
Abstract
Do all birds' sex chromosomes follow the same canonical one-way direction of evolution? We combined cytogenetic and genomic approaches to analyze the process of the W chromosomal differentiation in two selected Passeriform species, named the Pale-breasted Thrush Turdus leucomelas and the Rufous-bellied thrush T. rufiventris. We characterized the full catalog of satellite DNAs (satellitome) of T. leucomelas, and the 10 TleSatDNA classes obtained together with 16 microsatellite motifs were in situ mapped in both species. Additionally, using Comparative Genomic Hybridization (CGH) assays, we investigated their intragenomic variations. The W chromosomes of both species did not accumulate higher amounts of both heterochromatin and repetitive sequences. However, while T. leucomelas showed a heterochromatin-poor W chromosome with a very complex evolutionary history, T. rufiventris showed a small and partially heterochromatic W chromosome that represents a differentiated version of its original autosomal complement (Z chromosome). The combined approach of CGH and sequential satDNA mapping suggest the occurrence of a former W-autosomal translocation event in T. leucomelas, which had an impact on the W chromosome in terms of sequence gains and losses. At the same time, an autosome, which is present in both males and females in a polymorphic state, lost sequences and integrated previously W-specific ones. This putative W-autosomal translocation, however, did not result in the emergence of a multiple-sex chromosome system. Instead, the generation of a neo-W chromosome suggests an unexpected evolutionary trajectory that deviates from the standard canonical model of sex chromosome evolution.
Similar content being viewed by others
Introduction
Nearly all bird species share the same ZZ/ZW sex chromosome system (an exception to this rule is described by Ref.1, which is widely regarded as a stable sex system with males (ZZ) and females (ZW) representing the homogametic and heterogametic sex, respectively2,3. Both the Z and W sex chromosomes originated from an ancestral autosomal pair more than 110 Mya ago4,5. In most species (except the Ratites where the sex chromosomes are homomorphic), the W chromosome is small and mostly heterochromatic, whereas the Z is typically preserved in both morphology and gene content3,6,7,8,9,10. Despite the conservation of the Z chromosomes, they are subject to frequent intrachromosomal rearrangements, such as inversions, resulting in changes in their morphology within species2.
With over 6500 species, the order Passeriformes is the most varied group of birds10. Despite this remarkable variety, only ~ 460 of these species, or ~7.0% of the total in this order, have had their diploid number determined11. Most Passeriform species have small W chromosomes that have experienced dynamic processes of constitutive heterochromatin accumulation and sequence elimination throughout their evolutionary history12,13,14. Among them, with 88 species, the genus Turdus (Thrushes) stands out as the most diverse one10. While all Thrushes share common morphological characteristics, they exhibit a wide range of plumage colorations and ecological adaptations. This plasticity allows them to thrive in diverse biomes, including savannahs, alpine areas, and both tropical and temperate forests15.
Among the 88 species within the Turdus genus, 18 of them have had their karyotypes described, revealing a substantial degree of chromosome similarity among them. The diploid number (2n) ranges from 78 to 84, indicating slight variations11. Molecular cytogenetics studies using chicken macrochromosome probes (GGA1-10) have been conducted on a limited scale, encompassing only four species: T. merula, T. iliacus, T. rufiventris, and T. albicollis16,17,18. These investigations unveiled a sole interchromosomal rearrangement, specifically the fission of the ancestral chromosome one (GGA1), which is a common characteristic observed among Passeriforms2,11. In T. merula, except for chromosome 16, which has remained unstudied, no evidence of interchromosomal rearrangements in connection to the homologous chromosomes to GGA11-28 has been found in any of the microchromosomes19.
In recent years, the integration of molecular cytogenetics techniques with in silico data derived from the Next Generation Sequence (NGS) and novel software pipelines have provided significant advances in the comprehension of intricate chromosome rearrangements20,21,22 and in the evolution of sex chromosomes23,24. In particular, the characterization and the in situ mapping of the satelitome, which is a catalog of the most representative satellite DNAs (satDNAs) in a genome25, is capable of highlighting transpositions and translocation events, giving insights into the framework of karyotype evolution and chromosome speciation26,27,28,29. Moreover, although still incipient in birds (however see30,31), investigations in other vertebrates, such as mammals32,33,34,35,36,37 and amphibians38,39, demonstrated the fast-evolving nature of these in tandem repetitive DNAs and their putative role in the formation and composition of centromeres and in the evolution of sex chromosomes35,40,41.
Comparative genomic hybridization (CGH) (sometimes also referred as GISH) is a fine-scale molecular cytogenetic approach used to detect chromosomal rearrangements that has also been applied to discover the evolutionary origin and composition of sex chromosome systems42,43. This method allows us to recognize the high level of molecular differentiation of sex chromosomes, localize sex-specific chromosome regions, and to track early stages of sex chromosome differentiation in several groups44,45. In this way, repetitive DNAs and comparative genomic hybridization mapping, are an attempt to advance toward the knowledge of the processes that have shaped the evolution of sex chromosomes.
Here, we selected two Passeriform species belonging to the Turdidae family, named the Pale-breasted thrush T. leucomelas and the Rufous-bellied thrush T. rufiventris to analyze the process of their W chromosomal evolution. In that regard, we compared the intragenomic differences (focusing on their repetitive DNA content) between males and females of each species and used cytogenetic and genomic methods to analyze their satDNA composition and their putative involvement in their W chromosomal evolution.
Results
The aim of our work was to characterize and map the satellite DNA sequences present in the species T. leucomelas (TLE) and then compare these sequences isolated in a similar species, T. rufiventris (TRU). First, we investigated and confirmed that the 2n for both species investigated were 2n = 80 for T. leucomelas and 2n = 78 for T. rufiventris. These results corroborated earlier information for these species18,46. The next step in delving deeper into the previously mentioned issues was to describe T. leucomelas´ satellitome.
satDNA content of the T. leucomelas´ genome
After three iterations in TAREAN, 10 satDNA families (TleSatDNAs) were recovered. Table 1 presents the general characteristics of the T. leucomelas´ satellitome, such as the A + T content of the satellites, which ranged from 27.2 to 69.6%, with an average of 51.73%, and the length of the repeated units (RUL), which ranged between 21 and 1644 bp with 80% of the satDNAs families having monomers greater than 100 bp. By aligning each T. leucomelas satDNA in the RM_Homology version 1 (https://github.com/fjruizruano/satminer) and Geneious software version 8.0 (https://www.geneious.com), a superfamily relationship (50–80% similarity) was observed between the satDNAs TleSat02-145 and TleSat05-21, which are considered a high-order repeat (HOR). The repeat landscapes generated are shown in Supplementary Fig. S1. Comparing the satDNA catalogs of males and females, the presence of two satellites more abundant in females than in males was observed, these being TleSat06-645 (ratio of 3.94 between genders) and TleSat08-419 (ratio of 51.89 between genders) (Table 1).
