
Abstract
In this paper, the crystal geometry, electronic structure, lattice vibration, Infrared and Raman spectra of ternary layered borides M3AlB2 (M = Ti, Zr, Hf, Ta) are studied by using first principles calculation method based on the density functional theory. The electronic structure of M3AlB2 indicates that they are all electrical conductors, and the d orbitals of Ti, Zr, Hf, and Ta occupy most of the bottom of the conduction band and most of the top of the valence band. Al and B have lower contributions near their Fermi level. The lightweight and stronger chemical bonds of atom B are important factors that correspond to higher levels of peak positions in the Infrared and Raman spectra. However, the vibration frequencies, phonon density of states, and peak positions of Infrared and Raman spectra are significantly lower because of heavier masses and weaker chemical bonds for M and Al atoms. And, there are 6 Infrared active modes A2u and E1u, and 7 Raman active modes, namely A1g, E2g, and E1g corresponding to different vibration frequencies in M3AlB2. Furthermore, the Infrared and Raman spectra of M3AlB2 were obtained respectively, which intuitively provided a reliable Infrared and Raman vibration position and intensity theoretical basis for the experimental study.
Similar content being viewed by others


Introduction
It’s interesting to note that MAB phase materials have gained a lot of attention in recent years due to their similar structure to MAX phase1,2,3,4,5,6,7. These materials possess unique properties such as a nano-layered structure, diverse chemical bond types, high hardness, high elastic modulus, high conductivity, high thermal conductivity, and good resistance to high-temperature oxidation, making them highly suitable for various applications8,9,10,11. They can be used to manufacture high-temperature furnaces, aerospace spacecraft components, automotive engine components, nuclear reactor materials, and in other fields12,13,14,15. In addition, it's worth mentioning that the good thermal conductivity exhibited by MAB phase materials makes them highly suitable for the manufacturing of products like electronic components and radiators4,16. These materials also exhibit common characteristics of both ceramics and metals, making them a highly researched area in the field of structural ceramics. The unique properties of MAB phase materials generate significant interest in the ternary layered transition metal boride MAB phase, making it a subject of ongoing research17,18,19,20. At present, the orthorhombic MAB phases Ru2ZnB2, Ru3Al2B2, Mo2AlB2, and Cr4AlB2 have been successfully prepared. And MAlB (M = Ti, Hf, V, Nb, Ta, Cr, Mo, W, Mn, Tc), M2AlB2 (Sc, Ti, Zr, Hf, V, Cr, Mo, W, Mn, Tc, Fe, Rh, Ni), M3Al2B2 (M = Sc, T, Zr, Hf, Cr, Mn, Tc, Fe, Ru, Ni), M3AlB4 (M = Sc, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Mn, Fe), and M4AlB6 (M = Sc, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo) were successively studied21,22,23,24.
However, compared to MAX phases, the study of MAB phases is still in its early stages. Numerical simulation and theoretical analysis based on the first-principles method based on the density functional theory can play an important role in understanding and predicting the phase structure and properties of MAB. In order to gain a deeper understanding of the structure and properties of many MAB phase compounds, it is extremely urgent to employ the first-principles method based on density functional theory to study these materials. This approach will enable the rapid screening and theoretical design of materials with promising engineering application prospects7,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43. Currently, there has been research on Ti3AlB2 material, but there is still a lack of corresponding research on materials with similar structures or chemical formulas, such as Zr3AlB2, Hf3AlB2, Ta3AlB2, and other related materials. Judging from the limited theoretical studies, these material systems have similar electronic band structures, indicating that they are likely to exhibit similar physical properties44,45. Among these materials, Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 can form a series of layered materials with a similar structure and chemical composition.
Meanwhile, it is crucial to synthesize these materials and accurately determine their structure from experimental standpoint. And the Infrared or Raman spectroscopy is an effective method for distinguishing these similar crystal structures46. However, conducting experimental studies on their Infrared or Raman spectra is time-consuming, labor-intensive, and requires a large workload. We use the first-principles calculations to investigate the Infrared and Raman spectra of these four similar materials M3AlB2, which will provide us a theoretical reference for subsequent experimental work. Because the Infrared and Raman spectroscopy can provide a variety of information for the study of materials, it has been extended to a variety of disciplines and widely used. Nevertheless, the research on their Infrared and Raman spectrum is extremely few. Therefore, it is of great significance to study the Infrared and Raman spectrum and their vibration mode of M3AlB2.
Method and computational details
All first-principles calculations were performed by Vienna Ab-initio Simulation Package (VASP) with Plane-wave DFT methodology in this study47. The use of different exchange correlation functionals has been demonstrated to have a minor influence on the formation enthalpy48,49,50,51. And, the electronic exchange–correlation interactions were described by the generalized gradient approximation (GGA) with Perdew Burke Ernzerhof (PBE) functional52,53,54,55 in the paper.
In the calculation process, a plane wave cut-off energy of 700 eV was utilized. Additionally, integrations in the Brillouin zone were conducted using a 15 × 15 × 2 Monkhorst–Pack as specific k-points, while a K-mesh of 0.03 was chosen throughout the calculation56. Energy and force convergences are 1 × 10–8 eV and − 1 × 10–7 eV/Å. In calculations, the Density Functional Perturbation Theory (DFPT) approach was employed in both Phonopy57,58 and VASP to determine the second-order Interatomic Force Constants (IFCs). DFPT is a method for calculating the response of physical quantities to external fields using linear response theory. It can be used to calculate the force constant matrix. The Phono3py59,60 code generates the displacement structure with irreducible representation along the center q-point to produce positive and negative displacement structures. Finally, the dielectric constant is calculated. Only the structure with irreducible representation is selected for dielectric constant calculation to reduce the calculation burden61,62. And Phonopy-Spectroscopy software were utilized in the procedures to calculate the IR and Raman spectra63. Hessian matrix data and born effective charges also have to be taken into account. Moreover, the focus of this research was primarily on the atom vibration image of the structure's Γ point active Infrared and Raman spectra associated to phonons. Additionally, 3d24s2 for Ti, 4d25s2 for Zr, 5d26s2 for Hf, 5d36s2 for Ta, 3s23p1 for Al, and 2s22p1 for B were considered as valence states in the paper. The software packages Pymatgen and Vaspvis were used in the study64,65.
