
Abstract
In the present study, a series of aryl-substituted thioqunoline conjugated to thiosemicarbazide were rationally designed and synthesized. The formation of target compounds was confirmed by spectral characterization techniques such as IR, 1H-NMR, 13C-NMR, ESI–MS, and elemental analysis. Among the synthesized derivatives, compound 10g bearing para-chlorophenyl moiety was proved to be the most potent tyrosinase inhibitor with an IC50 value of 25.75 ± 0.19 µM. Compound 10g as the most potent derivative exhibited a noncompetitive inhibition pattern against tyrosinase in the kinetic study. Furthermore, the in silico cavity detection, as well as the molecular docking assessments, were performed to follow the behavior of 10g within the proposed binding site. Besides, the toxicity of 10g and its potency to reduce the melanin content on A375 cell lines were also measured. Consequently, aryl-substituted thioqunolines conjugated to thiosemicarbazide might be a promising candidate in the cosmetics, medicine, and food industry as tyrosinase inhibitors.
Similar content being viewed by others
Introduction
Melanin is a group of biopolymer pigments that is a mixture of dark brown or black eumelanin and lighter-colored pheomelanin in many organisms1. In humans, it protects the skin from UV radiation and other environmental challenges and gives the skin it's color2,3. However, the excessive accumulation of melanin might result in some disorders, such as melanoma, melasma, and freckles as well as increase the risk of cancer4,5 and Parkinson's disease6. Also, the undesirable phenomena of enzymatic browning in most fruits and vegetables as well as in seafood leading to brown or black pigments on products should not be neglected. Melanogenesis is the biochemical pathway leading to the synthesis of melanin in which tyrosinase as a key enzyme hydroxylates tyrosine to L-Dopa and oxidizes it to dopaquinone, which polymerizes spontaneously to form melanin2,7.
Tyrosinase (EC 1.14.18.1) is a copper-containing metalloenzyme that is widely distributed in microorganisms, plants, and animals8. Crystal structures of mushroom tyrosinase with a molecular mass of 120 kDa exhibited an H2L2 tetramer structure. The H subunit is the tyrosinase domain that possesses multiple loops, α-helices, and β-strands, and the L subunit has a lectin-like fold containing β-strands. The active site of tyrosinase located in two antiparallel α-helices of the H subunit contains two copper ions, which engage in strong interaction with three Histidine residues9,10. Noteworthy, the structure and function of the active site are similarly preserved in various types of organisms11.
Accordingly, the identification and development of novel anti-tyrosinase agents with anti-melanogenesis potential as an added value are currently the subject of interest in medicine as well as the cosmetic and food industries. Several natural and synthetic compounds have been introduced as tyrosinase inhibitors including flavonoids, polyphenols, phenylpropanoids, thioamides, thioureas, and kojic acid derivatives12,13. However, these derivatives face a lot of challenges and certain safety risks, as a result developing novel and potent inhibitors with low toxicity is highly needed.
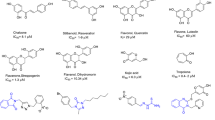
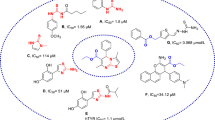
The quinoline framework is a well-known heterocyclic scaffold with a simple synthetic procedure that has drawn special interest because of its inherent and diverse biological response including antibacterial, antifungal, antimalarial, anthelmintic, anticonvulsant, cardiotonic, anti-inflammatory, and analgesic activities14,15. In addition, quinoline has been proven to be an excellent tyrosinase inhibitor. Jineol (A) dose-dependently inhibited mushroom tyrosinase activity with an IC50 of 39.46 ± 0.01 μM and 50.35 ± 0.05 μM in presence of L-tyrosine and L-Dopa as substrate, respectively. Jineol also reduced melanin content in melanoma cells by downregulating MITF expression through interference with ERK1/2 and p38 phosphorylation and suppressing the protein levels of tyrosinase, tyrosinase-related protein 1 (TYRP-1) and TYRP-216. Also, 2-(4-Fluorophenyl)-quinazolin-4(3H)-one (compound B) was introduced as a highly potent tyrosinase inhibitor with quinoline structure which demonstrated good activity compared to arbutin as the positive control (IC50 = 180 μM)17. In another study, thio-quinazolinones conjugated to kojic acid (compound C) were developed, synthesized, and evaluated for their inhibitory activity against mushroom tyrosinase. The most potent compound showed significant tyrosinase inhibitory activity and demonstrated 68.99% melanin content at 8 µM. Docking study showed that the substitutions on aromatic ring provide optimum bulkiness to participate in the different forms of interaction including H-bound, in π-π stacking, and π-aryl interactions with the binding site of tyrosinase18.