Minimmum spanning trees (MSTs)
We selected TleSat05-21 and TleSat07-103 to generate minimum spanning trees (MSTs) (Fig. 1). These satDNAs were selected due to their monomer sizes (< 150 bp), differential abundance between sexes (see Table 1), and clusterization after FISH results (Fig. 2). TleSat05-21 doesn’t demonstrate accumulation in the sex chromosomes of T. leucomelas (Fig. 2), and the MST is composed of six mainly haplotypes shared between males and females, following the observed ratio of abundance in males and females (1.02, Table 2). In contrast, TleSat07-103 shows a ratio of 0.70, with more abundance of this sequence in males than in females. The MST demonstrates a predominance of one haplotype, shared between sexes, and several less abundant haplotypes, and some of them are male-specific (Fig. 1), despite the absence of FISH signals in the Z chromosome.

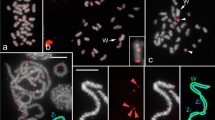
Linear MSTs of (A) TleSat05-21 and (B) TleSat07-103 obtained from female (red) and male (green) reads. Each circle represents one haplotype and the diameter is proportional to the abundance of the haplotype. Black dots represent a mutation event.
Chromosomal distribution of TleSatDNAs and microsatellites
Following the in situ investigations, we found that, Except for TleSat02 and TleSat04, all the remaining TleSatDNAs showed positive signals on female chromosome metaphases of T. leucomelas (Fig. 2). The TleSat01 displayed signals in the centromeric region of all chromosomes. TleSat03 was mapped in the centromeric region of two pairs of macrochromosomes, as well as in some microchromosomes. TleSat06 was located in the pericentromeric region of three macrochromosomes, one microchromosome, and on the W. TleSat05, TleSat07, and TleSat09 were exclusively mapped on microchromosomes, while TleSat10 was exclusively mapped on the pericentromeric region of the Z chromosomes (Fig. 2). The TleSat06 and TleSat08 displayed a variable number of sites among the individuals, indicating a polymorphism related to the satellites, which may involve W-autosomal translocation events (as will be further discussed) or also to transpositions of mobile elements.

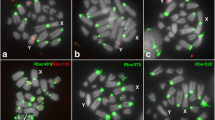
Chromosomal mapping of the eight TleSatDNAs hybridized on female metaphases of T. leucomelas (TLE ♀F01). The Z and W sex chromosomes are indicated. While the Z chromosome was identified by its distinct morphology (i.e., the only metacentric macrochromosome), the W chromosome was appropriately identified after a sequential hybridization with TleSat06, which provides a unique and distinctive pattern for this chromosome. Bar 10 μm.
In T. rufiventris, only six of the 10 TleSatDNA (TleSat01, TleSat05, TleSat06, TleSat07, TleSat08, and TleSat10) showed positive signals after in situ experiments (Fig. 3). The TleSat01 and TleSat10 present the same pattern found in T. leucomelas, being mapped in all centromeres and solely on the Z chromosome, respectively. However, TleSat05, TleSat06, TleSat07, and TleSat08 showed different accumulations in T. rufiventris. Although TleSat05 and TleSat07 exhibited hybridization signals only in the microchromosomes, like in T. leucomelas, no signals for TleSat06 were observed in the W chromosome of this species. TleSat08 only displayed hybridization clusters in a few pairs of microchromosomes (Fig. 3).

Chromosomal mapping of TleSatDNAs on metaphase plates of T. rufiventris (TRU ♀F01). While the Z chromosome was identified by its distinct morphology (i.e.: the only metacentric macrochromosome), the W chromosome was appropriately identified after a sequential C-banding, which provides a unique and distinctive pattern for this chromosome. The Z and W sex chromosomes are indicated. Bar 10 μm.
Of the total of 16 microsatellites tested in both Thrushes, only two had positive hybridization signals in T. leucomelas, named (GA)15 and (CGG)10, which demonstrated clusters in one and three pairs of microchromosomes, respectively (Fig. 4). On the other hand, T. rufiventris showed positive hybridization signals for three microsatellites, with (CGG)10 displaying signals in three pairs of microchromosomes, while (CAG)10 and (CAT)10 both accumulated in the telomeric region of Z chromosomes (Fig. 4).

Metaphase plates of T. leucomelas TLE♂M01; TLE♀F01; T. rufiventris TRU♀F01 and TRU♂M01 highlighting the chromosomal mapping of microsatellites (B–H) and TleSat10 (A,I). Bar 10 μm.
Comparative genomic hybridization
Lastly, after examining specific sequences for each sex, we found overlapping signals in the pericentromeric regions of almost all chromosomes, except for an exclusive strong female-specific region on the W chromosome, coincident with a C-positive heterochromatic block (Fig. 5, Supplementary Fig. S2E). Contrarily, four distinct hybridization patterns were identified in the T. leucomelas individuals (Fig. 6, Supplementary Figs. S2 and S3). In addition to overlapping signals in the centromeric region of all male and female chromosomes, the accumulation of female-biased hybridization signals in the entire W chromosome and half of a small autosome was evidenced in the two females (TLE♀F01 and TLE♀F02) analyzed (Fig. 6D, Supplementary Fig. S3). In turn, three different hybridization patterns were found in the four males analyzed. In the TLE♂M01 only overlapping signals of the male and female gDNA probes were detected in the centromeric region of all chromosomes (Fig. 6A, Supplementary Fig. S2A). This same pattern was also observed in the other three males, in addition to one copy (in TLE♂M02) or two copies (in both TLE♂M03 and TLE♂M04) of the same small autosome displaying female-biased hybridization signals (Fig. 6B,C, Supplementary Fig. S2B–D). We sampled individuals from different populations, thus ensuring that the patterns discovered were not exclusive to a specific population. In all cases, this small autosome also accumulated the TleSat06 and TleSat08 (Fig. 6). While the whole short arms of the W chromosome contain a weak C-positive heterochromatic block, prominent C-positive blocks are observed in the Z chromosome and in the short arms of the small autosome that exhibit the female-biased hybridization signals (Fig. 6).

