
Abstract
Drug-resistant tuberculosis (DR-TB) posed challenges to global TB control. Whole-genome sequencing (WGS) is recommended for predicting drug resistance to guide DR-TB treatment and management. Nevertheless, data are lacking in Taiwan. Phenotypic drug susceptibility testing (DST) of 12 anti-TB drugs was performed for 200 Mycobacterium tuberculosis isolates. WGS was performed using the Illumina platform. Drug resistance profiles and lineages were predicted in silico using the Total Genotyping Solution for TB (TGS-TB). Using the phenotypic DST results as a reference, WGS-based prediction demonstrated high concordance rates of isoniazid (95.0%), rifampicin (RIF) (98.0%), pyrazinamide (98.5%) and fluoroquinolones (FQs) (99.5%) and 96.0% to 99.5% for second-line injectable drugs (SLIDs); whereas, lower concordance rates of ethambutol (87.5%), streptomycin (88.0%) and ethionamide (84.0%). Furthermore, minimum inhibitory concentrations confirmed that RIF rpoB S450L, FQs gyrA D94G and SLIDs rrs a1401g conferred high resistance levels. Besides, we identified lineage-associated mutations in lineage 1 (rpoB H445Y and fabG1 c-15t) and predominant lineage 2 (rpoB S450L and rpsL K43R). The WGS-based prediction of drug resistance is highly concordant with phenotypic DST results and can provide comprehensive genetic information to guide DR-TB precision therapies in Taiwan.
Similar content being viewed by others

Introduction
According to the World Health Organization (WHO) global tuberculosis (TB) report, an estimated of 5.8 million new cases and 157,903 rifampicin (RIF)/multidrug-resistant (RR/MDR) TB cases in 20201. Nevertheless, approximately 33.3% of RR/MDR-TB cases were detected, and 59.0% are successfully treated1. Closing the gap in the detection of drug-resistance (DR)-TB cases requires universal and timely drug susceptibility testing (DST).
Conventional culture-based DST was the gold standard for DR-TB diagnoses, but it is time-consuming and labor intensive. Rapid molecular tests, such as the GeneXpert and line probe assays, have been adopted as diagnostic alternatives for Mycobacterium tuberculosis detection and DR prediction2. Nevertheless, these assays could only detect limited number of mutations and show low sensitivity for hetero-resistant strains with variant frequencies below 5–50%3.
Whole-genome sequencing (WGS) enables the identification of single-nucleotide polymorphisms (SNPs) and insertions and deletions (indels) in loci associated with drug resistance and are proven to have higher accuracy than phenotypic DST4. Since noncanonical mutations in known or unknown genes or other mechanisms still need to be identified in 10–40% of DR isolates, WGS can comprehensively identify drug resistance-associated genes to indicate drug susceptibility for clinical decision making5. Several bioinformatics tools have been developed for inferring drug resistance from WGS data, including KvarQ, PhyResSE, CASTB, Mykrobe, TB Profiler, and Total Genotyping Solution for TB (TGS-TB)6,7,8,9,10. The TGS-TB emphasis particularly on Beijing genotype M. tuberculosis, which is predominant in East Asia where Taiwan is located10. Previous study reported that performance of TGS-TB in predicting resistance to first-line drugs is comparable to other tools6. Nevertheless, data are lacking in Taiwan.
To strengthen DR-TB diagnosis, we report the performance of WGS with the TGS-TB to analyze (sub)lineages and prediction of M. tuberculosis drug resistance.
Materials and methods
Study population
We collected 200 isolates from approximately 30% of RR-/MDR-TB confirmed cases during 2013–2016. One M. tuberculosis isolate from each case was analyzed. Cultivation and processing of M. tuberculosis isolates were performed in a certified biosafety level 3 laboratory. Isolates were obtained by processing specimens with standard N-acetyl-L-cysteine (NALC)-NaOH method11, then inoculated onto Bactec MGIT 960 system. Information on the study cases was obtained from the National TB Registry.
Ethics statement
This study was approved by the Institutional Review Board of the Taiwan Centers for Disease Control (TwCDC IRB No. 106211). All methods were performed in accordance with the relevant guidelines and regulations. The study analyzed only archived isolates, and the need for the written informed consent of the participants was waived.