Thiosemicarbazide is known as a powerful anti-tyrosinase agent due to its capacity to reduce back o‐dopaquinone to L-Dopa and avoid melanin formation. Noteworthy it was well documented that thiosemicarbazide frequently appeared in tyrosinase inhibitors which are developed based on the structural modifications of linker type and length of L-tyrosine amino acid. In this context compounds, D-G bearing thiosemicarbazide moiety are good examples that showed high potency against tyrosinase (Fig. 1)19,20,21. Recently, compound G was developed as high potent tyrosinase inhibitor against tyrosinase with an IC50 value of 0.11 μM and 0.17 μM in the presence of L-tyrosine and L-Dopa as substrates, respectively. This side chain mimics the inherent structure of the native substrate and increases the affinity to tyrosinase.

The rational design of the target compound based on previously reported tyrosinase inhibitors.
Also, the biological activity of some designed compounds based on aryl-acetamide moieties is displayed in Fig. 1 (compound H–J)22,23,24. In silico studies confirmed that this linker is effectively involved in hydrogen bond interaction with the His residues and Cu ions of the tyrosinase active site and expanding the opportunity for further derivatization.
Regarding that thiosemicarbazide and aryl-acetamide presented in previous potent inhibitors and thioquinoline provide a suitable site to occupy the binding site and participate in several interactions with the enzyme, therefore, this study aimed to synthesize and evaluate the tyrosinase inhibitory potential of thiosemicarbazide-thioquinoline derivative bearing different aryl-acetamides. Moreover, the kinetics study and in silico evaluations were performed to determine the type of inhibition and binding pose within the enzyme. The cytotoxicity, as well as anti-melanogenesis potencies of the most potent derivative, were also executed.
Results and discussion
Chemistry
As presented in Fig. 2 to the mixture of N,N-dimethylformamide (1) and phosphoryl chloride in DMF at 0 °C, phenyl-acetamide (2) was added dropwise. After around 30 min, the mixture was heated to 80 °C for 12 h and the crude product was purified by recrystallization in ethanol to synthesize chloroquinoline-3-carbaldehyde (compound 3)25.

The synthetic path for the target compounds 10a-m.
Compound 4 in turn was prepared from the reaction of sodium sulfide with compound 3 in DMF at room temperature for 2 h. Next, amine derivatives (5a-m) were cooled to 0 °C in DMF and chloroacethyl chloride (6) was added. The reaction mixture was then stirred at room temperature for 12 h then cold water was added. The resulting solid was washed with water three times with petroleum ether giving a solid and pure product (7a-m). The yield of each derivative were presented in Table 1.
Next different bases, and solvents, as well as different temperatures, were examined to react compound 4 with 7a, and finally, potassium carbonate as a base in acetone at 25 °C for 3 h was chosen to afford the 8a–m (Table 2).
In the final step, different solvents, catalysts, and temperatures were used to examine the synthetic yield of 8a. As can be seen in Table 3, ethanol in presence of AcOH under the reflux conditions was chosen. As a result, 10a–m were prepared through the reaction of thiosemicarbazide with the appropriate 8a–m in presence of a catalytic amount of acetic acid in ethanol. The reaction mixture was refluxed for 3–4 h and then cooled to room temperature. The resulting precipitate was filtered, washed with ether, and recrystallized from ethanol to obtain the final product 10a–m derivatives26.
