were stimulated with CXCL12 (Fig. 6F). These results suggest that ACKR3 gene expression is modulated by specific brain microenvironments which does not only rely on CXCL12.

(A) T033-RFP-LUC cells were engrafted in the right striatum of nude mice, sacrificed 42 days post-engraftment. Two different brain regions were isolated (“SVZ in” and “SVZ out”) and cells were cultured for 7 days before RT-qPCR analyses were performed. (B) Quantitative RT-qPCR analysis allowed to detect ACKR3 mRNA expression normalized to HKG (GAPDH, EF1a, HPRT) in T033 (in vitro), T033 “SVZ in” (in vivo) and T033 “SVZ out” (in vivo). Statistical significance was determined by one-way ANOVA (*** p < 0.001, **** p < 0.0001). (C) ACKR3 mRNA expression relative to Ezrin used as HKG in T033 GSCs vs. T033 GSCs stimulated with CXCL12 (10nM).
.
Discussion
The chemokine receptor ACKR3 has gained increased therapeutic interest in the last years, along with the in-depth elucidation of its atypical mechanism of action, implication in different physiopathological processes and the development of new pharmacological modulators33,34,35,36,37,38,39,40,41. Together with its CXCL12/CXCR4 signaling partners, it has been suggested as an important player in cancer, including in brain tumors23. Additionally, our recent transcriptomic analysis of large patient cohorts showed a high expression of ACKR3 in glioma tissues12, which several reports have previously associated with disease prognosis. ACKR3 has therefore been endowed with significant therapeutic potential.
In the 2010’s, several studies described a high ACKR3 expression in glioma patient tissue and in GBM cells in vitro, mostly using RT-PCR, immunostainings and immunoblots25,42,43,44. Regrettably, not only did many of these studies focus on cell lines in vitro but they also made use of polyclonal antibodies against ACKR3 (e.g. ab12780 and ab72100), that had previously been suggested as unspecific45, and which we confirm here as not reliable for ACKR3 detection. These studies are nowadays still cited46,47 to support the biological relevance of ACKR3 in gliomas, setting the ground for clinical trials48, and we wished to confront current knowledge with an updated evaluation of this receptor in GBM tissues and cells. After verifying that the 11G8 and 8F11-M16 monoclonal antibodies consistently allow ACKR3 detection, as previously suggested45, we analysed the expression of ACKR3 in glioblastoma patient-derived tissue samples and cell cultures, using proper controls of ACKR3 overexpression (U87 ACKR3) and endogenous expression (e.g. MCF-7 cells). We report in-situ stainings, paying particular attention to ACKR3 expression pattern and to the type of cells that express it. ACKR3 appears associated with blood vessels in tumor tissue (“Type 2”), and present in GFAP+/SOX2 + cells, however not present in EGFR + cells (“Type 1”). We must consider that GFAP+/SOX2 + cells may include intra-tumoral reactive astrocytes, which were previously described as ACKR3 + in different models of brain pathologies49. In line with our results, Walters et al. used the 11G8 antibody to show that ACKR3 is expressed in tumor cells as well as endothelial cells, in GBM patient tissue50. In a very similar manner, Birner et al.51 describe ACKR3-positive cells as “vascular ACKR3 cells (vasACKR3 + cells detected in 58.1% of 320 cases) and much rarer “tumor cell ACKR3” (tcACKR3 cells in 11.6% of 320 cases). Interestingly, they described the number of vasACKR3 + cells as a bad prognosis marker in IDH mutant gliomas, the number of tcACKR3 + cells having no prognosis value51. The role of ACKR3 + in endothelial cells within GBM tumors warrants further investigation. In that line, Salazar et al. developed a scFv-based chimeric antibody against ACKR3 that triggers NK-mediated toxicity against both tumor and endothelial cells52. Elaborating an ACKR3-targeted therapeutic strategy in cancer indeed requires to identify the cell types of interest, and it is now of significant importance to orient future research endeavors considering ACKR3 in non-malignant cells from the TME (e.g. vascular cells, glial cells, putatively immune cells).