Phenotypic drug susceptibility testing
DST was conducted using the agar proportion method (APM) with 7H10 and 7H11 medium (Becton, Dickinson and Company, Spark, MD, USA). Drug resistance was defined as the growth of 1% of colonies in drug-containing medium. The critical concentrations of the tested drugs in 7H10 medium were as follows: rifampicin (RIF), 1 μg/mL; isoniazid (INH), 0.2 μg/mL; ethambutol (EMB), 5 μg/mL; streptomycin (STR), 2 μg/mL; ofloxacin (OFX), 2 μg/mL; and moxifloxacin (MFX), 0.5 μg/mL. The critical concentrations of the tested drugs in 7H11 medium were as follows: kanamycin (KM), 6 μg/mL; amikacin (AMK), 6 μg/mL; capreomycin (CM), 10 μg/mL; ethionamide (ETO), 10 μg/mL; and para-aminosalicylic acid (PAS), 8.0 μg/mL. Resistance to pyrazinamide (PZA) at 100 μg/mL was tested using Bactec MGIT 960 as described previously12. Inocula were cultured in a 37 °C incubator for 3 weeks. The DST results were categorized as resistant or susceptible, and the H37Rv (ATCC 27294) strain was used as the control. MDR is defined as an M. tuberculosis isolate resistant to at least INH and RIF. Pre-XDR is defined as an MDR isolate resistant to either fluoroquinolones (FQs) or second-line injectable drugs (SLIDs)13. XDR is defined as an MDR isolate resistant to both FQs and SLIDs14.
Minimum inhibitory concentration (MIC) testing
Phenotypic MIC testing was performed using the Sensititre™ Mycobacterium tuberculosis MYCOTB assay (Thermo Scientific™, TREK Diagnostic Systems, United Kingdom) following the manufacturer’s instructions. The 96-well microtiter plates of the assay containing RIF, INH, EMB, STR, rifabutin (RFB), OFX, MFX, KM, AMK, ETO, PAS and cycloserine (CS). The H37Rv (ATCC 27294) strain was used as the control. The plates were incubated at 37 °C for 2 weeks. The MIC values were recorded by 2 independent readers and a third reading was sought if a discrepant reading was found.
Whole-genome sequencing
Genomic DNA was extracted using the Gentra Puregene Yeast/Bact. Kit (QIAGEN GmbH, Hilden, Germany) following the manufacturer’s protocol, and was quantified using a Qubit 2.0 fluorometer (ThermoFisher Scientific, Waltham, MA, USA). WGS was performed as previously described15. Paired-end libraries were prepared using the QIAseq FX DNA Library Kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer’s protocol. The average fragment size (500–600 bp) of the DNA libraries was estimated by 2% agarose gel electrophoresis. Then, the fragments were eluted using the Wizard SV Gel and PCR Clean-Up System (Promega Corporation, Madison, WI, USA). The 24 purified DNA libraries were pooled (11 pM) were sequenced on an Illumina MiSeq system (Illumina, Inc., San Diego, CA, USA) with the MiSeq Reagent Kit ver. 3 (600 cycles).
Bioinformatic analysis
Sequence reads were checked using FastQC (www.bioinformatics.babraham. ac.uk/projects/fastqc/) for initial assessment of data quality. Drug resistance prediction and lineage analysis were performed using the web-based TGS-TB v210. The following drug-resistance associated genes were predicted: RIF (rpoB, rpoC), INH (katG, fabG1, ahpC, inhA), EMB (embA, embB, embC), PZA (pncA), FQs (gyrA, gyrB), STR (rpsL, rrs, gid), SLIDs (rrs, eis), ETO (ethA, ethR), and PAS (folC, thyA). A phylogenetic tree was constructed from reliable SNPs with respect to H37Rv (NC_000962.3) using the maximum likelihood method with the Tamura-Nei model in MEGA 7.016; 1,000 bootstrap replicates were conducted. The tree was annotated and visualized using iTOL v6 (https://itol.embl.de)17.
Statistical analysis
Descriptive statistics of demographics and clinical characteristics of study cases were presented as proportions. Odds ratios (ORs) and 95% confidence intervals (CIs) were calculated to estimate the correlation between the lineages and variables. The chi-squared test or Fisher’s exact test (when expected cell size < 5) was used for the univariate analysis of categorical variables. Statistical significance was considered as P < 0.05.
Results
Characteristics of the study population
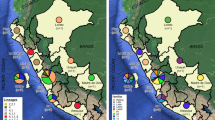
Among the 200 DR-TB cases, 146 (73.0%) cases were male, the median age was 66 (interquartile range = 55–78) years, 165 (82.5%) were new cases and 182 (91.0%) cases showed pulmonary TB (Table 1). The majority of DR-TB cases came from northern (74, 37.0%) Taiwan. Among the 200 DR M. tuberculosis isolates, the predominant lineages were lineage 2 East Asian (132, 66.0%) and lineage 4 Euro-American (52, 26.0%) (Table 1). Sublineage 2.2 isolates were isolated from eastern (11, 79.0%), central (31, 61.0%), northern (44, 59.0%) and southern (33, 54.0%) Taiwan. The sublineage 1.2.1 isolates mainly came from southern (11, 18.0%) Taiwan (Fig. 1).

Geographic distribution of lineages and sublineages of drug-resistant M. tuberculosis isolates in Taiwan. The distribution of each phenotype in each district is represented in the corresponding pie chart as indicated. Abbreviations: N, northern Taiwan; E, eastern Taiwan; C, central Taiwan; S, southern Taiwan.