The structures of all purified products, 10a–m, were confirmed by IR, 1H NMR, 13C NMR, ESI–MS, and elemental analysis.
Evaluation of tyrosinase inhibitory activity
The tyrosinase inhibitory activity of derivatives was determined by a colorimetric method and the results of the tyrosinase inhibitory assay were shown in Table 4 in the terms of IC50s.
As can be seen in Table 4, 10a as an unsubstituted derivative (R = phenyl) exhibited weak tyrosinase inhibition with an IC50 value of 155.46 µM. Next, to improve the inhibitory activity, different substitutions on this ring were performed.
The incorporation of para-methyl as a small electron-donating group (10b, R = 4-methyl phenyl; IC50 ≥ 200 µM) deteriorated the activity while para-ethyl moiety as a bulk electron-donating group (10c, R = 4-ethyl phenyl; IC50 = 85.26 µM) improved the inhibition to around two-fold compared to 10a. The same trend were seen in 10d bearing spacious 2,3 dimethyl moiety with improved potency (10d, 2,3-methyl phenyl; IC50 = 100.83 µM). Substitution of the methoxy as an electron-donating group on the phenyl ring resulted in an around the three-fold amend of potency compared to 10a (10e, R = 2,3-methyl phenyl; IC50 = 47.35 ± 0.13 µM). It seems that the presence of electron-donating heteroatom on the aromatic ring increased the inhibitory activities.
NO2 substitution (10f, R = 4-nitrophenyl) as strongly electron-withdrawing through both resonance and inductive effects demonstrated an IC50 value of 39.85 µM resulting in the second potent derivative.
The evaluations on 10g–j as the halogen-substituted group exhibited that 10g (R = 4-chlorophenyl) with an IC50 of 25.75 ± 0.19 μM was categorized as the top potent tyrosinase inhibitor in this group. Indeed addition of an extra Cl functional group at the ortho position of 10g was inferior the activity (10h, 2,3-dichlorophenyl; IC50 = 168.77 µM). Although 10i containing 4-fluorophenyl (IC50 = 66.28 µM) decreased the activity compared to its 4-chlorine counterpart, the activity was improved in comparison with 10a. Assessments on bromine substituted moiety showed that the ortho-bromide group deteriorated the potency (10j, 2-bromophenyl; IC50 ≥ 200 µM).
Comparison of 10k (R = benzyl; IC50 = 61.96 µM ) with 10a (R = phenyl; IC50 = 155.46 µM) showed the important role of linker elongation so that 10k disclosed around 2.5 fold improvement in the potency compared to 10a. The same trend were seen in 10l (R = 4-bromophenyl; IC50 = 78.54 µM) compared to (10b, 4-methylphenyl IC50 ≥ 200 µM) as well as 10m (IC50 = 53.34 ± 0.23 µM) in comparesion with 10i (IC50 = 66.28 ± 0.15 µM). From the screening data of the 10k–m bearing benzyl group compared to their counterpart in the phenyl set (10a–j), superior inhibitory activity against tyrosinase was seen.
The summary of SAR of 10a–m was presented in Fig. 3.

Summary of SAR of 10a-m derivatives.
Determining the inhibitory type of 10 g against mushroom tyrosinase
The enzyme inhibition mode by the most potent derivative, 10g, was studied by Lineweaver–Burk plot analysis. The results are presented in Fig. 4. Lineweaver–Burk plots (plot of 1/V versus 1/[S]) for the inhibition of tyrosinase were obtained with several concentrations of 10g (as the inhibitor) and L-Dopa (as the substrate). The plots of 1/V versus 1/[S] presented straight lines which crossed the x-axis at similar points. It was found that as the inhibitor concentration elevated, the value of Vmax was reduced, but Km was not affected by the concentration. Therefore, the results represented that compound 10g is a noncompetitive inhibitor of mushroom tyrosinase. The obtained values for the Km and Vm are summarized in Table 5.

Lineweaver–Burk plot for mushroom tyrosinase enzyme inhibition by different concentrations of 10g in the presence of L-Dopa. The reciprocal tyrosinase inhibitory activity was plotted against the reciprocal substrate concentration (double reciprocal plot, n = 3).