Turdus rufiventris male and female genomic DNA probes hybridized on female metaphase chromosomes of T. rufiventris (TRU♀F01) following the experimental design described in Table 3. The hybridization patterns of the probes derived from male (green), female (red), and the combined pictures are shown in (A), (B), and (C), respectively. The sequential C-banding highlighted a conspicuous C-positive heterochromatic block in the short arms of the W chromosome (D). Bar 10 μm.

Intraspecific genomic hybridization (CGH) in T. leucomelas males: (A) TLE♂M01; (B) TLE♂M02; (C) TLE♂M03 and female (D) TLE♀F01 specimens following the experimental design described in Table 3. The merged images displayed in the CGH column were obtained from those present in Supplementary Figs. S2 and S3. After the CGH, chromosomes were sequentially mapped with TleSat06 (second column) and TleSat08 (third column) probes, and then C-banded (fourth column). The small autosomes displaying female-biased hybridization signals, the accumulation of Tlesat06 and TleSat08, and a conspicuous C-positive block are indicated by the arrowheads. Bar 10 μm.
Discussion
The diploid numbers of both species, T. rufiventris and T. leucomelas, have already been characterized in previous works, like the morphology of their ZW sex chromosomes18,46. However, despite their similar sizes, no data on the molecular and heterochromatic content of their W chromosomes was currently available. Here, we provide further confirmation of the 2n number [i.e.: T. leucomelas (2n = 80) and T. rufiventris (2n = 78)] and molecular cytogenetic analyses. We showed that the W chromosome of T. rufiventris has a very strong C-positive band on its short arms (Fig. 5), while the W chromosome of T. leucomelas exhibited a faint block of heterochromatin encompassing just its entire short arms (Fig. 6). Although the occurrence of W chromosomes with unusual morphologies and scarce in heterochromatin has also been evidenced47,48, both these patterns seem atypical for W chromosomes of Passeriformes members, once most species up to now karyotyped have almost entirely heterochromatic W chromosomes, such as in the zebra finch (Taeniopygia guttata), the canary (Serinus canaria)14, and the Sooty-fronted Spinetail (Synallaxis frontalis)49.
To characterize the repetitive DNA fraction of these W chromosomes, we first isolated and characterized the satellitome of T. leucomelas and further in situ mapped the 10 TleSatDNAs obtained, together with 16 microsatellite motifs in both T. leucomelas and T. rufiventris species. The data confirm the previous findings obtained in some few bird species highlighting that avian satellites are usually composed of a small number of particularly large satDNAs rich in GC content30. However, this is the first case where the satellitomes were mapped in their respective chromosomes. The MSTs produced in this work demonstrate a prevalence of shared haplotypes between males and females to TleSat05 and TleSat07, due to their presence in autosomal microchromosomes, as demonstrated by FISH. The presence of TleSat07 haplotypes exclusive to males, together with its higher frequency in males, implies the presence of Z chromosome clusters that are not visible by FISH, possibly because of the small array sizes (Figs. 1 and 2).
Except for TleSat01, which is present in the centromeric region of all T. leucomelas and T. rufiventris chromosomes (probably representing their primary centromere component), and TleSat06, which accumulated exclusively in T. leucomelas' W chromosome, we did not detect any evidence of accumulation for the extant TleSatDNAs on the W chromosomes of both species (Figs. 2 and 3). Similarly, none of the microsatellites examined in this study were accumulated on any W chromosomes (Fig. 4). The heterogametic chromosomes (W and Y) tend to differentiate once recombination ceases and heterochromatization followed by the accumulation of repetitive elements begins50,51. In this pathway, Ref.52 proposed that the accumulation of satDNA sequences throughout the length of the sex-specific (Y and W) chromosome plays a significant role in generating its morphological differentiation from the X or Z, respectively. Likewise, microsatellite repeats are crucial for the differentiation of sex-specific chromosomes, as they may be the first type of repeat that accumulates during its early stages of differentiation53,54. Accordingly, reports from various taxa show the accumulation of repetitive sequences specifically on the Y or W chromosomes, which are enriched in high-, middle-, and low-copy repetitive sequences and contain only a few functional genes35,38,41,51,55,56. However, it is not a rule that most repetitive sequences are found exclusively in heterogametic chromosomes, as revealed by several groups57,58,59. Here, an exceptionally high number of repetitions accumulated on the Z chromosomes, including centromeric clusters of TleSat10 in both species as well as (CAG)n and (CAT)n in the terminal region of the q arms of the Z chromosomes of T. rufiventris (Figs. 2, 3, 4). This scenario is unusual among birds since very few cases of repeat accumulation on the Z chromosomes were documented59,60,61,62.
Instead, the great majority of TleSatDNAs was mapped in microchromosomes in both species. Experiments in other bird families, including Caprimulgidae and Picidae, have also demonstrated a high density of repetitive microsatellite and telomeric sequences in microchromosomes59,63,64. Similarly, in some species of turtles and lizards, the accumulation of these repeats in microchromosomes has also been shown65,66.
In both thrush species, the conventional chromosomal analysis, C-banding, and repetitive DNA mapping pointed to a specific W chromosome arrangement that differs from the majority of avian species up to now analyzed3,14, since it does not reveal many repeated sequences or significant blocks of heterochromatin accumulating on chromosomes. In addition, this particular scenario was shown to be even more complex when intraspecific CGH analyses were performed. While the T. rufiventris specimens presented the expected overall results after intraspecific-CGH experiments (i.e., the W chromosome showing the only particularly rich region in the female-biased hybridization signals), an unusual pattern was observed in the T. leucomelas individuals (Fig. 6). In the two females of T. leucomelas (TLE♀F01 and TLE♀F02) analyzed, besides the entire W chromosome, half of a small autosome is also enriched by the female-biased hybridization signals. Except for the TLE♂M01 specimen, the other males show a polymorphic state for this same small autosome, i.e., with only one copy (TLE♂M02) or two copies (both TLE♂M03 and TLE♂M04) of those female-biased hybridization signals (Fig. 6, Supplementary Figs. S2 and S3). So, it is likely that this portion of the autosome enriched by the female-biased hybridization signals was originally part of the W chromosome. Therefore, the occurrence of a W-autosomal reciprocal translocation (not involved in the creation of a multiple-sex chromosome system) is one of the hypotheses that best explains this complex scenario, where the W chromosome both gained and lost sequences67, as well as the small autosome (also present in males) which incorporated both TleSat06 and TleSat08 (the latter, being previously W-specific and now present in its short arms) (Fig. 7). Likely, this chromosomal rearrangement does not lead to a dosage composition problem for individuals exhibiting either the heteromorphic or homomorphic condition, as the translocated segment encompasses repetitive DNA sequences (as shown by our CGH-SatDNA-FISH analysis), which are usually transcriptionally silenced67,68. However, we cannot exclude an alternative hypothesis, as the presence of repetitive sequences may significantly change due to various parameters, such as copy number variation (expansions and contractions), their genome location, and sometimes even as a result of transposition events and/or major chromosomal rearrangements67,68,69.