Lattice dynamics has played a significant role in understanding certain physical characteristics of Infrared and Raman spectra. We employ the Phonopy package and VASP to obtain phonon dispersion and phonon density of states corresponding to lattice vibration in the paper. The phonon vibrational frequencies via DFPT method. Phonon frequencies and eigenvectors are computed in the harmonic limit using the second-order force constant matrix Φαβ(jl, j′l′), the elements of which are the change in force in the α Cartesian direction acting on atom j in unit cell l, in response to the displacement of atom j′ in unit cell l′ in the β direction. The phonon frequencies are the eigenvalues of the dynamical matrix at a given wavevector q:
mj refer to the atomic masses. The long-rang Coulomb interactions lead to non-analytic corrections in the limit q → 0, which cause the splitting of the longitudinal optic (LO) and transverse optic (TO) modes66:
\({Z}_{i,\alpha \beta }^{*}\) is the derivative of the cell polarization along the Cartesian direction (x, y, z) α with respect to the atomic displacement of atom i along β and defined as
The dynamical charges Z* are called “Born effective-charge tensors”, and can be calculated using perturbation theory. The calculations have been performed by using the LESPILON = True keyword of VASP. r is the displacement of the atom i. Ω is the volume of the cell and e is the electronic charge67. The Z* tensors were computed using DFPT as implemented in VASP. Ω represents the volume of the primitive unit cell, and e stand for the elementary charge constant. Within the dipole approximation, the Infrared intensity of an eigenmode s can be expressed as the square of the Born effective-charge and eignevector \({X}_{\beta }\left(s,j\right)\) as:
\({X}_{\beta }\left(s,j\right)\) represents the normalized vibrational eigenvector of the jth phonon mode of the sth atom in the unit cell. Raman activity tensors of an eigenmode are evaluated by taking the derivative of the high-frequency macroscopic dielectric constant ε∞ with respect to the normal mode amplitude Q(s) using a central difference scheme is:
ε∞ can be calculated by DFPT method and can be found in the OUTCAR with careful identification. At the same time, LESPILON = True is necessary. Q is the normal-mode coordinate at the Γ-point and is defined by
u(s, j) represents the atomic displacement. Phonopy and Phonopy-Spectroscopy were used to compute phonon frequencies, as well as Raman intensities68,69. The Raman intensities are then defined as:
In Eq. (8), the notation \({I}_{{\alpha \beta }}\) was used in place of \({I}_{\text{Raman},{\alpha \beta }}\left(s\right)\). With mj in amu, and Z* in e, the calculated IR intensities will have units e2 amu−1. With Q in amu1/2Å, the elements of the Raman-activity tensor have units of Å2 amu−1/2, and the units of the scalar Raman intensity. And the square of the Raman activity are Å4amu−1 at last 63,70.
Results and discussion
Structure
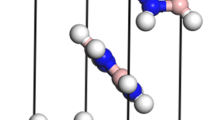
In Fig. 1, it is shown that the schematic graphs of crystal structure for M3AlB2, all of which belong to the P63/mmc (No. 194) space group. Each unit cell contains two primitive cells, resulting in a total of 12 atoms in each conventional unit cell. And the compounds Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 can be considered as 312 phases in the Mn+1AlBn phase structure, which is consistent with references71,72. In the M3AlB2 structure, the Al layer atoms are inserted between the adjacent M layer atoms, and the B atoms fill the octahedral interstitial positions of the former. The Al atom is located at the center of the quadrangular prism, surrounded by the M-layer atoms. Because the space at this position is larger than the octahedral gap, it can accommodate larger atoms such as Al.

Schematic graphs of M3AlB2 crystal structure.
Lattice constant
As shown in Table 1, the lattice constants of Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 have been calculated. It can be observed that the calculation results of Ti3AlB2 are in good agreement with the reference results72.
In general, the lattice constants “a” and “c” of the M3AlB2 phase decrease with the increase in the atomic radius of the transition metal element M, but have little relationship with the number of d electron layers. The lattice constant of the 3d compound is significantly smaller than that of the 4d and 5d compounds, but the difference between the 4d and 5d compounds is minimal.
At the same time, for a series of compounds with a given M3AlB2 value, the c/a ratio falls within a very narrow range and is less affected by M atoms. We assume that the MAX phase is an interstitial compound, in which A and X atoms fill the interstices between M atoms. In the 312 phase, the c-axis direction comprises 6 layers of M atoms. Without considering the ideal lattice distortion caused by the addition of A and X atoms, the c-axis length should be 6 times that of the a-axis length. That is to say, the c/a ratio of the 312 phase should be 6, and other researchers have also found a similar trend in their investigation34. However, in reality, the presence of Al and B atoms inevitably impacts the lattice, causing the actual c/a ratio to deviate slightly from the ideal value. As shown in Table 1, the c/a ratios of Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 are 6.231742, 6.114347, 6.074749, and 6.143366, respectively. This aligns with our speculation closely.
Electronic band structure
The projected band structures and density of states near the Fermi energy (Ef) for Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 is depicted in Fig. 2. It is reveal that the energy bands of these compounds intersect the Fermi surface, suggesting that Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 exhibit electrical conductivity9. This has typical characteristics of MAX phase. It can be seen from the energy band that Ti, Zr, Hf, and Ta are dominant in Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 materials near the Fermi level. It is also evident that the d orbitals of Ti, Zr, and Hf predominantly occupy the lower portion of the conduction band and the upper portion of the valence band. Al has a minor contribution at the top of the valence band, while it has a very small impact at the bottom of the conduction band, which is almost negligible. B also makes a specific contribution at the bottom of the conduction band and the top of the valence band, primarily in the p orbital. The Fermi level in Ta3AlB2 is predominantly occupied by the d orbital of the Ta element, while the contribution of Al and B near the Fermi level of Ta3AlB2 is minimal and negligible.