Drug resistance
Phenotypic drug resistance
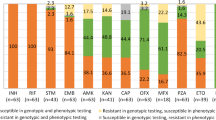
Supplementary Table S1 showed the drug resistance profiles of the 200 isolates. Excluding two pan-susceptible isolates with disputed rpoB mutations and one STR mono-resistant isolate, the remaining isolates were RR (61, 31.0%) and MDR (136, 69.0%). Among 136 MDR isolates, 28 (20.6%) and 1 (0.7%) were pre-XDR and XDR, respectively (Table 2). The resistance rates to the tested drugs were as follows: RIF (197, 98.5%), INH (136, 68.0%), EMB (77, 38.5%), PZA (40, 20.0%), STR (60, 30.0%), FQs (22, 11.0%), KM (11, 5.5%), AMK (8, 4.0%), CM (6, 3.0%), ETO (34, 17.0%) and PAS (6, 3.0%) (Supplementary Table S2).
Genotypic drug resistance
Using the phenotypic DST results as a reference, the drug resistance-associated mutations and MIC distributions of the isolates were shown in Supplementary Table S2 and Fig. 2, respectively. The confidence level for grading mutations was based on the 2021 WHO catalog of M. tuberculosis mutations18. In addition, the candidate mutations identified by the TGS-TB database, including fabG1 L203L, rpoB L430P, L452P, were classified as genotypically resistant according to the WHO mutations catalog.

Distribution of drug resistance-associated mutations with corresponding MICs. Each stacked column represents a collection of isolates colored according to their genetic background. The x-axes show the MICs in μg/mL. The dashed lines indicates the critical concentrations used for MYCOTB plates.
Rifampicin resistance
Among the 197 phenotypically RIF-resistant isolates, 120 (60.9%) isolates had high-confidence rpoB S450L and 25 (12.7%) isolates with H445Y, which showed MICs ≥ 4 μg/mL. Four isolates concurrently exhibited rpoB S450L and putative compensatory rpoC mutations, I491T, G332R, F452S, and L527V, with MICs > 16 μg/mL. Furthermore, six isolates presented disputed mutations, rpoB L430P or L452P, with concurrent mutations exhibited MICs > 16 μg/mL. In contrast, 69.2% of isolates with the single disputed mutation, rpoB L430P or L452P, exhibited MICs ≤ 1 μg/mL.
Isoniazid resistance
Among the 136 phenotypically INH-resistant isolates, 77 (56.6%) isolates had high-confidence katG S315T and 44 (32.4%) isolates with low-confidence fabG1 c-15t. We found that 60 (88.2%) isolates with single katG S315T showed MICs ≥ 0.5 μg/mL, while 22 (88.0%) isolates with single fabG1 c-15t showed MICs ≤ 0.5 μg/mL. The combination of katG S315T and fabG1 c-15t was associated with elevated MICs (≥ 4 μg/mL). In addition, six INH-resistant isolates with concurrent fabG1 c-15t and inhA I194T mutations also presented MICs ≥ 0.5 μg/mL. Five INH-susceptible isolates with katG S315T, katG W191R, fabG1 c-15t, or ahpC c-52t exhibited MICs ≤ 0.12 μg/mL. Furthermore, the novel mutations, katG D329Y, G370E and P375L, with MICs ≤ 0.5 μg/mL, were each found in three isolates.
Ethambutol resistance
Among the 77 phenotypically EMB-resistant isolates, 25 (32.5%) isolates had high-confidence embB M306V and 15 (19.5%) isolates with M306I. Of the 41 isolates with single embB M306V/I, 17 (41.5%) isolates presented MICs ≤ 4 μg/mL, and 7 (17.1%) of them exhibited an EMB-susceptible phenotype. All six isolates with single embA mutations also presented MICs ≤ 4 μg/mL, and four of them exhibited an EMB-susceptible phenotype. Notably, isolates concurrently harboring embA and embB mutations were associated with elevated MICs (≥ 8 μg/mL).
Pyrazinamide resistance
Among the 40 phenotypically PZA-resistant isolates, 39 isolates harbored 36 types of mutations scattered throughout the pncA gene and promoter; thus, high diversity of pncA mutations was observed without major hot spots.
Streptomycin resistance
Among the 60 STR-resistant isolates, 31 (51.7%) isolates had high-confidence rpsL K43R, 9 (15%) isolates with K88R, and 7 (11.7%) isolates with rrs a514c. All 31 isolates with rpsL K43R presented high MICs (≥ 32 μg/mL), while 9 isolates with rpsL K88R presented wide range of MICs (0.5 to > 32 μg/mL). Among 20 isolates with rrs mutations, 14 (70%) of them exhibited MICs ≤ 2 μg/mL. Besides, of 18 isolates with gid mutations, even though mutations in the gid gene were associated with STR resistance, 15 (83.3%) isolates exhibited MICs ≤ 2 μg/mL.