Idiogram representing one of the main hypotheses that involve a translocation event in T. leucomelas specimens and the resulting genotypes from possible crossings. Under this hypothesis, an ancestral female undergoes a translocation between the short arms of the W chromosome and an autosome (A). As a result, the W chromosome both acquired and lost sequences, while an autosome (A1) also lost sequences and integrated previously W-specific sequences, which included both TleSat06 and TleSat08, while its homologous remained untouched (A2). This pattern (ZW + A1A2) is observed in both T. leucomelas (TLE♀F01/F02). When crossed with a wild-type ZZ + A2A2 male (TLE♂M01), it produces an F1 offspring with four possible results: (i) ZW + A1A2 females (TLE♀F01/F02); (ii) ZW + A2A2 females (?); (iii) ZZ + A1A2 males (TLE♂M02), and (iv) ZZ + A2A2 males (TLE♂M01). A new crossing between TLE♀F01/F02 (ZW + A1A2) x TLE♂M02 (ZZ + A1A2) generates an F2 offspring with six possible results: (i) ZW + A1A2 females (TLE♀F01/F02); (ii) ZW + A2A2 females (?); (iii) ZW + A1A1 females (?); ZZ + A1A1 males (TLE♂M03/M04), and (iv) wild-type ZZ + A2A2 males (TLE♂M01). Individuals marked with (?) stand for those that we were unable to find in this work.
How do sex chromosomes evolve? Up until recently, it was widely believed that the sex chromosomes followed a canonical one-way direction of evolution, which was proposed by gathering information from multiple independent works6,70,71,72. This standard sex chromosome evolution model predicts that the Y and W chromosomes gradually differentiate and most of their genetic material is lost owing to a lack of recombination with the X or Z, respectively. This leads to the usual gradual loss of genes or gene function and structural modifications like deletions and heterochromatinization. As a result, the sex-specific chromosome might progressively shrink and ultimately be eliminated from the genome73,74,75. At first glance, since all Neognathae species, with a few exceptions, have small and heterochromatic W chromosomes, this seemed to be also the standard path taken by all bird sex chromosomes (reviewed in76 and 77. Besides, Ratite birds, which represent the basal avian lineage (paleognaths), present sex chromosomes at an early stage of differentiation, where Z and W chromosomes are still morphologically similar8,78,79,80. Our data in T. leucomelas, however, points to an unusual evolutionary pathway for the W chromosome that deviates from the standard canonical model of sex chromosome evolution.
Novel investigations (see for example81,82,83,84,85 are steadily describing new deviant models that differ from the canonical one-way direction of evolution. In particular, cases of new genetic material being added to the sex chromosomes are outstanding examples as they contradict the so-thought inevitable degeneration of the heteromorphic sex chromosome75,85. In these models, new linkage groups can be created by extensive amplification of sequence copy number, brought by molecular drive, and/or sex chromosome-autosome translocations85. In the latter, a reciprocal translocation (i.e., DNA segments are swapped mutually between chromosomes), results in two possible scenarios. The first, leads to a multiple-sex chromosome system, as the two linkage groups, being consequently whole chromosomes, remain in the form of the larger translocation product (e.g., Neo-sex chromosome)86. The second scenario, however, does not alter the 2n number, and consists of a non-homologous exchange between different sections of two or more chromosomes, thus generating independent Neo-chromosomes that share common DNA motifs86. Regarding W-autosome translocations, despite the unique multiple ♂Z1Z1Z2Z2/♀Z1Z2W sex chromosome system described1, recent studies have revealed the fusion of sex chromosomes and autosomes in different bird lineages, indicating that this type of rearrangements is more common than initially believed. For example, the fusion of ZW sex chromosomes with chromosome 11 has been proposed in the ancestor of parrots21. Additionally, in the parrot Myiopsitta monachus, chromosome 25 has been further fused to the sex chromosomes21. In the cuckoo species Crotophaga ani a Robertsonian translocation between the microchromosome 17 and the Z chromosome was found62. Among songbirds (Sylvoidea), a series of papers have indicated that autosomal material had been integrated into both Z and W87,88. Therefore, the evolution of bird W chromosomes is revealing more dynamic than previously thought as new data derived from cutting-edge sequencing and cytogenetic investigations (such as the ones described here) become available.
Conclusions
Why have birds´ W chromosomes endured for more than 100 Myr? We demonstrate that its evolution could be far more complex than previously thought. We showed that the W chromosomes of both Thrushes did not accumulate higher amounts of heterochromatin and repetitive sequences, as observed in most bird species. Besides, the W chromosome of the pale-breasted Thrush, instead of representing a straightforward "degenerated" version of its earlier homologous Z chromosomes, may represent a dynamic “patchwork” that includes deletions and the integration of new genomic material as a result of chromosomal rearrangements with autosomes. Specific satDNA families were directly associated with these rearranged regions. These findings challenge the unidirectional evolutionary process of W chromosomes widely proposed for birds.
Methods
Sampling, chromosomal preparation, and C-banding
The samplings of T. leucomelas and T. rufiventris were authorized by the Brazilian environmental agency ICMBio/SISBIO (Licenses 61047-4, 44173-1, and 68443-2) and SISGEN (A96FF09). Each individual of T. leucomelas and T. rufiventris was assigned a code (i.e., male—M; female—F) (Table 2). Mitotic chromosomes were obtained according to the protocols described by Refs.60,89, which utilized skin biopsies and bone marrow for fibroblast culture, respectively. The constitutive heterochromatin regions were evidenced following the protocol proposed by Ref.90. All experiments followed the guidelines and were approved by the Ethics Committee on Animal Experimentation of the Federal University of Pampa (018/2014 and 019/2020). The authors complied with ARRIVE guidelines.
The genomic DNAs (gDNAs) from T. leucomelas and T. rufiventris individuals were extracted following the protocol described by Ref.91. DNA samples from T. leucomelas♀F01 and T. leucomelas♂M01 were sequenced using the BGISEQ-500 platform (paired-end 2 × 150 bp) with a 3 × coverage normally required for satellite assembly25,37. The genomic reads obtained were deposited in the Sequence Read Archive (SRA) under accession numbers SRR26625300 (male) and SRR26625299 (female).