The putative clinical relevance of ACKR3 in glioma has also emerged from RNAseq data pointing to its high expression. However, bulk GBM tissue data stem from a large mixture of cell types, including peripheral blood, and do not allow to delineate the full picture of ACKR3 function within a tumor. More recently, single-cell RNAseq data helped to shed light on gene expression across different cell subgroups12. Although such data indicate ACKR3 as expressed in malignant cells, we firmly demonstrated here that in spite of expressionist presence at the transcript level, ACKR3 protein is undetected in patient-derived GBM cells in vitro, under various conditions. It strongly suggests that mRNA amount may not always be a reliable indicator of the protein abundance. In a different tumor model, Antonello et al. show that ACKR3 surface expression is detected only in a fraction of B-cell lymphoma cells, yet both ACKR3 + and ACKR3- cells have similar levels of mRNA53. Such results can partially be explained by the preferred localization of ACKR3 in intracellular compartments, e.g. endosomes54,55. It has also been shown that ACKR3 surface expression in the same lymphoma cells is increased upon in-vivo “conditioning” in subcutaneous grafts and further ex-vivo isolation, without any modification in ACKR3 gene expression, again suggesting a particular trafficking of the receptor from intracellular compartments to the plasma membrane56. Here however, we do not detect the receptor inside patient-derived GBM cells using immunofluorescence or western-blotting on whole cell lysates. It remains puzzling to observe that in vivo “conditioning” in orthotopic brain xenografts strikingly increases ACKR3 expression in ex vivo isolated cells although the protein was not detected in tumor tissue.
All in all, whether ACKR3 plays a key role in GBM tumors is still a matter of debate. Here, we concluded that patient-derived GSCs, widely used as in vitro models of GBM tumors in mechanistic studies or drug testing, do not express detectable levels of ACKR3 protein. However, we do not rule out that ACKR3 expression may be tightly regulated upon variable conditions. Given the fine-tuned chemokine-dependent cell-cell interactions, we now consider imperative to study ACKR3 as well as other chemokine receptor function in an exhaustive, well vascularized, immunocompetent tumor microenvironment.
Materials and methods
Human tissues – All human GBM samples were obtained from residual tumor tissue after surgical resection, in collaboration with the Neurosurgery department of the Liège University Hospital (CHU), and the University Hospital Biobank (BHUL, Liège, Belgium), in accordance with relevant guidelines and legal regulations on human body material. None of the included patients opposed to the use of residual human body material, according to the legal regulations in Belgium. Fresh tissue was further (1) dissociated to establish patient-derived glioblastoma stem-like cell cultures (GSCs), (2) formalin-fixed and paraffin-embedded (FFPE) to be used for immunohistofluorescence, (3) flash-frozen in liquid nitrogen for molecular biology analyses. Non-tumoral brain tissue was obtained from the BHUL, from brain donations or residual tissue following epileptic foci resections. none of the included patients opposed to the use of residual human body material, according to the legal regulations in Belgium.
Cell culture – In-house patient-derived GSC cultures (T08, T013, T018 and T033) were established from resected adult GBM tumors, and cultured as 3D tumorospheres in Dulbecco’s modified Eagle’s medium and Nutrient Mixture F-12 (DMEM/F12 with GlutaMAX, Gibco) supplemented with 2% of B27 without vitamin A (Gibco), 100 U/mL penicillin and 100 µg/mL streptomycin (ThermoFisher Scientific), 1 µg/mL of heparin (LEO pharma), 20 ng/mL of human EGF (Peprotech) and 20 ng/mL of bFGF (Peprotech). GCS cultures informations are listed in Table S1. T033 cells were stably transduced with a lentiviral vector LV-CMV-hACKR3 or LV-CMV-mRFP for the control vector. For in vivo monitoring, T033 were stably transduced with a lentiviral vector pLV-IRES-Luciferase-mRFP. Plasmid design and related experiments were carried out with the help of the GIGA Viral Vectors platform. The U87 human GBM cell line was obtained from ATCC. U87 cells were transfected with pIRES-puro-ACKR3-WT to overexpress the human ACKR3 or with pIRES-puro-CXCR4-WT to overexpress the human CXCR4, and further selected using puromycin (1 µg/mL and 0.5 µg/mL respectively). The MCF-7 human breast cancer cell line was obtained from ATCC. These cell lines were all cultivated as adherent cell monolayers in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS, Invitrogen), 100 U/mL penicillin and 100 µg/mL streptomycin (ThermoFisher Scientific). Mycoplasma tests were performed on a regular basis.