Fluoroquinolones resistance
Among the 22 FQs-resistant isolates, 14 (63.6%) isolates had high-confidence gyrA D94G. All 14 FQs-resistant isolates with gyrA D94G presented high MICs (≥ 4 μg/mL for ofloxacin (OFX) and ≥ 2 μg/mL for MFX). Other gyrA mutations were also associated with high MICs (≥ 4 μg/mL for OFX and ≥ 2 μg/mL for MFX). In addition, we identified one novel gyrB G522S mutation.
Second-line injectable drug resistance
Cross-resistance among injectable drugs was associated with the high-confidence mutation rrs a1401g, which was found in seven KM-resistant isolates (63.6%) with MICs > 40 μg/mL and seven AMK-resistant isolates (87.5%) with MICs > 16 μg/mL, respectively. Moreover, all six isolates with eis c-12t exhibited a KM-susceptible phenotype.
Ethionamide resistance
Among the 34 ETO-resistant isolates, 25 (73.5%) isolates had low-confidence fabG1 c-15t, which was cross-resistant to INH. Of the 33 isolates with single fabG1 c-15t, 28 (84.8%) isolates exhibited MICs ≤ 5 μg/mL, and 14 (42.4%) of them exhibited an ETO-susceptible phenotype. Besides, isolates with fabG1 c-15t with concurrent inhA I194T (n = 3, 50.0%) or ethR A95T (n = 5, 100.0%) exhibited MICs ≤ 5 μg/mL. In addition, of 11 isolates with single ethA frameshift mutations, 8 (72.7%) of them exhibited MICs ≤ 5 μg/mL.
Para-aminosalicylic acid resistance
Among six PAS-resistant isolates, one isolate carried folC E40G with MIC > 64 μg/mL, and the other five isolates harbored novel mutations, thyA L38S, L218P, R235W, and Y251stop with MICs = 2 to ≥ 64 μg/mL. One isolate with the folC S150G mutation was phenotypically PAS-susceptible with an MIC = 4 μg/mL.
Performance of WGS in drug resistance prediction
The performance of WGS for the prediction of drug resistance was shown in Table 3. The average concordance was 94.9%, ranging from 84.0% (ETO) to 99.5% (FQs, AMK and PAS). The overall sensitivity and specificity of WGS-based DST were 97.2% and 94.0%, respectively. The sensitivity of WGS to predict resistance to INH (96.3%), FQs (100.0%) and PAS (100.0%) were further improved by inclusion of novel mutations, katG D329Y, G370E, P375L, gyrB G522S, and thyA L38S, L218P, R235W, Y251stop (Table 3). Excluding SLIDs, the resistance predictive values of other tested drugs were higher than 95.0%. In addition, three isolates harboring rpoB L430P or L452P disputed mutations were phenotypically RIF susceptible, which resulted in low specificity.
Associations between lineages and drug-resistance
We constructed a maximum likelihood phylogenetic tree based on 12,015 SNP differences (Fig. 3). Lineage 2 isolates were significantly resistant to EMB and STR than lineage 1 and lineage 4 (P < 0.05) (Supplementary Table S3). Lineage 1 isolates were significantly resistant to ETO when compared to lineage 2 and lineage 4 (P < 0.05) (Supplementary Table S3). Furthermore, we identified lineage-specific variants, such as RIF rpoB S450L was predominant in lineage 2 (65.2%, P = 0.038); RIF rpoB H445Y was significantly associated with lineage 1 (31.3%, P = 0.034); INH fabG1 c-15t was significantly associated with lineage 1 (50.0%, P = 0.011); STR rpsL K43R was significantly associated with lineage 2 (20.5%, P = 0.007) (Fig. 3, Table 4).

Maximum likelihood phylogenetic tree of the 200 DR-TB isolates from Taiwan. The tree was constructed based on 12,015 genome-wide SNPs. Lineages are represented by different colored blocks. Mutations are represented by filled (presence of mutation) or empty (absence of mutation) symbols. The figure was generated using iTOL v6 (https://itol.embl.de). The scale bar indicates the genetic distance proportional to the total number of SNPs. Abbreviations: RIF, rifampicin; INH, isoniazid; EMB, ethambutol; PZA, pyrazinamide; STR, streptomycin; FQs, fluoroquinolones; SLIDs, second-line injectable drugs; ETO, ethionamide; MDR, multidrug resistant; RR, rifampicin resistant; PXDR, pre-extensively drug resistant; XDR, extensively drug resistant; MSTM, mono-streptomycin resistant; PS, pansusceptible.