Bioinformatic analyses: construction of T. leucomelas satellite DNA catalogs and additional analyses
The genomic libraries were subjected to a process of quality trimming using the software Trimmomatic version 0.36 (https://github.com/usadellab/Trimmomatic)92. After, the satellitome of a female of T. leucomelas was characterized using the TAREAN tool93, following the SatMiner pipeline25. Then, the outputs containing the putative consensus sequences of satDNAs were used to filter the genomic libraries using the software Deconseq version 0.4.3 (https://deconseq.sourceforge.net)94, and other iterations of TAREAN were performed until no satDNAs were found. After the characterization of all consensus sequences, we filtered and removed other tandemly repeated elements, such as multigene families, and a homology search using RepeatMasker95 was performed to group the sequences as the same variant (similarity greater than 95%), variants of the same satDNA (similarity between 80 and 95%), and superfamilies (similarity between 50 and 85%), following the patterns established by25. The abundance and divergence of each satDNA were estimated in females and males using RepeatMasker software version 3.0 (https://www.repeatmasker.org)95, with a random selection of 2 × 5,000,000 reads. After that, satDNA families were named according to their abundance in T. leucomelas. Considering the particularities of the sex chromosome system of T. leucomelas, the quotient between the abundance of each satDNA in females and males (F/M) was calculated to verify putatively accumulated satDNAs in the sex chromosomes. TleSatDNAs were deposited in GenBank with accession numbers OR675141.1–OR675150.
Besides, we selected TleSat05-21 and TleSat07-103 to construct minimum spanning trees (MSTs). Only these satDNAs were selected due to technological limitations, in which it is only possible to use satDNAs whose monomer size is smaller than the read size (< 150 bp in this case). We extracted monomers of the cited satDNAs from genomic libraries of both sexes, followed by alignment of the reads against each satDNA, to select only full reads. After that, we discarded singletons using CD-Hit software version V4.8.1 (https://sites.google.com/view/cd-hit/home?authuser=0)96. Finally, the MSTs were constructed using PHILOVIZ 2.0 software version 2.0 (https://www.phyloviz.net)97, and Inkscape was utilized to produce the final image.
Primer design and amplification using polymerase chain reaction (PCR)
A total of ten satDNA sequences (hereafter named TleSatDNAs) were isolated (Table 1), for which eight were designed primers (TleSat01, TleSat02, TleSat03, TleSat06, TleSat07, TleSat08, TleSat09, and TleSat10). The remaining two (TleSat04 and TleSat05) were synthesized and labeled with Cy3 at the 5' end by ThermoFisher (ThermoFisher Scientific), since they are smaller than 30 bp. The PCR reactions followed the conditions optimized according to35. To confirm the amplification of each satDNA, the PCR products were subjected to electrophoresis in a 1% or 2% agarose gel, and subsequently quantified by the ThermoFisher NanoDrop spectrophotometer (ThermoFisher Scientific).
Fluorescence in situ hybridization (FISH)
All TleSatDNAs were labeled using a nick translation Kit from Jena Bioscience (Jena, Germany) incorporating the fluorophore Atto488-dUTP or Atto550-dUTP according to the instructions in the manufacturer's manual. Microsatellite sequences (GAA)10, (GAC)10, (CGG)10, (CAC)10, (CAG)10, (CAT)10, (GAG)10, (TAA)10, (TAC)10, (CAA)10, (GA)15, (CA)15, (GC)15, (TA)15, (C)30, and (A)30 were labeled directly with Cy3 at the 5' end during synthesis (VBC Biotech, Vienna, Austria) and also used in the hybridization experiments. We performed the fluorescence in situ hybridization experiments following the protocol described by98. The slides were dehydrated in a 70%, 85%, and 100% ethanol solution and the metaphases were stained with 4',6-diamidino-2-phenylindole (DAPI).
Comparative genomic hybridization (CGH)
We performed intraspecific CGH in both T. leucomelas and T. rufiventris individuals following the experimental designs described in Table 3. For this purpose, gDNAs from males and females of each species were respectively labeled using a nick-translation labeling kit with Atto488-dUTP (green) and Atto550-dUTP (red), from Jena Bioscience (Jena, Germany). To block common genomic repetitive regions, we used Cot-1 DNA derived from the male gDNA of each species, produced according to99. Each hybridization was composed of 3 µg of male-derived Cot-1 DNA and 500 ng of each labeled male and female gDNAs. After using ethanol-precipitation, the pellet was air-dried and well mixed with 20μL of hybridization buffer (Denhardt's buffer, pH 7.0), composed of 50% formamide, 2% 2xSSC, 10% SDS, 10% dextran sulfate. The CGH experiments followed the methodology detailed in100. After the CGH experiments, the T. leucomelas chromosomal slides were washed 3 times in a 4SSC-Tween solution at 42 °C, and sequentially in situ mapped with TleSat06 (red) and TleSat08 (green) probes. Finally, the material was sequentially C-banded using the abovementioned probes and protocols.
Microscopic analysis and image processing
To corroborate the 2n, karyotype structure, FISH, and CGH results, at least 30 metaphase spreads per individual were examined. Images were captured using an Olympus BX50 microscope (Olympus Corporation, Ishikawa, Japan), with CoolSNAP, and the images were processed using Image-Pro Plus software version 4.1 (https://mediacy.com/image-pro) (Media Cybernetics, Silver Spring, MD, USA). Chromosomes were classified according to101.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. The catalog of satellite DNAs was deposited on the GenBank with accession numbers OR675141.1- OR675150 and raw reads are available in Sequence Read Archive (SRA-NCBI) under accession numbers SRR26625300 (male) and SRR26625299 (female).
References
Gunski, R. J. et al. Multiple sex chromosome system in penguins (Pygoscelis, Spheniscidae). Comp. Cytogenet. 11, 541–552 (2017).
Griffin, D. K., Robertson, L. B. W., Tempest, H. G. & Skinner, B. M. The evolution of the avian genome as revealed by comparative molecular cytogenetic. Cytogenet. Genome. Res. 117, 64–77 (2007).
Kretschmer, R., Ferguson-Smith, M. A. & De Oliveira, E. H. C. Karyotype evolution in birds: from conventional staining to chromosome painting. Genes 9, 181 (2018).
Marshall Graves, J. A. & Shetty, S. Sex from W to Z: evolution of vertebrate sex chromosomes and sex-determining genes. J. Exp. Zool. 290, 449-462 (2001).