Immunofluorescent stainings - FFPE human brain tissue sections were heated at 60 °C, dewaxed with 100% xylene (2 × 20 min), and rehydrated through a set of alcohol baths: 100% ethanol (2 × 5 min), 95% ethanol, 80% ethanol, 75% ethanol and water (1 × 2 min each). An antigen retrieval step was performed using Tris–EDTA buffer (10 mM Tris-base, 1 mM EDTA solution, 0.05% Tween 20, pH 9.0). Slices were heated in a pressure cooker for 3 min then were left to cool down at room temperature. Slices were permeabilized with PBS + 0,2% Triton-X100 for 10 min, incubated 30 s with TrueBlack Lipofuscin Autofluorescence Quencher (Biotium), blocked with PBS + 10% normal donkey serum, then incubated with antibodies overnight at 4 °C. ACKR3 (R&D systems, #MAB42273, clone 11G8) was co-stained with SOX2 (Cell Signaling Technology, #3579), PDGFRβ (R&D systems, #AF385), Iba1 (Abcam, #ab5076), GFAP (Abcam, #ab4674), EGFR (Cell Signaling Technology, #4267), Rab5 (Cell Signaling Technology, #3547) and CXCR4 (Abcam, #ab124824). After washing steps, slides were incubated for 1 h with conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) and nuclei were counterstained with DAPI (Sigma). Cells coated on coverslips were fixed in 4% PFA for 10 min at room temperature, permeabilized with PBS + 0.1% Triton-X100, incubated with primary antibodies overnight at 4 °C, then with fluorescently labelled secondary antibodies (Jackson ImmunoResearch Laboratories) for 1 h at 4 °C. Image acquisition was performed with an epifluorescence microscope (Zeiss Apotome) and analyzed on ZENlite and ImageJ 2 (Fiji) softwares. Antibodies are listed in Table S2.
Flow cytometry - Cells were collected by centrifugation at 300 g for 5 minutes. The supernatant was removed, and cells were washed with PBS. Cells were dissociated with 1 mL Accutase (StemCell Technologies) and incubated at 37 °C for 5 minutes. Twice the volume of flow buffer (PBS with 1% BSA, 1 mM EDTA, 0.1% Azide) were added to quench Accutase action. Cells were collected by centrifugation at 300 g for 5 minutes. Cell pellet was resuspended in 1 mL of flow buffer and were counted to prepare 3 × 105 cells in 100 µL. APC-conjugated ACKR3 antibody (Biolegend, #331114, clone 8F11-M16) or mouse-anti ACKR3 (R&D systems, #42273, clone 11G8) were added and incubated for 1 h at 4 °C, in the dark. Cells were washed three times by adding 1 mL of flow buffer. A centrifugation step at 300 g for 4 min at 4 °C was performed. After the third wash, supernatant was removed, and cells were resuspended in flow buffer to a final volume of 300 µL. Samples were recorded on a flow cytometer Canto II (BD Biosciences). Percentage of positive cells as well as Mean Fluorescence Intensity values (ΔMFI = MFI of stained cells – MFI of unstained cells) were analyzed using FlowJo 10 software.