Fluorescence quenching spectra of tyrosinase
Tyrosinase intrinsic fluorescence was studied as a means of elucidating the inhibitory mechanism of 10g. As shown in Fig. 5. tyrosinase emitted strong fluorescence with a maximum wavelength of 330 nm after being excited at 280 nm. Tyrosinase fluorescence intensity reduced as the concentration of inhibitor 10g increased.

Fluorescence intensity of Enzyme in presence of different concentrations of inhibitor 10g. (From top to down concentration of 10g increased).
The quenching of the intrinsic fluorescence provided unambiguous evidence that the 10g was capable of binding to tyrosinase, and the binding of 10g to tyrosinase resulted in a change in the microenvironment around the fluorophore. Table 6 summarizes the Ksv values obtained from Eq. (1).
One reliable method for classifying the quenching process is to examine how temperature affects drug-enzyme interaction. The slope of the fluorescence curves in Fig. 6 was used to determine the fluorescence quenching constant, KSV, and after that kq for temperatures of 298 K, and 304 K. From Table 6, it was observed that Kq of tyrosinase by inhibitor 10g were all higher than 2 × 1010 L⋅mol−1⋅s−1. Since it has been reported, that kq in different types of quenchers with biopolymer is generally 2 × 1010 L⋅mol-1⋅s-1, compound 10g is presumed to quench the intrinsic fluorescence of biomacromolecules through a static process. Ksv values for compound 10g were inversely related to temperature, suggesting that 10g and tyrosinase interact in a way that is impeded by static quenching.

Sterm-Volmer plots for the quenching of tyrosinase by 10g at 298 K and 304 K.
Binding constants and binding sites
If there were identical and independent binding sites in a protein during the process of static quenching, the binding constant (KA) and the binding site (n) might be calculated using the Double Logarithm of Log ((F0 – F)/F) = log KA + n Log [Q] Eq. (2).
As implied by Eq. (2), there is a straight-line relationship between log ((F0 − F)/F) versus log [Q], with the slope equal to n and the intercept equal to log KA (Fig. 7). Table 7 displays that the n values for inhibitor-tyrosinase complexes were close to 1, indicating that a single inhibitor 10g coupled to a single tyrosinase molecule. Additionally, when the temperature increased, the value of KA decreased, which was consistent with the reliance of ksv on temperature. The findings are consistent with a static quenching process.

Double-Logarithmic plots of 10g quenching effect on tyrosinase fluorescence at 298 K and 304 K.
Molecular docking study
The molecular binding analysis was then performed to gain insight into the interactions and binding mode of 10g in the tyrosinase active site. First docking validation was performed through the docking of tropolone as a crystallographic inhibitor against tyrosinase (PDB code: 2Y9X). MoleDock scoring functions were examined and the RMSD value was less than 2.00 Å. According to the kinetic study, 10g is a noncompetitive inhibitor and 10g binds to the enzyme at a location other than the active site. As a result, MolDock cavity detection was applied to find the possible cavity of the enzyme. As presented in Fig. 8, three possible binding sites were detected on the surface of the enzyme which can be suitable for noncompetitive inhibition (regardless of the active site). Next 10g as the potent inhibitor was docked on all of the potential binding sites of the enzyme. Considering the Moledock score and interactions, site 3 demonstrated the highest affinity in comparison to other identified sites (Fig. 9).

Potential binding sites for tyrosinase noncompetitive inhibitor colored as green.

3D (a) and 2D (b) binding orientation and interactions of compound 10g into the tyrosinase binding site.
As can be seen in Fig. 8, compound 10g fitted well in the tyrosinase allosteric site through various interactions including hydrogen bonding, electrostatic and hydrophobic interactions which enhanced its potency. The detailed interaction information of 10g is presented in Table 8.
Cell viability assessments
First, the toxicity of all derivatives against HEK 293 as a normal cell line was evaluated by MTT assay, and the results were presented in Table 9. The dose-dependent reduction of viability upon the increase in the concentration of derivatives can be seen.