Nanda, I. et al. 300 million years of conserved synteny between chicken Z and human chromosome 9. Nat. Genet. 21, 258–259 (1999).
Ohno, S., Muramoto, J., Christian, L. & Atkin, N. B. Sex chromosome and sex-linked genes. Chromosoma. 23, 1–9 (1967).
Nanda, I. & Schmid, M. Conservation of avian Z chromosomes as revealed by comparative mapping of the Z-linked aldolase B gene. Cytogenet. Genome. Res. 96, 176–178 (2002).
Nishida-Umehara, C., Tsuda, Y. & Ishijima. J. The molecular basis of chromosome orthologies and sex chromosomal differentiation in palaeognathous birds. Chromosome Res. 15, 721–734 (2007).
Nanda, I., Schlegelmilch, K., Haaf, T., Schartl, M. & Schmid, M. Synteny conservation of the Z chromosome in 14 avian species (11 families) supports a role for Z dosage in avian sex determination. Cytogenet. Genome. Res. 122, 150–156 (2008).
Gill, F., Donsker, D. & Rasmussen, P. IOC World Bird List (v13.1). https://www.worldbirdnames.org/new/ (2023).
Degrandi, T. et al. Introducing the bird chromosome database: An overview of cytogenetic studies in birds. Cytogenet. Genome. Res. 160, 199–205 (2020).
Correia, V. et al. Alta similaridade cariotípica na família Emberezidae (Aves: Passeriformes). Biosci. J. 25, 99–111 (2009).
Barbosa, M. et al. Nucleolar organizer regions in Sittasomus griseicapillus and Lepidocolaptes angustirostris (Aves, Dendrocolaptidae): Evidence of a chromosome inversion. Genet. Mol. Biol. 36, 70–73 (2013).
Dos Santos, M. et al. Comparative cytogenetics between two important songbird, models: The Zebra Finch and the Canary. PLoS ONE. 12, e0170997 (2017).
del Hoyo, J. D. et al. Handbook of the birds of the world alive (Lynx Edicions, 2019).
Guttenbach, M. et al. Comparative chromosome painting of chicken autosomal paints 1–9 in nine different bird species. Cytogenet. Genome. Res. 103, 173–184 (2003).
Derjusheva, S. et al. High chromosome conservation detected by comparative chromosome painting in chicken, pigeon, and passerine birds. Chromosome Res. 12, 715–723 (2004).
Kretschmer, R. et al. Molecular cytogenetic characterization of multiple intrachromosomal rearrangements in two representatives of the genus Turdus (Turdidae, Passeriformes). PLoS ONE 9, e103338 (2014).
O’Connor, R. et al. Patterns of microchromosome organization remain highly conserved throughout avian evolution. Chromosoma 128, 21–29 (2019).
Furo, I. et al. Chromosomal evolution in the phylogenetic context: A remarkable karyotype reorganization in neotropical parrot Myiopsitta monachus (psittacidae). Front. Genet. 11, 721 (2020).
Huang, Z. et al. Recurrent chromosome reshuffling and the evolution of neo-sex chromosomes in parrots. Nat. Commun. 13, 944 (2022).
Höök, L. et al. High-density linkage maps and chromosome-level genome assemblies unveil direction and frequency of extensive structural rearrangements in wood-white butterflies (Leptidea spp.). Chromosome Res. 31, 2 (2023).
Stöck, M. et al. A brief review of vertebrate sex evolution with a pledge for integrative research: Towards ‘sexomics’. Philos. Trans. R. Soc. b. 376, 20200426 (2021).
Štundlová, J. et al. Sex chromosome differentiation via changes in the Y chromosome repeat landscape in African annual killifishes Nothobranchius furzeri and N. kadleci. Chromosome Res 30, 309–333 (2022).
Ruiz-Ruano, F. J., López-León, M. D., Cabrero, J. & Camacho, J. P. M. High-throughput analysis of the satellitome illuminates satellite DNA evolution. Sci. Rep. 6, 28333 (2016).
Potter, S. et al. Chromosomal speciation in the genomics era: disentangling phylogenetic evolution of rock-wallabies. Front. Genet. 8, 10 (2017).
Garrido-Ramos, M. A. Satellite DNA: An evolving topic. Genes 8, 230 (2017).
Ahmad, S. et al. Consequence of paradigm shift with repeat landscapes in reptiles: Powerful facilitators of chromosomal rearrangements for diversity and evolution. Genes 11, 827 (2020).
Talbert, P. B. & Henikoff, S. The genetics and epigenetics of satellite centromeres. Genome Res. 32, 608–615 (2022).
Peona, V. et al. Satellite DNA evolution in Corvoidea inferred from short and long reads. Mol. Ecol. 32, 1288–1305 (2023).
Kulak, M. et al. Genome organization of major tandem repeats and their specificity for heterochromatin of macro-and microchromosomes in Japanese quail. Genome 65, 391–403 (2022).
Sena, R. et al. Identification and characterization of satellite DNAs in two-toed sloths of the genus Choloepus (Megalonychidae, Xenarthra). Sci. Rep. 10, 19202 (2020).
Valeri, M. et al. First description of a satellite DNA in manatees’ centromeric regions. Front. Genet. 12, 694866 (2021).
Gutiérrez, J. et al. Satellitome Analysis on Talpa aquitania Genome and Inferences about the satDNAs Evolution on Some Talpidae. Genes 14, 117 (2022).
Kretschmer, R. et al. Satellitome analysis illuminates the evolution of ZW sex chromosomes of Triportheidae fishes (Teleostei: Characiformes). Chromosoma 131, 29–45 (2022).
Toma, G. et al. Cytogenetics meets genomics: Cytotaxonomy and genomic relationships among color variants of the Asian Arowana Scleropages formosus. Int. J. Mol. Sci. 24, 9005 (2023).
Utsunomia, R. et al. Satellitome landscape analysis of Megaleporinus macrocephalus (Teleostei, Anostomidae) reveals intense accumulation of satellite sequences on the heteromorphic sex chromosome. Sci. Rep. 9, 5856 (2019).
Gatto, K. et al. Sex chromosome differentiation in the frog genus Pseudis involves satellite DNA and chromosome rearrangements. Front. Genet. 9, 301 (2018).
Da Silva, J. et al. Analysis in Proceratophrys boiei genome illuminates the satellite DNA content in a frog from the Brazilian Atlantic forest. Front. Genet. 14, 1101397 (2023).
Ferretti, A. et al. High dynamism for neo-sex chromosomes: Satellite DNAs reveal complex evolution in a grasshopper. Heredity 125, 124–137 (2020).