Projected band structures and density of states for Ti3AlB2 (a), Zr3AlB2 (b), Hf3AlB2 (c), Ta3AlB2 (d).
It can be observed from these figures that the fundamental characteristics in the density of states for the same series of compounds, Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2, are very similar. Since it is far from the Al atom, the d orbitals of M atom primarily hybridize with the B-p state, and there is some hybrid state with Al atoms. Thus, the M atom forms a covalent bond with the B atom. At the same time, these compounds contain weak M–M covalent bonds. The hybrid state is in a high-energy range, the M–M bond is weaker than the M–B bond.
Combined with Fig. 3, it can be observed that the lower energy band primarily originates from the B-s state. The spikes in this range are primarily caused by the strong hybridization between the M-d state and the B-p state, specifically the formation of the M–B covalent bond. The energy band at the top of the valence band is the result of hybridization between the M-d state and the Al-p state, which corresponds to M–Al covalent bond. The energy range corresponding to the M–B bond is significantly lower than that of the M–M bond. And, M–B, M–Al bond is stronger than M–M bond.

Orbital-weighted band structures and projected density of states of Ti3AlB2 (a), Zr3AlB2 (b), Hf3AlB2 (c), and Ta3AlB2 (d).
As shown in Fig. 3, we find that all the d orbitals (dxy, dxz, dyz, dx2-y2, and dz2) in Ti near the Fermi surface of Ti3AlB2 are given from the band and projected density of states of Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2. And the contribution of Ti(dyz) and Ti(dx2-y2) is particularly pronounced. The contribution of Al is very small here, and only pz of B contributes to the density of states. All the d orbitals (dxy, dxz, dyz, dx2-y2, and dz2) in Zr near the Fermi surface of Zr3AlB2 contribute. Among these, Zr(dyz) and Ti(dx2-y2) contribute more significantly, while Al contributes very little. Additionally, only the pz orbital of B contributes here. All the d orbitals (dxy, dxz, dyz, dx2-y2, and dz2) in Hf near the Fermi surface of Hf3AlB2 contribute. Among these, Hf(dyz) and Ti(dx2-y2) contribute more significantly, while Al contributes very little. B has py and pz orbitals that contribute, with a larger contribution. All the d orbitals (dxy, dxz, dyz, dx2-y2, and dz2) in Ta near the Fermi surface of Ta3AlB2 contribute, with Ta(dx2-y2), Ta(dz2), and Ta(dxz) making the most significant contributions, while Al and B contribute minimally.
Lattice vibration
The lattice vibration spectra of Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 are depicted along the direction of high symmetry points in the Brillouin zone as shown in Fig. 4. Firstly, there is no phonon vibration with negative frequency in the phonon dispersion in the studied compounds, which indicates that there is no imaginary frequency and Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 are stable.

Phonon dispersion of Ti3AlB2 (a), Zr3AlB2 (b), Hf3AlB2 (c), Ta3AlB2 (d).
Secondly, it can be observed from these figures that Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 compounds have phonon band gaps. For Ti3AlB2, the phonon band gap is 306.97 cm−1–465.33 cm−1. To Zr3AlB2, the phonon band gap is 276.67 cm−1–406.63 cm−1. And there are phonon band gaps of 277.33 cm−1–441.66 cm−1 and 340.66 cm−1–589.00 cm−1 for Hf3AlB2, and Ta3AlB2 respectively. Furthermore, the phonon vibrations of M and Al are below the phonon band gap, while the phonon vibrations above belong to B atoms. Similar to the electronic state, the flat band in the phonon spectrum corresponds to the peak in the phonon state density as shown in Fig. 5. This indicates that the lattice vibration exhibits clear localization characteristics. The frequencies of the optical branches near the long-wave (Γ point) are significantly different, indicating a noticeable ionic character in these compounds.

Phonon density of states of Ti3AlB2 (a), Zr3AlB2 (b), Hf3AlB2 (c), and Ta3AlB2 (d).
Thirdly, compared to M and Al atoms, the mass of B atoms is significantly lower. More importantly, B atoms are connected to the lattice through very strong M–B bonds. This results in the vibration frequency of the B atom being significantly higher than that of other atoms. The phonon energy intervals corresponding to M and Al atoms are more complex. On the one hand, based on the electronic structure and chemical bonding mentioned above, M atoms are connected to the lattice through strong M–B bonds and weak M–Al bonds. Therefore, the binding of M atoms to the lattice should be stronger than that of Al atoms. On the other hand, the M atom is much heavier than the Al atom. These factors result in the phonon states corresponding to M and Al atoms being at lower energy levels, but the relative energy level changes of the two are more complicated. And the B atoms are at higher energy levels and form phonon band gaps with them.
In the M3AlB2 compound, the phonon density of states (PHDOS) below the phonon band gap primarily corresponds to the lattice vibration dynamics of M and Al atoms, while above the band gap, it relates to the lattice vibration of B atoms. There is a sharp peak in the middle in the PHDOS of Zr3AlB2, Hf3AlB2, and Ta3AlB2 due to the vibration of Al atoms. And the proportion of Ti, Zr, Hf, and Ta in PHDOS of Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 respectively, increases at low frequencies.
It is assumed that an increase in the number of d electron layers results in and its atom mass, leading to a decrease in the lattice vibration frequency for M atom. For Al and B atoms, although the changing trend of the phonon frequency of the Al atom is similar to that of the M atom, the decisive factor should be the bond strength of M–Al, and the changing trend of the phonon frequency of the B atom is determined by the bond strength of M–B. Among the M3AlB2 phase compounds, M–Al bond exhibits the lowest chemical bond stiffness in each system, with a stiffness value approximately 1/3–1/2 of the corresponding M-B bond stiffness. Additionally, the M-B bond closest to the Al atom demonstrates the highest stiffness. The results in Fig. 5 also indirectly indicate the changing trend of M–Al bond and M–B bond strength.