Discussion
This is the first study to demonstrate that WGS/TGS-TB had excellent performance in drug resistance prediction and the genetic diversity identification of M. tuberculosis in Taiwan. The good concordance rates in the detection of drug resistance against RIF, INH, PZA and FQs ranged from 95.0 to 99.5%, which were comparable to 96.4–100.0% reported in a previous study15. Together with the MIC measurements, the novel mutations katG D329Y, G370E and thyA L38S might confer low resistance levels. The predominant lineage 2 East Asian (particularly Beijing 2.2.1) was associated with drug resistance, as previously suggested19. Besides, rpoB S450L and rpsL K43R were significantly prevalent in lineage 2. Collective information is useful for DR-TB diagnosis and care.
WGS and MICs data provided informative insights on MTBC drug resistance. Nevertheless, suboptimal agreement in predictions of resistance to EMB (87.5%), STR (88.0%) and ETO (84.0%) was mainly attributed to mutations conferring low resistance levels, clinical breakpoint artifacts in pDST, incomprehensive mutation catalogs, and unknown resistance mechanisms20. False-susceptible pDST results for EMB, STR, and ETO might occur because some mutations cause slight MIC increases close to the critical concentration (CC). Thus, the overlap between the MICs of mutant and wild-type isolates would result in misclassification based on pDST. These elevated MICs below current CCs may still be clinically meaningful due to a chance of higher drug resistance acquisition and risk treatment failure20.
The fabG1 c-15t and inhA I194T mutation were associated with low-level INH resistance. In this study, six isolates with concurrent c-15t and I194T showed elevated MICs (≥ 0.5 μg/mL) and a previous study revealed that conferred high resistance levels and exhibited a synergistic effect on INH resistance21. In addition, fabG1 c-15t was associated with cross-resistance between INH and the structurally related ETO. It is worth noting that two isolates with the fabG1 L203L silent mutation were INH resistant (MDR_17 and MDR_40). This might occur through the upregulation of fabG1 resulting from the creation of an alternative promoter for fabG1 expression22. We found that isolates with frameshift and nonsense mutations in the ethA gene, encoding the EthA monooxygenase, might not be phenotypically resistance to ETO. The presence of other monooxygenases in M. tuberculosis might be able to compensate the inactivation of EthA23.
The low specificity and NPV were due to all three RIF-susceptible isolates carrying disputed rpoB L430P or L452P mutation, which exhibited low MICs (≤ 1 μg/mL). Previous studies have reported that isolates with disputed rpoB mutations, L430P, D435Y, H445C/L/N/S, and L452P, confer low levels of RIF resistance24. However, isolates harboring disputed mutations concurrent with R62C, Q67R/H, M434L, or D435G mutations presented high MIC values (≥ 16 μg/mL), as mentioned in our previous study24.
Isolates with embB mutations combined with EMB embC-embA intergenic region (IGR) mutations, such as embA c-11a, c-12t, and c-16t, could show increased MIC values. Mutations in the embC-embA IGR might enhance the binding of EmbR to the promoter region of embAB and increase the transcription of embAB, thus contributing to EMB resistance25. Mutations in embB M306V/I and G406D/S were found in both EMB-resistant and EMB-susceptible isolates. Previous studies reported that embB M306V/I mutations cause slight MIC increases close to the CC 26. The inconsistency of EMB between WGS and pDST may also be due to inappropriate CCs and poor repeatability of pDST26. In addition, the embABC operon is involved in the decaprenylphosphoryl-β-D-arabinose (DPA) biosynthetic and utilization pathway, which might alter cell wall permeability and cause variability in EMB MICs27. This implies that the embB306 mutation results in varying degrees of EMB resistance but does not cause high-level EMB resistance on its own27.
Mutations in the pncA gene leading to a reduction in pyrazinamidase (PZase) activity are the main mechanism of PZA resistance28. We found a high diversity of pncA gene mutations without major hot spots in the PZA-resistant isolates, consistently with previous studies29,30. Although the reason for this diversity is still unclear, it might be due to adaptive mutagenesis or deficiency in DNA mismatch repair mechanisms31. Mutations in the gyrA or gyrB genes are associated with FQs resistance28. In particular, isolates with gyrA D94G show high MICs. Our study revealed that gyrA D94G was the predominant mutation associated with high MIC values for OFX and MFX (Supplementary Table S2, Fig. 2) as previously reported32.
Mutations in the rrs gene, encodes the 16S rRNA, confer moderate levels of STR resistance33. Whereas, mutations in the gid gene, encodes a 7-methylguanosine methyltransferase, reduce 16S rRNA methylation, thereby interfering with STR binding and consequently conferring low levels of STR resistance34. Besides, eis promoter mutations, g-10a and c-14t, accounted for 33% of KM resistance35. The eis c-14t mutation conferred a higher level of KM resistance than the g-10a, g-37t, and c-12t mutations36. Nevertheless, no eis c-14t mutants were identified in this study.