Crepaldi, C. & Parise-Maltempi, P. P. Heteromorphic sex CHROMOSOMES AND THEIR DNA Content in Fish: An insight through satellite DNA accumulation in Megaleporinus elongatus. Cytogenet. Genome Res. 160, 38–46 (2020).
Traut, W. et al. Molecular differentiation of sex chromosomes probed by comparative genomic hybridization. Chromosoma 108, 173–180 (1999).
Symonová, R., Sember, A. & Majtánová, Z. et al. Characterization of fish genomes by GISH and CGH. In Fish Cytogenetic Techniques (Ozouf-Costaz, C., Pisano, E., Foresti, F., de Almeida, L., Eds), pp. 118–131 (2015).
De Freitas, N. L. et al. Early stages of XY sex chromosomes differentiation in the fish Hoplias malabaricus (Characiformes, Erythrinidae) revealed by DNA repeats accumulation. Curr. Genomics. 19, 216–226 (2018).
Giardini, M. et al. Comparative Genomic Hybridization (CGH) in Anastrepha fraterculus sp. 1 reveals active divergence between sex chromosomes (2023).
Giannoni, M. L., Ferrari, I. & Giannoni, M. A. Cytogenetic analysis of Thrush (Turdus, Muscicapidae) species. Revista Brasileira de Genética 13, 33–46 (1990).
Gerbault-Seureau, M., Fuchs, J. & Dutrillaux, B. High BrdU sensitivity of Passeriformes chromosomes: Conservation of BrdU-sensitive fragile sites on their Z chromosomes during evolution. Cytogenet. Genome. Res. 157, 158–165 (2019).
Costa, A. et al. Comparative cytogenetics in three species of Wood-Warblers (Aves: Passeriformes: Parulidae) reveal divergent banding patterns and chromatic heterogeneity for the W chromosome. Caryologia. 74, 43–51 (2021).
Kretschmer, R. et al. Multidirectional chromosome painting in Synallaxis frontalis (Passeriformes, Furnariidae) reveals high chromosomal reorganization, involving fissions and inversions. Comp. Cytogenet. 12, 97–110 (2018).
Charlesworth, B. The evolution of chromosomal sex determination. Novartis. Found. Symp. 244, 207–219 (2002).
Poltronieri, J., Marquioni, V. & Bertollo, L. A. Comparative chromosomal mapping of microsatellites in Leporinus species (Characiformes, Anostomidae): Unequal accumulation on the W chromosomes. Cytogenet. Genome. Res. 142, 40–45 (2014).
Singh, L., Purdom, I. F. & Jones, K. W. Sex chromosome associated satellite DNA: Evolution and conservation. Chromosoma 79, 137–157 (1980).
Cermak, T. et al. Survey of repetitive sequences in Silene latifolia with respect to their distribution on sex chromosomes. Chromosome Res. 16, 961–976 (2008).
Kubat, Z., Hobza, R., Vyskot, B. & Kejnovsky, E. Microsatellite accumulation on the Y chromosome in Silene latifolia. Genome 51, 350–356 (2008).
Pokorná, M., Kratochvíl, L. & Kejnovský, E. Microsatellite distribution on sex chromosomes at different stages of heteromorphism and heterochromatinization in two lizard species (Squamata: Eublepharidae: Coleonyx elegans and Lacertidae: Eremias velox). BMC. Genetics. 12, 1–7 (2011).
Palacios-Gimenez, O. et al. High-throughput analysis of the satellitome revealed enormous diversity of satellite DNAs in the neo-Y chromosome of the cricket Eneoptera surinamensis. Sci. Rep. 7, 6422 (2017).
Cioffi, M. B., Kejnovsky, E. & Bertollo, L. A. The chromosomal distribution of microsatellite repeats in the genome of the wolf fish Hoplias malabaricus, focusing on the sex chromosomes. Cytogenet. Genome. Res. 132, 289–296 (2010).
Cioffi, M. B. et al. Differentiation of the XY sex chromosomes in the fish Hoplias malabaricus (Characiformes, Erythrinidae): Unusual accumulation of repetitive sequences on the X chromosome. Sex. Dev. 4, 176–185 (2010).
De Oliveira, T. et al. Genomic Organization of Repetitive DNA in Woodpeckers (Aves, Piciformes): Implications for Karyotype and ZW Sex Chromosome Differentiation. PLoS One 12, e0169987 (2017).
Furo, I. et al. Chromosomal Mapping of Repetitive DNAs in Myiopsitta monachus and Amazona aestiva (Psittaciformes, Psittacidae), with emphasis on the sex chromosomes. Cytogenet. Genome. Res. 151, 151–160 (2017).
Barcellos, S. et al. Karyotype evolution and distinct evolutionary history of the W chromosomes in Swallows (Aves, Passeriformes). Cytogenet. Genome. Res. 158, 98–105 (2019).
Kretschmer, R. et al. Chromosomal analysis in Crotophaga ani (Aves, Cuculiformes) reveals extensive genomic reorganization and an unusual Z-autosome robertsonian translocation. Cells 10, 4 (2021).
International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695–716 (2004).
De Souza, M. et al. Repeat sequence mapping shows different W chromosome evolutionary pathways in two Caprimulgiformes Families. Birds 1, 19–34 (2020).
Yamada, K., Nishida-Umehara, C. & Matsuda, Y. Molecular and cytogenetic characterization of site-specific repetitive DNA sequences in the Chinese soft-shelled turtle (Pelodiscus sinensis, Trionychidae). Chromosome. Res. 13, 33–46 (2005).
Srikulnath, K. et al. Chromosomal localization of the 18S–28S and 5S rRNA genes and (TTAGGG) n sequences of butterfly lizards (Leiolepis belliana belliana and Leiolepis boehmei, Agamidae, Squamata). Genet. Mol. Biol. 34, 583–586 (2011).
Charlesworth, B., Sniegowski, P. & Stephan, W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 371, 215–220 (1994).
Jurka, J. et al. Repetitive sequences in complex genomes: structure and evolution. Annu. Rev. Genomics Hum. Genet. 8, 241–259 (2007).
Biscotti, M. A., Olmo, E. & Heslop-Harrison, J. S. Repetitive DNA in eukaryotic genomes. Chromosome. Res. 23, 415–420 (2015).
Muller, H. Genetic variability, twin hybrids, and constant hybrids, in a case of balanced lethal factors. Genetics 3, 422–499 (1918).
Rice, W. Sex chromosomes and the evolution of sexual dimorphism. Evolution 735–742 (1984).