IR and Raman spectra
Theoretically, Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 belongs to point group D6h and has 36 vibration modes at the center of the Brillouin zone respectively. The irreducible representation of their vibration modes73,74,75 at the Γ point is in Eq. (9):
The acoustic branch has two irreducible representation. E1u is doubly degenerate, and the acoustic branch contains 3 lattice waves. And the acoustic branch is as following Eq. (10):
There are 22 types of irreducible representation in the optical branch, among which E1g, E2g, E1u, and E2u are doubly degenerate, corresponding to 33 lattice waves. The optical branch is as following Eq. (11):
Therefore, there are a total of 36 lattice waves. And Table 2 presents the frequencies of phonon vibration modes at the center of the Brillouin zones for Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2. E1g, E2g, E1u, and E2u are doubly degenerate, and the frequencies of the corresponding phonon vibration modes are identical.
As shown in Table 2, the highest phonon frequency of Ti3AlB2 is approximately 609.0 cm−1, while Zr3AlB2 has a highest phonon frequency of about 536.5 cm−1. Hf3AlB2 exhibits a highest phonon frequency of around 591.1 cm−1, and Ta3AlB2 has the highest phonon frequency at approximately 673.2 cm−1. Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2. Although the atom mass of Ti, Zr, Hf, and Ta increases in turn, the highest phonon frequency may be related to atom mass, bond length, and bond angle. Therefore, the highest phonon frequency of Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 does not increase completely in accordance with the atom mass of Ti, Zr, Hf, and Ta. It can be seen from the table that for similar crystal structures (Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2), the Mulliken symbol order corresponding to the characteristic table is not necessarily the same. This discrepancy may be attributed to the distinct nature of the Ti, Zr, Hf, and Ta elements, leading to differences in the fine structure of the crystal. This may be one of the reasons why people use Raman spectroscopy as a “fingerprint spectrum” to distinguish between different crystals and even very similar crystal fine structures.
According to the character table of the point group, A2u and E1u are Infrared-active modes in the optical properties of Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2, totaling 6 modes: 3A2u + 3E1u. The Raman-active modes include A1g, E2g, and E1g, totaling 7 modes: 2A1g + 3E2g + 2E1g. Based on structural optimization, we calculated the atom vibration modes, as well as the Infrared and Raman spectra of Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 materials, as depicted in Figs 6, 7, 8, and 9.

Atom vibration diagram (a) (‘I’ represents Infrared activity, ‘R’ represents Raman activity), Infrared and Raman spectra of Ti3AlB2 (b and c).

Atom vibration diagram (a), Infrared and Raman spectra of Zr3AlB2 (b and c).

Atom vibration diagram (a), Infrared and Raman spectra of Hf3AlB2 (b and c).

Atom vibration diagram (a), Infrared and Raman spectra of Ta3AlB2 (b and c).
The atom vibration diagrams of the Infrared and Raman modes in the Ti3AlB2 crystal are depicted in Fig. 6a. It can be seen from Fig. 6a that Ti3AlB2 has 6 Infrared modes: E1u(150.2)I, A2u(238.2)I, E1u(252.5)I, A2u(306.3)I, E1u(482.7)I, A2u(588.6)I. In addition to E1u(150.2)I, the low-frequency Infrared vibration is primarily attributed to the vibration of Ti and Al atoms. In contrast, the high-frequency vibration is primarily attributed to the vibration of B atoms. At the same time, the B atom participates in all Infrared vibrations.
E1u(150.2)I corresponds to the anti-direction bending vibration in the basal plane of the B atom, E1u(482.7)I corresponds to the anti-direction shear vibration in the basal plane of the B atom, and A2u(588.6)I corresponds to the longitudinal vibration in the same direction in the basal plane of the B atom. The A2u(238.2)I and A2u(306.3)I modes represent the longitudinal vibration of Ti, Al, and B atoms along the c-axis direction, while the E1u(252.5)I mode corresponds to the shear vibration of Ti, Al, and B atoms along the c-axis direction. Because titanium Ti and Al atoms are relatively heavy, the frequency of these three vibration modes is always low. The E1u(482.7)I and A2u(588.6)I modes correspond to the vibration of the B atom, respectively. The mass of the B atom is lower, resulting in a higher corresponding frequency.
It can also be seen that Ti3AlB2 has 7 Raman active modes: E2g(101.3)R, E1g(161.0)R, E2g(180.1)R, A1g(227.2)R, E2g(519.2)R, E1g(527.4)R, A1g(609.0)R from Fig. 6a. It is evident that the atom motion associated with the Raman vibration mode primarily involves elongation and bending vibration of covalent bond chains of M–Al–B. Among them, the vibrations of Ti and Al atoms commonly result in the E2g(101.3)R, E1g(161.0)R, E2g(180.1)R, and A1g(227.2)R vibrations at low frequency. The lower frequency is due to the greater mass of Ti and Al atoms and the deformation of the weaker M–Al bond during its bending vibration. At high frequencies, the E2g(519.2)R, E1g(527.4)R, and A1g(609.0)R modes are mainly affected by the vibration of B atoms, with the vibration of Ti and Al playing a minor role. This could be attributed to the strong M–B bond and the relatively low mass of the B atom, which results in higher vibration frequencies in these stretching vibration modes. These findings are consistent with the phonon density of states diagram Fig. 5a. The frequencies E2g(101.3)R, E1g(161.0)R, E2g(180.1)R, E2g(519.2)R, and E1g(527.4)R indicate that atoms in the Ti3AlB2 crystal vibrate in opposite directions perpendicular to the c axis. On the other hand, A1g(227.2)R and A1g(609.0)R show that the atoms in the Ti3AlB2 crystal vibrate in opposite or same directions along the c axis, respectively. Figure 6b shows the position and vibration intensity of the specific peak of the Infrared active vibration mode. Figure 6b illustrates that the A2u(238.2)I, E1u(252.5)I, E1u(482.7)I, A2u(588.6)I modes in the Ti3AlB2 structure exhibit strong Infrared vibration intensity, while the E1u(150.2)I and A2u(306.3)I modes have lower Infrared vibration intensity. And Fig. 6c provides their specific peak positions and intensities. The higher vibration intensities can be observed in the Raman vibration modes of Ti3AlB2, including E2g(101.3)R, E1g(161.0)R, E2g(180.1)R, A1g(227.2)R, E2g(519.2)R, E1g(527.4)R, and A1g(609.0)R, as shown in Fig. 6c.