MTT assessments of all derivatives at 16 µM exhibited no significant toxicity. The exception in this trend came back to 10i (R = para fluorophenyl), 10k (R = benzyl), and 10l (R = para-methyl benzyl) with 75.30 ± 2.36%, 77.28 ± 2.55% and 75.12 ± 1.99% cell viability, respectively. Derivatives 10f, 10g, 10j and 10m exhibited limited toxicity (viability > 85% ) at 32 µM. Evaluation among the phenyl set of compounds exhibited that the incorporation of fluorine group at the para position of phenyl ring (10b) reduced the bioavailability to around 46.59% followed by compound 10c bearing para ethyl moiety with 58.33 ± 1.03 cell viability at 64 µM. Among benzyl analogs, it was understood that elongation of the linker reduced the bioavailability. This trend can be seen in 10a vs 10k as well as 10b vs 10l. The exception in this trend came back to 10m which showed 71.25% cell viability compared to 10i 46.59%.
Next, the MTT assessment on 10g as the most potent tyrosinase inhibitor on the A375 cell line was performed and this compound did not show significant toxicity up to 32 μM (Table. 10).
Melanin content assay
The potency of 10g to reduce the melanin content on the A375 cell line was evaluated. As can be seen in Fig. 10, 10g reduced the melanin content in skin melanoma cells at tested concentrations compared to kojic acid as the positive control.

Effect of 10g on melanin content in the A375 cell line.
ADME-Toxicity profiles and physicochemical properties
The physicochemical properties and pharmacokinetic profile of the new synthesis derivatives were summarized in Table 1127. The results good human intestinal absorption of all compounds which caused fast absorption from the intestine to the bloodstream. All derivatives showed low metabolism with cytochrome p400 with low clearance and limited rat acute toxicity (value less than 0.5 categorized as high toxic).
Conclusion
Following our expertise in the rational design of tyrosinase inhibitors; herein, a series of thiosemicarbazide-thioquinoline derivatives bearing different aryl-acetamides were designed and synthesized. The most potent derivative 10g bearing 4-chlorophenyl ring demonstrated an IC50 value of 25.75 ± 0.19 μM compared to that of kojic acid as the positive control (IC50 = 34.93 ± 0.06 μM). SAR study revealed that the presence of one electron-withdrawing group at the para position of the aromatic ring improved the activity compared to the rest of the derivatives. Moreover, it is worth mentioning that 10g showed a noncompetitive inhibition mode of action in the enzymatic assay. In this context, in silico cavity detection was performed to extract the possible allosteric site and determine the binding pose of 10g with the binding site. 10g demonstrated several hydrophobic and hydrophilic interactions with the proposed binding site. In addition, cell toxicity assessments against HEK293 cell lines were executed and most derivatives exhibited no toxicity at 16 µM. Also, 10g derivative was tested against A375 cell lines, and it exhibited a significant reduction of melanin content on A375 cell lines at tested concentrations. It can be understood that this set of compounds can serve as structural outlines to design and expand potential tyrosinase inhibitors.
Material and methods
Chemistry
All the reagents were purchased from commercial sources. 1H and 13C NMR spectra were determined by a Bruker Advance spectrometer 400 MHz spectrometer. All the chemical shifts were reported as (δ) values ppm. Multiplicities were indicated by s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and coupling constant J was reported in hertz (Hz). CHNOS analysis was performed using Costech Company. IR spectra were obtained with a Nicolet, FR -IR Magna 550. Melting-point were also recorded using Kofler hot-stage apparatus. All the chemicals were purchased from Merck, Germany, and Sigma, Germany.
Synthesis of 2-chloroquinoline-3-carbaldehyde (3)
To stir DMF (3.6 mL, 46 mmol), 12.5 mL POCl3 (134 mmol) was added dropwise at 0 °C, then the mixture was allowed to stir for 30 min at room temperature. Next, acetanilide 1 (Compound 2, 18.5 mmol) was added and the resulting mixture was heated for 12 h at 80–90 °C. The mixture was poured into ice-cold water and stirred for 10 min. The obtained yellow precipitate 2-chloroquinoline-3-carbaldehyde 2 after filtration, was washed with cold water, was dried and without purification28.