Charlesworth, B., Jordan, C. Y. & Charlesworth, D. The evolutionary dynamics of sexually antagonistic mutations in pseudoautosomal regions of sex chromosomes. Evolution 68, 1339–1350 (2014).
White, M. J. D. Animal cytology and evolution, 3rd edn, pp. 961 (Cambridge University Press, Cambridge, 1973).
Graves, J. Sex chromosome specialization and degeneration in mammals. Cell. 124, 901–914 (2006).
Charlesworth, D. Plant contributions to our understanding of sex chromosome evolution. New Phytol. 208, 52–65 (2015).
Pigozzi, M. I. & Solari, A. J. The ZW pairs of two paleognath birds from two orders show transitional stages of sex chromosome differentiation. Chromosome. Res. 7, 541–551 (1999).
Wang, Z. et al. Temporal genomic evolution of bird sex chromosomes. BMC. Evol. Biol. 14, 1–12 (2014).
Ogawa, A., Murata, K. & Mizuno, S. The location of Z-and W-linked marker genes and sequence on the homomorphic sex chromosomes of the ostrich and the emu. Proc. Natl. Acad. Sci. 95, 4415–4418 (1998).
Nishida-Umehara, C. et al. Differentiation of Z and W chromosomes revealed by replication banding and FISH mapping of sex-chromosome-linked DNA markers in the cassowary (Aves, Ratitae). Chromosome. Res. 7, 635–640 (1999).
Tsuda, Y. et al. Comparison of the Z and W sex chromosomal architectures in elegant crested tinamou (Eudromia elegans) and ostrich (Struthio camelus) and the process of sex chromosome differentiation in palaeognathous birds. Chromosoma 116, 159–173 (2007).
Perrin, N. Sex reversal: A fountain of youth for sex chromosomes?. Evolution 63, 3043–3049 (2009).
Furman, B. L. et al. A frog with three sex chromosomes that co-mingle together in nature: Xenopus tropicalis has a degenerate W and a Y that evolved from a Z chromosome. PLoS Genet. 16, e1009121 (2020).
Meisel, R. P. Evolution of sex determination and sex chromosomes: A novel alternative paradigm. BioEssays 42, 1900212 (2020).
Perrin, N. Sex-chromosome evolution in frogs: What role for sex-antagonistic genes?. Philos. Trans. R. Soc. b 376, 20200094 (2021).
Kratochvíl, L. et al. Expanding the classical paradigm: What we have learnt from vertebrates about sex chromosome evolution. Philos. Trans. R. Soc. b: Biol. Sci. 376, 20200097 (2021).
Schubert, I. & Lysak, M. A. Interpretation of karyotype evolution should consider chromosome structural constraints. Trends Genet. 27, 207–216 (2011).
Pala, I. et al. Evidence of a neo-sex chromosome in birds. Heredity 108, 264–272 (2012).
Sigeman, H. et al. Repeated sex chromosome evolution in vertebrates supported by expanded avian sex chromosomes. Proc. R. Soc. B: Biol. Sci. 286, 20192051 (2019).
Garnero, A. V. & Gunski, R. J. Comparative analysis of the karyotype of Nothura maculosa and Rynchotus rufescens (Aves: Tinamidae). A case of chromosomal polymorphism. The Nucleus 43, 64–70 (2000).
Sumner, A. T. A simple technique for demonstrating centromeric heterochromatin. Exp. Cell. Res. 75, 304–306 (1972).
Sambrook, J. & Russel, D. W. Molecular cloning: A laboratory manual. Cold Spring Harb. 3, 2100 (2001).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Novák, P., Neumann, P. & Macas, J. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat. Protoc. 15, 3745–3776 (2020).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864 (2011).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-3.0 1996–2010 http://www.repeatmasker.org (2020).
Fu, L. et al. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Nascimento, M. et al. PHYLOViZ 2.0: Providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 33, 128–129 (2017).
Kretschmer, R. Santos, M. S., Furo, I. O., Oliveira, E. H. C. & Cioffi, M.B. FISH–in birds. In Cytogenetics and Molecular Cytogenetics (ed. Liehr, T.) (CRC Press, 2023).
Zwick, M. et al. A rapid procedure for the isolation of C0t–1 DNA from plants. Genome 40, 138–142 (1997).
Yano, C. F., Bertollo, L. A. C. & Cioffi, M. B. Fish-FISH: Molecular cytogenetics in fish species. In Fluorescence In Situ Hybridization (FISH)- Application Guide (ed. Liehr, T.) 429–444 (Springer, 2017).
Levan, A., Fredga, K. & Sandberg, A. Nomenclature for centromeric position on chromosomes. Hereditas 52, 201–220 (1964).
Acknowledgements
The authors would like to thank Dr. Anderson J. B. Gomes for his support in collecting the Turdus leucomelas individual from Belém, Pará, Brazil, and Dr. Thales Renato Ochotorena de Freitas for his support in performing the cell culturing.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by São Paulo Research Foundation (FAPESP) grant 2022/14584-3 (GMS); 2022/00427-3 (GAT), 2023/00955-2 (MBC); by the state of Rio Grande do Sul research support foundation (FAPERGS) (22/2551-0000572-1) (RK) and Brazilian National Council for Scientific and Technological Development (CNPq), grant number 302928/2021-9 (MBC), 407285/2021-0 and 306717/2022-0 (ADVG), and 304781/2022-3 (EHCO).
Author information
Authors and Affiliations
Contributions
GMS, RK, RU, and MBC conceived and designed research. GMS, GAT, AMO, RK, GAD, PGS, RZS, and CAGG conducted experiments. GMS, RK, GAT, GAD, RU, TL and MBC analyzed the data. GMS, RK, GAT, AMO, GAD, PGS, RZS, CAGG, RJG, ADVG, EHCO, FPF, RU, TL and MBC wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Sample was approved by the Brazilian Environmental Agency ICMBIO/SISBIO (Licenses 61047-4, 44173-1, and 68443-2) and SISGEN (A96FF09). All experiments followed the guidelines and were approved by the Ethics Committee on Animal Experimentation of the Federal University of Pampa (018/2014 and 019/2020). The authors complied with ARRIVE guidelines.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Souza, G.M., Kretschmer, R., Toma, G.A. et al. Satellitome analysis on the pale-breasted thrush Turdus leucomelas (Passeriformes; Turdidae) uncovers the putative co-evolution of sex chromosomes and satellite DNAs. Sci Rep 14, 20656 (2024). https://doi.org/10.1038/s41598-024-71635-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-71635-5
- Springer Nature Limited