The atom vibration of Infrared and Raman modes are shown in Fig. 7a. Zr3AlB2 has 6 Infrared modes: E1u(138.8)I, E1u(178.7)I, A2u(187.6)I, A2u(271.4)I, E1u(405.9)I, A2u(503.7)I. The B atom also participates in all the Infrared vibrations. In addition to E1u(138.8)I, the low-frequency Infrared vibration is dominated by the vibration of Zr and Al atoms, while the high-frequency vibration is dominated by the vibration of B atoms. E1u(138.8)I corresponds to the anti-direction bending vibration in the basal plane of B atom, E1u(405.9)I corresponds to the anti-direction shear vibration in the basal plane of B atom, and A2u(503.7)I corresponds to the longitudinal vibration in the same direction in the basal plane of B atom. A2u(187.6)I, A2u(271.4)I are the longitudinal vibration of Ti, Al and B atoms along the c-axis direction, and E1u(178.7)I is the shear vibration of Zr, Al and B atoms along the c-axis direction. Due to the relatively heavy Zr and Al atoms, the frequency of these three vibration modes is always not high. E1u(405.9)I and A2u(503.7)I correspond to the vibration of B atom, respectively. The mass of B atom is lower, so the corresponding frequency is higher.
It can also be seen from Fig. 7a that Zr3AlB2 has 7 Raman active modes: A1g(139.7)R, E2g(153.8)R, E2u(175.0)R, E1u(178.7)R, E1g(459.4)R, E2g(467.1)R and A1g(534.4)R. Among them, the modes of A1g(139.7)R, E2g(153.8)R, E2u(175.0)R, E1u(178.7)R at low frequencies are the common results of Zr and Al atom vibrations. The E1g(459.4)R, E2g(467.1)R and A1g(534.4)R modes at high frequencies are dominated by the vibration of B atoms, and the vibration of Zr and Al is almost negligible. This may be due to the higher strength of the M-B bond and the significantly smaller mass of the B atom, which makes these stretching vibration modes have higher vibration frequencies. These results correspond to the phonon density of states Fig. 5b. E2g(153.8)R, E2u(175.0)R, E1u(178.7)R, E1g(459.4)R, E2g(467.1)R show that the atoms in Zr3AlB2 crystal vibrate in opposite directions perpendicular to the c axis, while A1g(139.7)R, A1g(534.4)R show that the atoms in Zr3AlB2 crystal vibrate in opposite and same directions along the c axis, respectively.
Figure 7b,c shows specific peak positions and intensities of Infrared and Raman spectra in Zr3AlB2. Figure 7b shows that E1u(178.7)I, A2u(187.6)I, and E1u(405.9)I in the Ti3AlB2 structure have strong Infrared vibration intensity. The Infrared vibration intensity of A2u(187.6)I is low, and the Infrared vibration intensity of E1u(138.8)I, A2u(271.4)I, and A2u(503.7)I is very low, which may be difficult to observe experimentally. While, it can be seen from Fig. 7c that in the Raman vibration mode of Zr3AlB2, A1g(139.7)R, E2g(153.8)R, E2u(175.0)R, E1u(178.7)R, E1g(459.4)R, E2g(467.1)R, A1g(534.4)R have higher vibration intensity, but A1g(139.7)R, E2g(153.8)R, E1u(178.7)R peaks are annihilated between other peaks, which may be difficult to identify experimentally. And it is interesting that the E1u(178.7)peak has both Infrared activity and Raman activity.
The schematic diagram of atom vibration, Infrared and Raman spectra of Hf3AlB2 are given in Fig. 8a,b,c, respectively. It can be seen from Fig. 8a that Hf3AlB2 has 6 Infrared modes: E1u(124.4)I, A2u(138.3)I, E1u(153.3)I, A2u(274.0)I, E1u(441.7)I, A2u(531.9)I. Figure 8b shows the position and vibration intensity of the specific peak of the Infrared active vibration mode. E1u(124.4)I, A2u(138.3)I, A2u(274.0)I is clear in the Hf3AlB2 Infrared spectra in Fig. 8b. E1u(441.7)I, A2u(531.9)I have strong Infrared vibration intensity and larger broadening. However, the Infrared vibration intensity of E1u(153.3)I is very low and not observable in the study. It can also be seen from Fig. 8a that Ti3AlB2 has 7 Raman active modes: E2g(70.8)R, E1g(85.7)R, A1g(129.5)R, E2g(151.2)R, E1g(510.3)R, E2g(511.7)R, A1g(591.1)R. Figure 8c gives their specific peak positions and intensities. It can be seen from Fig. 8c that the Raman vibration mode of Hf3AlB2, Hf3AlB2 has E2g(70.8)R, E1g(85.7)R, A1g(129.5)R, E2g(151.2)R, E1g(510.3)R, E2g(511.7)R, and A1g(591.1)R all have high vibration intensity. And the broadening of A1g(591.1)R peak is larger. While, E1g(510.3)R E2g(511.7)R peak is very close and almost unrecognizable.