Synthesis of 2-mercaptoquinoline-3-carbaldehyde (4)
The reaction was initiated by stirring the mixture of 2-chloroquinoline-3-carbaldehyde 2 (Compound 3, 1 mmol) and sodium sulfide (1 mmol) for 2 h at room temperature in dry DMF (5 mL). Then, the reaction mixture was poured into crushed ice and made acidic with acetic acid. The product was filtered off, washed with water, and dried to give the desired 2-mercaptoquinoline-3-carbaldehyde (Compound 4) that was further purified by recrystallization in ethanol28.
Synthesis of 7a-m derivatives
A mixture of aniline derivatives (5a–m, 1 mmol) and chloro acetyl chloride 2 (compound 6, 1 mmol) in DMF (5 ml) was stirred at room temperature for 30 min. Then, the obtained mixture was diluted with cold water, poured into ice, and formed a white precipitate that was filtered off. The residue was washed with cold water to obtain pure N‐phenyl‐2‐chloroacetamides29.
Synthesis of 8a-m derivatives
A mixture of 2-mercaptoquinoline-3-carbaldehyde (compound 4, 1 mmol) and 2-chloro-N-substituted acetamide derivatives (8a–m, 1 mm mol) in dry acetone (10 mL) and anhydrous K2CO3 (1 mmol) was stirred at room temperature for 8 h, filtered and the solid product formed was crystallized from ethanol to give 2-((3-formylquinolin-2-yl)thio)-N-phenylacetamide derivatives29.
Synthesis of 10a-m derivatives
The appropriate thiosemicarbazide (compound 9, 1 mmol) and the 8a–m (1 mmol) were dissolved in ethanol (10 mL). To this solution, a catalytic amount of acetic acid was added. The reaction mixture was refluxed for 3–4 h and then cooled to room temperature. The resulting precipitate was filtered, washed with ether, and recrystallized from ethanol to obtain the corresponding final products (10a–m).
(E)-2-((3-((2-carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)-N-phenylacetamide (10a)
Brown solid; Yield: 93%; MP = 180–182° C; IR (KBr, vmax) 3400(NH2 ), 3150(NH), 3025 (C-H Aromatic), 2970(CH2 Aliphatic), 1675(C = O), 1520(C = N) Cm-1; 1H NMR (400 MHz,DMSO-d6) δ 11.86 (s, 1H, NH Thio amid), 10.44 (s, 1H, CH Hydrazone), 8.86 (s, 1H, NH Amid), 8.56, 7.95 (s, 2H, NH2), 8.45 (s, 1H, H4), 7.91(d, J = 7.90 Hz, 1H, H5), 7.83 (d, J = 8.40 Hz 1H, H8), 7.72 (t, J = 8.30 Hz, 1H, H7), 7.62 (d, J = 7.70 Hz, 1H, H6), 7.62 (d, J = 7.70 Hz, 2H, H2, , H6,), 7.53 (t, J = 7.50 Hz, 1H, H6), 7.31 (t, J = 7.90 Hz, 2H, H3, , H5,), 7.05 (t, J = 7.40 Hz, 1H, H4,), 4.25 (s, 2H, CH2) ppm. 13C NMR (101 MHz, DMSO-d6): δ 178.72, 167.22, 156.70, 147.28, 139.68, 137.88, 135.18, 131.38, 129.26, 128.86, 127.61, 126.69, 126.12, 125.80, 123.73, 119.52, 35.67 ppm; ESI–MS (C19H17N5OS2): calculated m/z 395.09 [M + H]+, observed m/z 395.12 [M + H]+Anal. Calcd: C19H17N5OS2 ; C, 57.70; H, 4.33; N, 17.71; Found C, 57.90; H, 4.50; N, 17.90.