CXCL11 and CXCL12 stimulation - Cells were collected by centrifugation at 300 g for 5 minutes. The supernatant was removed, and cells were washed with PBS. Cells were dissociated with 1 mL Accutase (StemCell Technologies) and incubated at 37 °C for 5 minutes. 3 × 105 cells were plated in a 6-well plate with 1 mL of medium and 2.5 nM or 10 nM human CXCL12 (Peprotech) or 10 nM of human CXCL11 (Peprotech) were added for 24 to 48 h. Flow cytometry analysis was performed using FlowJo 10 software as described above.
Quantitative RT-PCR – Total RNA was isolated using the RNA isolation Nucleospin kit (Macherey-Nagel). RNA was reverse transcribed by using ProtoScript II First Strand cDNA Synthesis Kit (New England Biolabs) with random primers mix. For qRT-PCR reaction samples, a mix of a total volume of 5 µL was prepared. qRT-PCR Mix contained 2 µL of the diluted cDNA (10 ng per reaction), 2.5 µL of SYBRGreen (Eurogentec) and 0.25 µL of each primer (reverse and forward). Quantitative RT-PCR was performed using LightCycler 480 Roche. Primer sequences are listed in Table S2.
Cell counting – 1 × 105 cells were seeded in 1 mL of medium and incubated at 37 °C for 4 and 7 days. Then, cells were dissociated with trypsin-EDTA (Gibco) and counted with trypan blue staining by using an automatic cell counter (Countess™ II Automated Cell Counter, Thermo Fisher Scientific).
Cell Trace/CFSE – GSCs were stained with CellTrace Violet Stain (Invitrogen, 5 µM /106 cells) and incubated for 15 min at 37 °C in the dark. Medium was added to quench the excess of dye and centrifuged at 300 g for 3 min. Supernatant was removed and cells were resuspended in culture medium and incubated for 30 min at 37 °C. Then, a cell suspension of 3 × 105 cells was prepared for each condition. The CXCL12 condition was treated with 10 nM human recombinant CXCL12. Cells were incubated for 4 days at 37 °C in the dark. Before flow cytometry analysis, cells were stained with 100 µL of a 1/1000 Zombie NIR fixable viability dye (SONY) and incubated for 15 min at RT. Cells were analyzed with FACS Canto II (Becton Dickinson).
U87 and U87 ACKR3 cells were stained with CellTrace CFSE Cell Proliferation kit (Invitrogen, 0.25 µM/2.106 cells) for 15 min at 37 °C in the dark. The excess of free dye was quenched with medium during 5 minutes at 37 °C in the dark. The cells were washed with complete medium and incubated for additional 10 min at 37 °C to allow the reagent to undergo acetate hydrolysis. Then, part of the cells (2.5 × 105 cells per condition) were washed with PBS and stained with Zombie NIR fixable viability dye (Biolegend, dilution 1/3000) during 30 min at 4 °C. The cells were again washed with PBS and analyzed by FACS using a NovoCyte Quanteon Flow Cytometer (Agilent) (condition day 0). The remaining cells (7.5 × 105 cells per condition) were seeded in 6 cm culture dishes either in complete medium (untreated control) or in complete medium supplemented with 10 nM of CXCL12. The cells were incubated for 5 days at 37 °C in the dark. After 5 days, the cells were detached using Versene (Gibco), washed with PBS and stained with Zombie NIR fixable viability dye (Biolegend, dilution 1/3000) during 30 min at 4 °C. The cells were washed with PBS and analyzed by FACS using a NovoCyte Quanteon Flow Cytometer (Agilent). Flow cytometry analysis was performed using FlowJo 10 software as described above.
Invasion assay – For in vitro invasion assays, transwell chambers (Boyden chambers with 8 μm pore diameter, Thincert, Greiner) were coated with a 1:1 mixture of 0.05 mg/mL collagen type I (Gibco) and 0.5 mg/mL protein of ECM gel (Sigma-Aldrich) in 1:1 PBS-DMEM for 2 h at 37 °C. U87, U87 ACKR3 and U87 CXCR4 cells were detached using Versene (Gibco), washed with PBS and seeded in the upper compartment of the previously coated inserts (3 × 104 cells) in DMEM supplemented with 100 U/mL penicillin and 100 µg/mL streptomycin (Gibco). DMEM was added as chemoattractant in the lower compartment. After 24 h, cells were fixed with 4% formaldehyde (VWR) in the presence of 1 µg/mL of Hoechst 33,342 (Thermo Fisher Scientific) for 15 min at room temperature. Non-invading cells were removed, and the invasion was assessed by counting the cells on the lower side of the membrane under fluorescence microscope (Axio Observer Z1, Zeiss). The cell number was determined using QuPath software.