The vibration diagram of atoms, Infrared and Raman spectra of Ta3AlB2 are shown in Fig. 9a,b,c, respectively. From Fig. 9a, we can see that Ta3AlB2 also has 6 Infrared modes: E1u(146.4)I, E1u(193.3)I, A2u(202.6)I, A2u(336.1)I, A2u(596.9)I, E1u(659.7)I. Figure 9b shows the position and vibration intensity of the specific peak of the Infrared active vibration mode. E1u(193.3)I, A2u(336.1)I, E1u(659.7)I in Hf3AlB2 structure has strong Infrared vibration intensity. A2u(202.6)I, A2u(596.9)I have lower Infrared vibration intensity, and the Infrared vibration intensity of E1u(146.4)I is very low and almost difficult to identify. From Fig. 9a, it can also be seen that Ti3AlB2 has 7 Raman active modes: E2g(83.9)R, E1g(107.8)R, A1g(142.9)R, E2g(192.2)R, E1g(662.7)R, E2g(663.7)R, A1g(673.2)R. Figure 9c gives their specific peak position and intensity. From Fig. 9c, it can be seen that all of peaks of Raman spectra in Ta3AlB2 have high vibration intensity, while E1g(662.7)R, E2g(663.7)R is very close and need to be carefully distinguished in the experiment.
Conclusion
In summary, the study provided detailed insights into the structural, electronic, and mechanical properties of M3AlB2 compounds using first-principles calculations. The results contribute to the understanding of these materials and their potential applications in various fields. The findings can guide further experimental investigations and the design of new materials with tailored properties. Combined with the calculation results of the intensity and distribution of Infrared and Raman vibrations of M3AlB2, it can be seen that the Infrared and Raman vibrations of M and Al atoms account for a large proportion at low frequencies, while the vibration of lighter B atoms accounts for a large proportion at high frequencies. Some of the Infrared or Raman peaks of Zr3AlB2, Hf3AlB2, and Ta3AlB2 are difficult to distinguish, probably because the frequencies of the two vibration modes are very close, or because the weak Infrared or Raman peaks are easily annihilated in the substrate spectral lines and are difficult to identify. This paper predicts that the crystal Infrared and Raman spectra of Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 provide a good theoretical basis for future experiments, but the hyper-Raman spectrum and the change of Raman characteristic spectrum under stress have the potential for further research.
Data availability
All data included in this study are available upon request by contact with the corresponding author Shengzhao Wang (shengzw@nyist.edu.cn).
References
Barsoum, M. W. & Radovic, M. Elastic and mechanical properties of the MAX phases. Annu. Rev. Mater. Res. 41, 195–227 (2011).
Gogotsi, Y. & Anasori, B. The rise of MXenes. ACS Nano 13, 8491–8494 (2019).
Mathis, T. S. et al. Modified MAX Phase synthesis for environmentally stable and highly conductive Ti3C2 MXene. ACS Nano 15, 6420–6429 (2021).
Nie, J., Liu, S., Zhan, X., Ao, L. & Li, L. First-principles study of Hf/Nb/Zr-doped MAX phases Ti3AlC2 and Ti3SiC2. Phys. B 571, 105–111 (2019).
Ding, H. et al. Chemical scissor-mediated structural editing of layered transition metal carbides. Science 379, 1130–1135 (2023).
Wuchina, E. et al. Designing for ultrahigh-temperature applications: The mechanical and thermal properties of HfB2, HfCx, HfNx and αHf(N). J. Mater. Sci. 39, 5939–5949 (2004).
Li, X., Cui, H. & Zhang, R. First-principles study of the electronic and optical properties of a new metallic MoAlB. Sci. Rep. 6, 39790 (2016).
Bai, Y., He, X., Zhu, C. & Chen, G. Microstructures, electrical, thermal, and mechanical properties of bulk Ti2AlC synthesized by self-propagating high-temperature combustion synthesis with pseudo hot isostatic pressing. J. Am. Ceram. Soc. 95, 358–364 (2012).
Miao, N. et al. Computational prediction of boron-based MAX phases and MXene derivatives. Chem. Mater. 32, 6947–6957 (2020).
Liu, S. G. et al. Effect of microstructure evolution on mechanical properties of a TiZrAlB alloy rolled by different processes. Mater. Sci. Eng. A 766, 138348 (2019).
Li, S. et al. Pushing the limit of thermal conductivity of MAX borides and MABs. J. Mater. Sci. Technol. 97, 79–88 (2022).
Zhang, D. et al. Experimental and theoretical investigation of the damage evolution of irradiated MoAlB and WAlB MAB phases. J. Alloy Compd. 942, 169099 (2023).
Wang, S. & Shen, J. Fabrication of sol–gel derived ZrO2 thin film for HR coatings via rapid thermal annealing process. J. Sol-Gel Sci. Technol. 67, 339–343 (2013).
Alameda, L. T., Moradifar, P., Metzger, Z. P., Alem, N. & Schaak, R. E. Topochemical deintercalation of Al from MoAlB: stepwise etching pathway, layered intergrowth structures, and two-dimensional MBene. J. Am. Chem. Soc. 140, 8833–8840 (2018).
Ali, M. A. et al. DFT insights into new B-containing 212 MAX phases: Hf2AB2(A = In, Sn). J. Alloys Compd. 860, 158408 (2021).
Xu, T. et al. A rising 2D star: Novel MBenes with excellent performance in energy conversion and storage. Nano-Micro Lett. 15, 6 (2023).
Dai, F.-Z., Xiang, H., Sun, Y. & Zhou, Y. M2M’AlB4 (M = Mn, Fe Co, M’ = Cr, Mo, W): Theoretical predicted ordered MAB phases with Cr3AlB4 crystal structure. J. Mater. Sci. Technol. 35, 1432–1438 (2019).
Dai, F.-Z., Zhang, H., Xiang, H. & Zhou, Y. Theoretical investigation on the stability, mechanical and thermal properties of the newly discovered MAB phase Cr4AlB4. J. Mater. Sci. Technol. 39, 161–166 (2020).