Suboptimal prediction of resistance to KM (81.8%), AMK (87.5%), and CM (83.3%) might be a few resistant isolates analyzed, the presence of additional resistance mutations in genes not assessed, or to unknown resistance mechanisms. The mechanisms of drug resistance have yet to be fully elucidated. The strain genetic background, clonal interference, epistatic interactions, efflux pump mutations, target modification and mimicry could contribute to various levels of drug resistance37. Rv1258c encodes the homologous Tap protein in M. tuberculosis, which is regulated by transcriptional activator WhiB738. An increase in whiB7 expression, resulting from mutations located in the 5’ untranslated region, leads to upregulation of eis and tap, conferring low-level resistance to aminoglycosides39.
Lineage 2 and lineage 4 M. tuberculosis isolates were predominant in Taiwan (Fig. 1). Sublineage 2.2 and sublineage 4.5 were predominant in Taiwan as well as in East Asia40. In addition, lineage 1 isolates, particularly sublineage 1.2.1, were prevalent in south and southeast Asia41. Notably, we found geographic disparities in sublineage 1.2.1 isolates mainly found in southern Taiwan, where the majority of migrants live, and none were identified in eastern Taiwan. Phylogenetic analysis showed that drug resistance mutations, RIF rpoB H445Y, was associated with lineage 1, as observed in a previous study42; whereas, RIF rpoB S450L and STR rpsL K43R, were associated with lineage 2, as observed in previous studies19,43. Higher mutation rates of lineage 2 isolates might account for increased adaptation abilities and drug resistance rates44.
Several software tools were available for predicting the drug resistance of M. tuberculosis, including PhyResSE, MyKrobe Predictor, KvarQ, TB profiler and TGS-TB6. However, performance of drug resistance prediction varies between the different tools and anti-TB drugs tested6,45. A previous study revealed that the sensitivity of PZA resistance prediction was higher using TGS-TB (87.0%) than that using TB profiler (< 65.0%)45. The major difference between TGS-TB and TB profiler in PZA resistance prediction was due to the inclusion of insertions and deletions associated with PZA resistance46. Additionally, the performances of PhyResSE, MyKrobe Predictor and KvarQ were unsatisfactory for predicting resistance to PZA and EMB6. Notably, TGS-TB was much more user friendly as compared to other tools for WGS data analysis and could process online batch analysis for multiple samples.
The study has some limitations. Firstly, due to the low resistant rates (< 10%) of certain study drugs, KM, AMK, CM, and PAS and few fully susceptible isolates were analyzed. As results, biases on performance might occur. Secondly, there was no MIC testing for PZA and CM to compare WGS with the level of phenotypic resistance. Thirdly, lineages may affect the prediction of drug resistance by WGS and was not take into account. Lastly, besides the genetically-encoded determinants, changes in transcription or translation may also mediate antibiotic tolerance and persistence state, which also impact the efficacy of antibiotics in vivo47.
Phenotypic DST for the prediction of TB drug resistance has limitations, hampering timely personalized precision therapy and comprehensive surveillance. To strengthen and revolutionize the DR-TB control program, WGS provides a solution for genetic drug resistance prediction and surveillance of existing, new, and repurposed TB drugs with satisfactory accuracy. This pilot study demonstrated the feasibility of the application of WGS for TB control programs in Taiwan. Notably, a diagnostic policy to streamline and integrate WGS into our routine TB laboratory services for analyzing M. tuberculosis isolated from all new RR/MDR cases has been established since 2019. We expect to expand the services to DR-TB cases with other drug-resistant patterns if the resource is available. In line with some high-income countries, this study reassures that WGS is a valuable tool to inform clinical and public health actions. Our results could serve as a guide to facilitate the uptake of new technology in the TB control program.
Data availability
Sequencing reads have been submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under BioProject ID PRJNA879962.
References
WHO. Global Tuberculosis Report 2021. (WHO, 2021).
Iacobino, A., Fattorini, L. & Giannoni, F. Drug-resistant tuberculosis 2020: Where we stand. Appl. Sci. 10, 2153 (2020).
Engström, A. Fighting an old disease with modern tools: Characteristics and molecular detection methods of drug-resistant Mycobacterium tuberculosis. Infect. Dis. 48, 1–17 (2016).
Papaventsis, D. et al. Whole genome sequencing of Mycobacterium tuberculosis for detection of drug resistance: A systematic review. Clin. Microbiol. Infect. 23, 61–68 (2017).
Cohen, K. A., Manson, A. L., Desjardins, C. A., Abeel, T. & Earl, A. M. Deciphering drug resistance in Mycobacterium tuberculosis using whole-genome sequencing: Progress, promise, and challenges. Genome Med. 11, 45 (2019).
van Beek, J., Haanperä, M., Smit, P. W., Mentula, S. & Soini, H. Evaluation of whole genome sequencing and software tools for drug susceptibility testing of Mycobacterium tuberculosis. Clin. Microbiol. Infect. 25, 82–86 (2019).