Western blotting – Whole cell lysates were treated with RIPA (Thermo Fisher Scientific) buffer, and extracted proteins were then denatured for 5 min at 95 °C. 20 µg of proteins were loaded on a 10% acrylamide/bis-acrylamide gel for SDS-PAGE, then transferred onto a PVDF membrane. The membrane was later incubated with blocking solution for 1 h at room temperature, and with primary antibody overnight at 4 °C. Horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson ImmunoResearch) were then incubated for 1 h at room temperature and signals were revealed with a chemiluminescent HRP substrate before being imaged using the ImageQuant LAS 4000. Antibodies are listed in Table S2.
In vivo orthotopic xenografts – Adult female immunodeficient mice (Crl: NU-Foxn1nu) were obtained from Charles River Laboratories and were used for xenograft experiments. Mice were anesthetized in a cage containing isoflurane. Mice were placed in a stereotactic frame and kept under isoflurane anesthesia. After a precise bone drill, 1 × 105 T033 (GSCs) cells suspended in 2 µL of PBS were slowly infused into the right striatum (from Bregma: -0.5 mm AP, + 2 mm VL, + 2.5 mm DV). Monitoring of tumor growth was performed with in vivo bioluminescence imaging (IVIS, Xenogen), mice health status was evaluated daily, and body weight was recorded every week. Mice were sacrificed when they showed first clinical signs of significant discomfort or suffering. All animal experiments were approved by the ethical committee of the University of Liège (#2290). All experiments were performed in accordance with ARRIVE guidelines.
Mouse brain tissue processing and immunohistochemistry– Mice were euthanized with intraperitoneal injection of 400 mg/kg of Euthasol vet in NaCl 0.9% and immediately perfused intracardially with ice-cold NaCl 0.9% solution containing heparin (LEO Pharma) and then with 4% paraformaldehyde (PFA) in PBS. Brains were removed and postfixed in 4% PFA for 24 h. Brains were cryoprotected in PBS + 30% sucrose for 24 h before being frozen at -80 °C. Coronal brain slices of 14 μm were generated with a cryostat and stored at -20 °C. For the detection of T033 cells in mouse brain tissue, sections were stained with human Vimentin antibody (MAB3400, Millipore), with the Enzo PolyView IHC kit, according to the manufacturer’s instructions.
Ex vivoisolation of cells after tumor growth- Mice were sacrificed by cervical dislocation and brain were collected. Brains were dissected into thick coronal sections at the level of the dorsal horn of the lateral ventricles. In both hemispheres, restricted tissue areas were removed, (1) in the close proximity of the lateral wall of the lateral ventricle (“SVZ in”) and (2) in the temporal cortex, distant from the lateral ventricle (“SVZ out”). Tissue pieces were mechanically and enzymatically dissociated in a solution of Hibernate-A (Fisher Scientific) containing 10 U/mL DNase and 2.5 U/mL papain and then incubated for 10 min at 37 °C under gentle agitation. DMEM/F12 was then added to dilute the enzymes and the solution was passed through a 100 μm strainer to remove debris. Cells were collected by centrifugation (160 g for 10 min) and resuspended in DMEM/F12 supplemented with 2% of B27 without vitamin A (Gibco), 100 U/mL penicillin and 100 µg/mL streptomycin (ThermoFisher Scientific), 1 µg/mL of heparin (LEO pharma), 20 ng/mL of human EGF (Peprotech) and 20 ng/mL of bFGF (Peprotech). Cells were maintained in culture for 7 days, enriched in RFP-positive tumor cells, then collected before a qRT-PCR analysis.