Zhou, Y., Xiang, H., Zhang, H. & Dai, F.-Z. Theoretical prediction on the stability, electronic structure, room and elevated temperature properties of a new MAB phase Mo2AlB2. J. Mater. Sci. Technol. 35, 2926–2934 (2019).
Hanner, L. A. et al. Synthesis, characterization and first principle modelling of the MAB phase solid solutions: (Mn1-xCrx)2AlB2 and (Mn1-xCrx)3AlB4. Mater. Res. Lett. 9, 112–118 (2021).
Bai, Y. et al. Density functional theory insights into ternary layered boride MoAlB. Acta Mater. 132, 69–81 (2017).
Ade, M. & Hillebrecht, H. Ternary borides Cr2AlB2, Cr3AlB4, and Cr4AlB6: The first members of the series (CrB2)nCrAl with n = 1, 2, 3 and a unifying concept for ternary borides as MAB-phases. Inorg. Chem. 54, 6122–6135 (2015).
Radziuk, D. & Möhwald, H. Ultrasonically treated liquid interfaces for progress in cleaning and separation processes. Phys. Chem. Chem. Phys. 18, 21–46 (2016).
Richardson, P. J., Keast, V. J., Cuskelly, D. T. & Kisi, E. H. Theoretical and experimental investigation of the W-Al-B and Mo-Al-B systems to approach bulk WAlB synthesis. J. Eur. Ceram. Soc. 41, 1859–1868 (2021).
Zhang, H. et al. Defect behavior and radiation tolerance of MAB phases (MoAlB and Fe2AlB2) with comparison to MAX phases. Acta Mater. 196, 505–515 (2020).
Lepakova, O. K., Karakchieva, N. I., Golobokov, N. N., Galchenko, N. K. & Afanasev, N. I. High-temperature synthesis of Ti–Si–B and Ti–Al–B composites and coatings. Int. J. Self-Propag. High-Temp. Synth. 29, 150–156 (2020).
Babak, V. P. & Shchepetov, V. V. Wear resistance of amorphous-crystalline coatings with lubricants. J. Frict. Wear 39, 38–43 (2018).
Zhou, Y., Xiang, H., Dai, F.-Z. & Feng, Z. Electrical conductive and damage-tolerant nanolaminated MAB phases Cr2AlB2, Cr3AlB4 and Cr4AlB6. Mater. Res. Lett. 5, 440–448 (2017).
Mo, Y., Rulis, P. & Ching, W. Y. Electronic structure and optical conductivities of 20 MAX-phase compounds. Phys. Rev. B 86, 165122 (2012).
Lu, Y.-C. et al. First-principles study on the mechanical properties of M2CT2 (M = Ti, Zr, Hf; T = O, F, OH) MXenes. Nucl. Sci. Tech. 30, 172 (2019).
Li, F. & Tang, Q. First-principles calculations of TiB MBene monolayers for hydrogen evolution. ACS Appl. Nano Mater. 2, 7220 (2019).
Shein, I. R. & Ivanovskii, A. L. Graphene-like titanium carbides and nitrides Tin+1Cn, Tin+1Nn (n=1, 2, and 3) from de-intercalated MAX phases: First-principles probing of their structural, electronic properties and relative stability. Comput. Mater. Sci. 65, 104–114 (2012).
Yang, Z.-J. et al. Magnetic moment collapse induced axial alternative compressibility of Cr2TiAlC2 at 420 GPa from first principle. Sci. Rep. 6, 34092 (2016).
Bai, Y. et al. Phase stability and weak metallic bonding within ternary-layered borides CrAlB, Cr2AlB2, Cr3AlB4, and Cr4AlB6. J. Am. Ceram. Soc. 102, 3715–3727 (2019).
Swapnalin, J. et al. Surface modification of Ti3C2Tx using terminal groups and heteroatoms with excellent electrochemical performance in supercapacitors. Appl. Phys. Lett. 122, 161902 (2023).
Khatun, R. et al. Physical properties of MAX phase Zr2PbC under pressure: Investigation via DFT scheme. Phys. B 620, 413258 (2021).
Nishat, M. et al. Pressure effect on the physical properties of 413-type MAX phase compound Nb4AlC3: Insights from DFT simulation. Mater. Sci. Eng. B-Adv. Funct. Solid-State Mater. 299, 116940 (2024).
Wang, X. H. & Zhou, Y. C. Solid-liquid reaction synthesis and simultaneous densification of polycrystalline Ti2AlC. Z. Metallk. 93, 66–71 (2002).
Dahlqvist, M., Alling, B. & Rosen, J. Stability trends of MAX phases from first principles. Phys. Rev. B 81, 220102 (2010).
Wei, X., Li, L., Wu, Y. & Liu, F. Structure and physical properties of predicted MAX phase V2SiC under pressure: A first principle calculation. Mater. Today Commun. 35, 105907 (2023).
Majed, A. et al. Transition metal carbo-chalcogenide “TMCC:” A new family of 2D materials. Adv. Mater. 34, 2200574 (2022).
Barsoum, M. W. et al. Thermal and electrical properties of Nb2AlC, (Ti, Nb)2AlC and Ti2AlC. Metall. Mat. Trans. A 33, 2775–2779 (2002).
Ouadha, I. et al. Thermomechanical analysis of the new ferromagnetic MAX-phase compound Mn2VSnC2: Insights from DFT calculations. Pramana-J. Phys. 97, 57 (2023).
Fatima, S., Tahir, R. & Rizwan, S. Ferroelectric-controlled all MXene nonvolatile flexible memory devices for data storage application. Appl. Phys. Lett. 123, 013503 (2023).
Yamada-Kaneta, H. Vibrational energy levels of oxygen in silicon up to one-A2u-phonon plus one-A1g-phonon states. Phys. B Condens. Matter 302–303, 172–179 (2001).
Bai, Y., He, X. & Wang, R. Lattice dynamics of Al-containing MAX-phase carbides: A first-principle study. J. Raman Spectrosc. 46, 784–794 (2015).