Steiner, A., Stucki, D., Coscolla, M., Borrell, S. & Gagneux, S. KvarQ: Targeted and direct variant calling from fastq reads of bacterial genomes. BMC Genomics 15, 881 (2014).
Feuerriegel, S. et al. PhyResSE: A Web tool delineating Mycobacterium tuberculosis antibiotic resistance and lineage from whole-genome sequencing data. J. Clin. Microbiol. 53, 1908–1914 (2015).
Iwai, H., Kato-Miyazawa, M., Kirikae, T. & Miyoshi-Akiyama, T. CASTB (the comprehensive analysis server for the Mycobacterium tuberculosis complex): A publicly accessible web server for epidemiological analyses, drug-resistance prediction and phylogenetic comparison of clinical isolates. Tuberculosis 95, 843–844 (2015).
Sekizuka, T. et al. TGS-TB: Total genotyping solution for Mycobacterium tuberculosis using short-read whole-genome sequencing. PLoS ONE 10, e0142951 (2015).
Kent, P. T. & Kubica, G. P. Public Health Mycobacteriology: A Guide for the Level III Laboratory (Public Health Service, Centers for Disease Control, 1985).
WHO. Technical Manual for Drug Susceptibility Testing of Medicines Used in the Treatment of Tuberculosis. (WHO, 2018).
Banerjee, R. et al. Extensively drug-resistant tuberculosis in California, 1993–2006. Clin Infect Dis 47, 450–457 (2008).
WHO. Report of the meeting of the WHO Global Task Force on XDR-TB: Geneva, Switzerland, 9–10 October 2006. (WHO, 2007).
Takii, T. et al. Whole-genome sequencing-based epidemiological analysis of anti-tuberculosis drug resistance genes in Japan in 2007: Application of the Genome Research for Asian Tuberculosis (GReAT) database. Sci. Rep. 9, 12823 (2019).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
WHO. Catalogue of Mutations in Mycobacterium tuberculosis Complex and Their Association with Drug Resistance. (WHO, 2021).
Gupta, A. et al. Detection of Beijing strains of MDR M. tuberculosis and their association with drug resistance mutations in katG, rpoB, and embB genes. BMC Infect. Dis. 20, 752 (2020).
Heyckendorf, J. et al. What is resistance? Impact of phenotypic versus molecular drug resistance testing on therapy for multi- and extensively drug-resistant tuberculosis. Antimicrob. Agents Chemother. 62, e01550-e11517 (2018).
Machado, D. et al. High-level resistance to isoniazid and ethionamide in multidrug-resistant Mycobacterium tuberculosis of the Lisboa family is associated with inhA double mutations. J. Antimicrob. Chemother. 68, 1728–1732 (2013).
Ando, H., Miyoshi-Akiyama, T., Watanabe, S. & Kirikae, T. A silent mutation in mabA confers isoniazid resistance on Mycobacterium tuberculosis. Mol. Microbiol. 91, 538–547 (2014).
Ushtanit, A. et al. Molecular determinants of ethionamide resistance in clinical isolates of Mycobacterium tuberculosis. Antibiotics 11, 133 (2022).
Lin, W. H., Lee, W. T., Tsai, H. Y. & Jou, R. Disputed rpoB Mutations in Mycobacterium tuberculosis and tuberculosis treatment outcomes. Antimicrob. Agents Chemother. 65, e0157320 (2021).
Cui, Z. et al. Mutations in the embC-embA intergenic region contribute to Mycobacterium tuberculosis resistance to ethambutol. Antimicrob. Agents Chemother. 58, 6837–6843 (2014).
Li, M. C. et al. Detecting ethambutol resistance in Mycobacterium tuberculosis isolates in China: A comparison between phenotypic drug susceptibility testing methods and DNA sequencing of embAB. Front. Microbiol. 11, 781 (2020).
Safi, H. et al. Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-β-D-arabinose biosynthetic and utilization pathway genes. Nat. Genet. 45, 1190–1197 (2013).
Palomino, J. C. & Martin, A. Drug resistance mechanisms in Mycobacterium tuberculosis. Antibiotics 3, 317–340 (2014).
Miotto, P. et al. Mycobacterium tuberculosis pyrazinamide resistance determinants: A multicenter study. MBio 5, e01819-e11814 (2014).
Ramirez-Busby, S. M. & Valafar, F. Systematic review of mutations in pyrazinamidase associated with pyrazinamide resistance in Mycobacterium tuberculosis clinical isolates. Antimicrob. Agents Chemother. 59, 5267–5277 (2015).
Pandey, S. et al. Characterization of pncA mutations in multi-drug and pyrazinamide resistant Mycobacterium tuberculosis isolates cultured from Queensland migrants and Papua New Guinea residents. Tuberculosis 111, 109–113 (2018).