Statistical analyses - The GraphPad Prism 8 software was used for generating graphs and for statistical analysis. The normal distribution of data was verified, and independent comparisons were further performed using unpaired t-tests, Kruskall-Wallis or parametric one-way ANOVA tests. Data were represented as mean ± SD, with the n representing the number of independent experiments. A p-value ≤ 0.05 was considered as statistically significant.
Data availability
All data used and/or analyzed during the current study available from the corresponding author upon reasonable request.
References
Louis, D. N. et al. The 2021 WHO classification of tumors of the Central Nervous System: A summary. Neuro Oncol.23, 1231–1251 (2021).
Ostrom, Q. T. et al. CBTRUS Statistical Report: Primary brain and other Central Nervous System tumors diagnosed in the United States in 2016–2020. Neuro Oncol.25, IV1–IV99 (2023).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med.352, 987–996 (2005).
Wen, P. Y. & Kesari, S. Malignant gliomas in adults. N. Engl. J. Med.359, 492–507 (2008).
Patel, A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science344, 1396 (2014).
Neftel, C. et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell178, 835–849e21 (2019).
Dirkse, A. et al. Stem cell-associated heterogeneity in glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun.10 (2019).
Yabo, Y. A., Niclou, S. P. & Golebiewska, A. Cancer cell heterogeneity and plasticity: A paradigm shift in glioblastoma. Neuro Oncology. https://doi.org/10.1093/NEUONC/NOAB269 (2021).
Zlotnik, A. & Yoshie, O. Chemokine Superfamily Revisit. Immun.36, 705 (2012).
Zlotnik, A. Chemokines and cancer. Int. J. Cancer119, 2026–2029 (2006).
Poeta, V. M., Massara, M., Capucetti, A. & Bonecchi, R. Chemokines and chemokine receptors: New targets for cancer immunotherapy. Front. Immunol.10, 379 (2019).
Isci, D. et al. Patient-oriented perspective on chemokine receptor expression and function in glioma. Cancers14, 130 (2022).
Goffart, N. et al. Adult mouse subventricular zones stimulate glioblastoma stem cells specific invasion through CXCL12/CXCR4 signaling. Neuro Oncol.17, 81–94 (2015).
Kroonen, J. et al. Human glioblastoma-initiating cells invade specifically the subventricular zones and olfactory bulbs of mice after striatal injection. Int. J. Cancer129, 574–585 (2011).
Balabanian, K. et al. The chemokine SDF-1/CXCL12 binds to and signals through the Orphan receptor RDC1 in T lymphocytes. J. Biol. Chem.280, 35760–35766 (2005).
Yen, Y. C. et al. Structures of atypical chemokine receptor 3 reveal the basis for its promiscuity and signaling bias. Sci. Adv.8, 8063 (2022).
Shimizu, S., Brown, M., Sengupta, R., Penfold, M. E. & Meucci, O. CXCR7 protein expression in human adult brain and differentiated neurons. PLoS ONE6 (2011).
Gerrits, H. et al. Early postnatal lethality and cardiovascular defects in CXCR7-deficient mice. Genesis46, 235–245 (2008).
Sierro, F. et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci.104, 14759–14764 (2007).
Wang, Y. et al. CXCR4 and CXCR7 have distinct functions in regulating Interneuron Migration. Neuron69, 61 (2011).
Fumagalli, A. et al. The atypical chemokine receptor 3 interacts with Connexin 43 inhibiting astrocytic gap junctional intercellular communication. Nat. Commun.11 (2020).
Koenen, J., Bachelerie, F., Balabanian, K., Schlecht-Louf, G. & Gallego, C. Atypical chemokine receptor 3 (ACKR3): A comprehensive overview of its expression and potential roles in the immune system. Mol. Pharmacol.96, 809–818 (2019).
Neves, M. et al. The role of ACKR3 in breast, lung, and brain cancer. Mol. Pharmacol.96, 819–825 (2019).