Saikia, U., Sahariah, M. B., González, C. & Pandey, R. Vacancy assisted He-interstitial clustering and their elemental interaction at fcc-bcc semicoherent metallic interface. Sci. Rep. 8, 3844 (2018).
Poulou, A., Mellan, T. A. & Finnis, M. W. Stability of Zr-Al-C and Ti-Al-C MAX phases: A theoretical study. Phys. Rev. Mater. 5, 033608 (2021).
Yao, Y., Miao, N., Gong, Y. & Wang, J. Theoretical exploration of quaternary hexagonal MAB phases and two-dimensional derivatives. Nanoscale 13, 13208–13214 (2021).
Khazaei, M. et al. Novel MAB phases and insights into their exfoliation into 2D MBenes. Nanoscale 11, 11305–11314 (2019).
Carlsson, A., Rosen, J. & Dahlqvist, M. Theoretical predictions of phase stability for orthorhombic and hexagonal ternary MAB phases. Phys. Chem. Chem. Phys. 24, 11249–11258 (2022).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Shimojo, F., Hoshino, K. & Zempo, Y. Ab initio molecular-dynamics simulation method for complex liquids. Comput. Phys. Commun. 142, 364–367 (2001).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1998).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Togo, A., Chaput, L. & Tanaka, I. Distributions of phonon lifetimes in Brillouin zones. Phys. Rev. B 91, 094306 (2015).
Togo, A., Chaput, L., Tanaka, I. & Hug, G. First-principles phonon calculations of thermal expansion in Ti3SiC2, Ti3AlC2, and Ti3GeC2. Phys. Rev. B 81, 174301 (2010).
Togo, A., Chaput, L., Tadano, T. & Tanaka, I. Implementation strategies in phonopy and phono3py. J. Phys. Condes. Matter 35, 353001 (2023).
Togo, A. First-principles phonon calculations with phonopy and Phono3py. J. Phys. Soc. Jpn. 92, 012001 (2023).
Ikeda, Y., Carreras, A., Seko, A., Togo, A. & Tanaka, I. Mode decomposition based on crystallographic symmetry in the band-unfolding method. Phys. Rev. B 95, 024305 (2017).
Koermann, F., Ikeda, Y., Grabowski, B. & Sluiter, M. H. F. Phonon broadening in high entropy alloys. npj Comput. Mater. 3, 36 (2017).
Skelton, M. J. et al. Lattice dynamics of the tin sulphides SnS2, SnS and Sn2S3: Vibrational spectra and thermal transport. Phys. Chem. Chem. Phys. 19, 12452–12465 (2017).
Ong, S. P. et al. Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Yu, M. et al. Dependence of the electronic structure of the EuS/InAs interface on the bonding configuration. Phys. Rev. Mater. 5, 064606 (2021).
Insixiengmay, L. & Stixrude, L. Hydrogen bond symmetrization and high-spin to low-spin transition of ε-FeOOH at the pressure of Earth’s lower mantle. Am. Mineral. 108, 2209–2218 (2023).
Palumbo, M. et al. Ion conductivity in a magnesium borohydride ammonia borane solid-state electrolyte. J. Phys. Chem. C 126, 15118–15127 (2022).
Smith, K. A. et al. Pressure-induced phase transition and phonon softening in h—Lu0.6Sc0.4FeO3. Phys. Rev. B 104, 094109 (2021).
Lai, H.-E., Yoo, R. M. S., Djire, A. & Balbuena, P. B. Investigation of the vibrational properties of 2D titanium nitride MXene using DFT. J. Phys. Chem. C 128, 3327–3342 (2024).
Porezag, D. & Pederson, M. R. Infrared intensities and Raman-scattering activities within density-functional theory. Phys. Rev. B 54, 7830–7836 (1996).
Alam, M. S. et al. Creation of a boron carbide-based Ti3AlBC (312) MAX phase: A route to novel MXenes for energy storage. Chem. Commun. 60, 4585–4588 (2024).
Feng, S. et al. Ab initio study of hypothetical Ti3AlB2 and two new designed layered ordered double-transition metals MAX compound Ti2ZrAlB2. Int. J. Quantum Chem. 122, e26994 (2022).
Niranjan, M. K. Significance of Coulomb interaction in interlayer coupling, polarized Raman intensities, and infrared activities in the layered van der Waals semiconductor GaSe. Phys. Rev. B 103, 195437 (2021).
Liu, Y. et al. Atomic-size dependence of the cohesive energy, bandgap, Young’s modulus, and Raman frequency in different MA2Z4: A bond relaxation investigation. Appl. Phys. Lett. 121, 244105 (2022).
Kroumova, E. et al. Bilbao crystallographic server: Useful databases and tools for phase-transition studies. Phase Transit. 76, 155–170 (2003).
Acknowledgements
The authors gratefully acknowledge the financial supported by The Science and Technology Department of Henan Province (No. 212102210464) and Interdisciplinary Sciences Project of Nanyang Institute of Technology (No. 230072). The authors gratefully acknowledge the assistance and instruction of respectable and professional Prof. Jun Shen of Tongji University during the progress of the research. The author thanks Dr. Erik Streed, the other editors and reviewers for their careful help and guidance.
Author information
Authors and Affiliations
Contributions
Dr. S.Z. Wang wrote the manuscript. Prof. L.L. Chen performed the formal analysis and methodology. Prof. H.S. Hao performed the formal analysis, conceptualization. Dr. C. Qiao performed the formal analysis. Prof. J.F. Song performed the validation. Prof. C.J. Cui performed the methodology, investigation. Prof. B. Liu performed the data analysis. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, S., Chen, L., Hao, H. et al. Study on the Infrared and Raman spectra of Ti3AlB2, Zr3AlB2, Hf3AlB2, and Ta3AlB2 by first-principles calculations. Sci Rep 14, 15030 (2024). https://doi.org/10.1038/s41598-024-65980-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-65980-8
- Springer Nature Limited