(E)-2-((3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)thio)-N-(p-tolyl)acetamide (10b)
Cream solid;Yield:91%;MP = 180–185 °C; IR (KBr, vmax) 3380(NH2), 3120(NH), 3040(C-H Aromatic), 2900(CH-Aliphatic), 1670(C = O), 1510 (C = N)Cm-1; 1H NMR (400 MHz,DMSO-d6) δ 11.81 (s, 1H, NH Thio amid), 10.34 (s, 1H, CH Hydrazone), 8.86 (s, 1H, NH Amid), 8.56, 7.91 (s, 2H, NH2), 8.45 (s, 1H, H4), 7.91(d, J = 7.80 Hz, 1H, H5), 7.83 (d, J = 8.40 Hz 1H, H8), 7.72 (t, J = 8.20 Hz, 1H, H7), 7.55–7.48 (m, 3H, H6, H2, , H6,),7.10 (d, J = 8.30 Hz, 2H, H3, , H5, ), 4.25 (s, 2H, CH2), 2.23 (s, 3H, CH3) ppm. 13C NMR (101 MHz, DMSO-d6): δ 178.72, 166.96, 156.72, 147.28, 137.90, 137.17, 135.16, 132.63, 131.37, 129.62, 128.84, 127.62, 126.67, 126.12, 125.78, 125.70, 119.54, 35.64, 20.91 ppm; ESI–MS (C20H19N5OS2): calculated m/z 409.10 [M + H]+, observed m/z 440.8 [M + H]+, Anal. Calcd : C20H19N5OS2 ; C, 58.67; H, 4.68; N, 17.10; Found C, 58.69; H, 4.66; N, 17.12.
(E)-2-((3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)thio)-N-(4-ethylphenyl)acetamide (10c)
Brown solid;Yield:93%;MP = 178–180 °C IR (KBr, vmax) 3390(NH2 ), 3130(NH), 3020(C-H Aromatic), 2975(CH2 Aliphatic), 1670(C = O), 1530 (C = N) Cm-1;1H NMR (400 MHz,DMSO-d6) δ 11.87 (s, 1H, NH Thio amid), 10.36 (s, 1H, CH Hydrazone), 8.85 (s, 1H, NH Amid), 8.56, 7.96 (s, 2H, NH2), 8.45 (s, 1H, H4), 7.91(d, J = 7.81 Hz, 1H, H5), 7.84 (d, J = 8.40 Hz 1H, H8), 7.72 (t, J = 8.30 Hz, 1H, H7), 7.57–7.50 (m, 3H, H6, H2, , H6,),7.13 (d, J = 8.40 Hz, 2H, H3, , H5, ), 4.25 (s, 2H, CH2), 2.57–2.51(m.2H, CH2ethyl),1.14(t, J = 7.50 Hz,3H,CH3ethyl), ppm. 13C NMR (101 MHz, DMSO-d6): δ 178.73, 166.97, 156.72, 147.29, 139.12, 137.89, 137.36, 135.16, 131.37, 128.84, 128.43, 127.63, 126.67, 126.13, 125.79, 119.64, 35.62, 28.06, 16.17 ppm; Anal. Calcd : C21H21N5OS2 ; C, 59.55; H, 5.00; N, 16.53; Found C, 59.69; H, 5.16; N, 16.47.
(E)-2-((3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)thio)-N-(2,3-dimethylphenyl)acetamide (10d)
Brown solid;Yield:93%;MP = 185–187 °C IR (KBr, vmax) 3430(NH2 ), 3140(NH), 3020(C-H Aromatic), 2975(CH2 Aliphatic) 1675(C = O), 1540 (C = N) Cm-1;1H NMR (400 MHz,DMSO-d6) δ 11.86 (s, 1H, NH Thio amid), 9.78 (s, 1H, CH Hydrazone), 8.89 (s, 1H, NH Amid), 8.55, 7.97 (s, 2H, NH2), 8.46 (s, 1H, H4), 7.95–7.89 (m, 2H, H5, H8),7.77 (t, J = 8.2 Hz, 1H, H7), 7.56 (t, J = 7.70 Hz, 1H, H6), 7.11(d, J = 7.3 Hz, 1H, H6,), 7.07–6.96 (m, 2H, H4,, H5’), 4.25 (s, 2H, CH2),2.21 (s.3H, CH3),2.02(s,3H,CH3) ppm. 13C NMR (101 MHz, DMSO-d6): δ 178.71, 167.17, 156.76, 147.38, 137.85, 137.42, 136.55, 135.07, 131.73, 131.36, 128.88, 127.72, 127.44, 126.70, 126.22, 125.86, 125.64, 123.86, 34.83, 20.59, 14.48 ppm; Anal. Calcd : C21H21N5OS2 ; C, 59.57; H, 5.03; N, 16.55; Found C, 59.57; H, 5.10; N, 16.47.