Hattermann, K. et al. Effects of the chemokine CXCL12 and combined internalization of its receptors CXCR4 and CXCR7 in human MCF-7 breast cancer cells. Cell. Tissue Res.357, 253–266 (2014).
Hattermann, K. et al. The chemokine receptor CXCR7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res.70, 3299–3308 (2010).
Levoye, A., Balabanian, K., Baleux, F., Bachelerie, F. & Lagane, B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood113, 6085–6093 (2009).
Berahovich, R. D. et al. Endothelial expression of CXCR7 and the regulation of systemic CXCL12 levels. Immunology141, 111 (2014).
Naumann, U. et al. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE5, e9175 (2010).
Gonçalves, T. L. & de Araújo, L. P. Pereira Ferrer, V. Tamoxifen as a modulator of CXCL12-CXCR4-CXCR7 chemokine axis: A breast cancer and glioblastoma view. Cytokine170, 156344 (2023).
Ehrlich, A. T. et al. Ackr3-Venus knock-in mouse lights up brain vasculature. Mol. Brain14 (2021).
Goffart, N. et al. CXCL12 mediates glioblastoma resistance to radiotherapy in the subventricular zone. Neuro Oncol.19, 66–77 (2017).
Goffart, N. et al. Adult mouse subventricular zones stimulate glioblastoma stem cells specific invasion through CXCL12/CXCR4 signaling. Neuro Oncol.https://doi.org/10.1093/neuonc/nou144 (2015).
Cebo, M. et al. Platelet ACKR3/CXCR7 favors antiplatelet lipids over an atherothrombotic lipidome and regulates thromboinflammation. Blood139, 1722–1742 (2022).
Del Barrio, I. M., Wilkins, G. C., Meeson, A., Ali, S. & Kirby, J. A. Breast Cancer: An examination of the potential of ACKR3 to modify the response of CXCR4 to CXCL12. Int. J. Mol. Sci.19 (2018).
Song, B. et al. Stromal cell-derived factor-1 exerts opposing roles through CXCR4 and CXCR7 in angiotensin II-induced adventitial remodeling. Biochem. Biophys. Res. Commun.594, 38–45 (2022).
Walters, M. J. et al. Inhibition of CXCR7 extends survival following irradiation of brain tumours in mice and rats. Br. J. Cancer110, 1179–1188 (2014).
Smit, M. J. et al. The CXCL12/CXCR4/ACKR3 Axis in the Tumor Microenvironment: Signaling, crosstalk, and therapeutic targeting. Annu. Rev. Pharmacol. Toxicol.61, 541–563 (2021).
Bayrak, A. et al. Discovery and Development of First-in-class ACKR3/CXCR7 superagonists for platelet degranulation modulation. J. Med. Chem.65, 13365–13384 (2022).
Szpakowska, M. et al. The natural analgesic conolidine targets the newly identified opioid scavenger ACKR3/CXCR7. Signal. Transduct. Target. Ther.6 (2021).
Meyrath, M. et al. The atypical chemokine receptor ACKR3/CXCR7 is a broad-spectrum scavenger for opioid peptides. Nat. Commun.11 (2020).
Sjöberg, E. et al. The diverse and complex roles of atypical chemokine receptors in cancer: From molecular biology to clinical relevance and therapy. Adv. Cancer Res.145, 99–138 (2020).
Bianco, A. M. et al. CXCR7 and CXCR4 expressions in Infiltrative Astrocytomas and their interactions with HIF1α expression and IDH1 mutation. Pathol. Oncol. Res.21, 229–240 (2015).
Calatozzolo, C. et al. Expression of the new CXCL12 receptor, CXCR7, in gliomas. Cancer Biol. Ther.11, 242–253 (2011).
Esencay, M., Sarfraz, Y. & Zagzag, D. CXCR7 is induced by hypoxia and mediates glioma cell migration towards SDF-1α. BMC Cancer. https://doi.org/10.1186/1471-2407-13-347 (2013).