Uddin, M. K. et al. Correlation of gyr mutations with the minimum inhibitory concentrations of fluoroquinolones among multidrug-resistant Mycobacterium tuberculosis isolates in Bangladesh. Pathogens 10, 1422 (2021).
Nasr Esfahani, B., Mirhendi, H., Riyahi Zaniani, F., Salehi, M. & Karimi, S. Genetic patterns of rpsL and rrs genes in clinical isolates of Mycobacterium tuberculosis, Isfahan, Iran. Mycobact. Dis. 7, 235 (2017).
Wong, S. Y. et al. Mutations in gidB confer low-level streptomycin resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 55, 2515–2522 (2011).
Georghiou, S. B. et al. Evaluation of genetic mutations associated with Mycobacterium tuberculosis resistance to amikacin, kanamycin and capreomycin: A systematic review. PLoS ONE 7, e33275 (2012).
Pholwat, S. et al. eis Promoter C14G and C15G mutations do not confer kanamycin resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 60, 7522–7523 (2016).
Swain, S. S., Sharma, D., Hussain, T. & Pati, S. Molecular mechanisms of underlying genetic factors and associated mutations for drug resistance in Mycobacterium tuberculosis. Emerg. Microbes. Infect. 9, 1651–1663 (2020).
Morris Rowan, P. et al. Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. 102, 12200–12205 (2005).
Reeves, A. Z. et al. Aminoglycoside cross-resistance in Mycobacterium tuberculosis due to mutations in the 5’ untranslated region of whiB7. Antimicrob. Agents Chemother. 57, 1857–1865 (2013).
Stucki, D. et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat. Genet. 48, 1535–1543 (2016).
Netikul, T., Palittapongarnpim, P., Thawornwattana, Y. & Plitphonganphim, S. Estimation of the global burden of Mycobacterium tuberculosis lineage 1. Infect. Genet. Evol. 91, 104802 (2021).
Shanmugam Siva, K. et al. Mycobacterium tuberculosis lineages associated with mutations and drug resistance in isolates from India. Microbiol. Spectr. 10, e01594–e11521 (2022).
San, L. L. et al. Insight into multidrug-resistant Beijing genotype Mycobacterium tuberculosis isolates in Myanmar. Int. J. Infect. Dis. 76, 109–119 (2018).
Hakamata, M. et al. Higher genome mutation rates of Beijing lineage of Mycobacterium tuberculosis during human infection. Sci. Rep. 10, 17997 (2020).
Macedo, R. et al. Dissecting whole-genome sequencing-based online tools for predicting resistance in Mycobacterium tuberculosis: Can we use them for clinical decision guidance? Tuberculosis 110, 44–51 (2018).
Iwamoto, T. et al. Overcoming the pitfalls of automatic interpretation of whole genome sequencing data by online tools for the prediction of pyrazinamide resistance in Mycobacterium tuberculosis. PLoS ONE 14, e0212798 (2019).
Harms, A., Maisonneuve, E. & Gerdes, K. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science 354, aaf468 (2016).
Acknowledgements
This study was supported by grants MOHW106-CDC-C-315-113134, MOHW107-CDC-C-315-123118, MOHW108-CDC-C-315-133119, MOHW109-CDC-C-315-113114 and MOHW110-CDC-C-315-123114 from the Taiwan Centers for Disease Control, Ministry of Health and Welfare, Taiwan. The authors thank the team of Dr. Satoshi Mitarai at the Research Institute of Tuberculosis, Japan Anti-Tuberculosis Association, for performing whole-genome sequencing.
Author information
Authors and Affiliations
Contributions
R.J. designed the research. Y.X.X., K.H.L., W.H.L., and T.H.C. performed the experiments. R.J., Y.X.X., and K.H.L. analyzed the results. R.J., Y.X.X., and K.H.L. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, YX., Liu, KH., Lin, WH. et al. Whole-genome sequencing-based analyses of drug-resistant Mycobacterium tuberculosis from Taiwan. Sci Rep 13, 2540 (2023). https://doi.org/10.1038/s41598-023-29652-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-29652-3
- Springer Nature Limited
This article is cited by
-
New insight in molecular detection of Mycobacterium tuberculosis
AMB Express (2024)
-
Large-scale statistical analysis of Mycobacterium tuberculosis genome sequences identifies compensatory mutations associated with multi-drug resistance
Scientific Reports (2024)
-
Incipient tuberculosis: a comprehensive overview
Infection (2024)
-
Precision Medicine Strategies to Improve Isoniazid Therapy in Patients with Tuberculosis
European Journal of Drug Metabolism and Pharmacokinetics (2024)
-
Genotypic and phenotypic comparison of drug resistance profiles of clinical multidrug-resistant Mycobacterium tuberculosis isolates using whole genome sequencing in Latvia
BMC Infectious Diseases (2023)