Berahovich, R. D., Penfold, M. E. T. & Schall, T. J. Nonspecific CXCR7 antibodies. Immunol. Lett.133, 112–114 (2010).
Neves, M. et al. Special section: From insight to modulation of CXCR4 and ACKR3 (CXCR7) function-minireview the role of ACKR3 in breast, lung, and brain cancer. https://doi.org/10.1124/mol.118.115279
Samus, M. & Rot, A. Atypical chemokine receptors in cancer. Cytokine176, 156504 (2024).
Giordano, F. A. et al. L-RNA aptamer-based CXCL12 inhibition combined with radiotherapy in newly-diagnosed glioblastoma: Dose escalation of the phase I/II GLORIA trial. Nat. Commun.15(1), 1–14 (2024).
Puchert, M. et al. Astrocytic expression of the CXCL12 receptor, CXCR7/ACKR3 is a hallmark of the diseased, but not developing CNS. Mol. Cell. Neurosci.85, 105–118 (2017).
Walters, M. J. et al. Inhibition of CXCR7 extends survival following irradiation of brain tumours in mice and rats. Br. J. Cancer. https://doi.org/10.1038/bjc.2013.830 (2014).
Birner, P., Tchorbanov, A., Natchev, S., Tuettenberg, J. & Guentchev, M. The chemokine receptor CXCR7 influences prognosis in human glioma in an IDH1-dependent manner. J. Clin. Pathol.68, 830–834 (2015).
Salazar, N. et al. A chimeric antibody against ACKR3/CXCR7 in combination with TMZ activates Immune responses and extends survival in mouse GBM models. Mol. Ther.26, 1354–1365 (2018).
Antonello, P. et al. ACKR3 promotes CXCL12/CXCR4-mediated cell-to-cell-induced lymphoma migration through LTB4 production. Front. Immunol.13, 1067885 (2023).
Szpakowska, M. et al. Human herpesvirus 8-encoded chemokine vCCL2/vMIP-II is an agonist of the atypical chemokine receptor ACKR3/CXCR7. Biochem. Pharmacol.114, 14–21 (2016).
Neves, M., Marolda, V., Mayor, F. & Penela, P. Crosstalk between CXCR4/ACKR3 and EGFR signaling in breast Cancer cells. Int. J. Mol. Sci.23 (2022).
Puddinu, V. et al. ACKR3 expression on diffuse large B cell lymphoma is required for tumor spreading and tissue infiltration. Oncotarget8, 85068–85084 (2017).
Acknowledgements
The authors would like to thank the patients and the Neurosurgery department of the Liège University Hospital Belgium. We also would like to thank the GIGA-ULiège Flow cytometry and Cell Imaging (S. Ormenese and colleagues), the NCP platform from LIH (Fanny Hedin, Giovanna Radicchio and Dominique Revets) and Viral Vector (E. Di Valentin and colleagues) platforms for their expert advice and technical support.
Funding
This work was supported by the National Fund for Scientific Research (FNRS, Télévie) (grants 7.4593.19, 7.4529.19 and 7.8504.20) and the Leon Frédéricq Foundation (Liège, Belgium), the Luxembourg Institute of Health (LIH) through the NanoLux platform, Luxembourg National Research Fund (INTER/FNRS grant 20/15084569 and CORE C23/BM/18068832).
.
Author information
Authors and Affiliations
Contributions
DI and VN designed the study. Entire experiments and data analysis were mainly performed by DI and partially by other authors. The manuscript was written by DI and revised by VN, BR, AC and MS. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
All animal experiments were approved by the ethical committee of the University of Liège (#2290) and were performed in accordance with relevant guidelines and regulations. All animal research reported in this paper was in accordance with the ARRIVE guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Isci, D., Kuppens, A., Scalisi, J. et al. Heterogeneous expression of the atypical chemokine receptor ACKR3 in glioblastoma patient-derived tissue samples and cell cultures. Sci Rep 14, 21925 (2024). https://doi.org/10.1038/s41598-024-73064-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-73064-w
- Springer Nature Limited