(E)-2-((3-((2-carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)-N-(4-methoxyphenyl)acetamide (10e)
Cream solid;Yield:93%;MP = 191–193 °C; IR (KBr, vmax) 3390(NH2 ), 3140 (NH), 3030 (C-H Aromatic), 2910 (CH-Aliphatic), 1680 (C = O), 1530(C = N)Cm-1;1H NMR (400 MHz,DMSO-d6) δ 11.85 (s, 1H, NH Thio amid), 10.30 (s, 1H, CH Hydrazone), 8.85 (s, 1H, NH Amid), 8.56, 7.74 (s, 2H, NH2), 8.45 (s, 1H, H4), 7.91(d, J = 7.70 Hz, 1H, H5), 7.85 (d, J = 8.30 Hz 1H, H8), 7.73 (t, J = 8.30 Hz, 1H, H7), 7.56–7.51 (m, 3H, H6 , H2, , H6,), 6.88 (d, J = 9.10 Hz, 2H, H3, , H5,), 4.23 (s, 2H, CH2), 3.71(s, 3H, CH3) ppm. 13C NMR (101 MHz, DMSO-d6): δ 178.72, 16.68, 156.74, 155.67, 147.30, 137.90, 135.13, 132.82, 131.37, 128.85, 127.64, 126.67, 126.13, 125.79, 121.10, 114.35, 55.59, 35.54 ppm; Anal. Calcd : C20H19N5O2S2; C, 56.45; H, 4.50; N, 16.46; Found C, 56.65; H, 4.70; N, 16.65.
(E)-2-((3-((2-carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)-N-(4-nitrophenyl)acetamide (10f)
Pale yellow solid;Yield:93%;MP = 180–182 °C IR (KBr, vmax) 3420 (NH2), 3150 (NH), 3020 (C-H Aromatic), 2965 (CH2 Aliphatic), 1670 (C = O), 1555–1350(NO2 ) Cm-1 ;1H NMR (400 MHz,DMSO-d6) δ, 11.12 (s, 1H, CH Hydrazone), 10.20 (s, 1H, NH Amid), 8.96 (s, 1H, H4), 8.24 (d, J = 9.30 Hz, 2H, H3, , H5,), 8.10 (d, J = 8.00 Hz, 1H, H5), 7.90 (d, J = 9.30 Hz, 2H, H2, , H6,), 7.87–7.82 (m, ,1H, H8), 7.79 (t, J = 8.10 Hz, 1H, H7), 7.59 (t, J = 8.10 Hz, 1H, H6), 4.25 (s, 2H, CH2) ppm. 13C NMR (101 MHz, DMSO-d6): δ 192.18, 168.76, 157.53, 148.76, 148.66, 146.52, 145.96, 142.56, 134.08, 130.11, 127.53, 127.19, 127.03, 125.58, 124.80, 119.09, 35.63 ppm; ESI–MS (C19H16N6O3S2): calculated m/z 440.07 [M + H]+, observed m/z 440.10 [M + H]+ ,Anal. Calcd : C19H16N6O3S2, C,51.81.29; H, 3.66; N, 19.08; Found C, 51.85; H, 3.64.; N, 19.15.
(E)-2-((3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)thio)-N-(4-chlorophenyl)acetamide (10g)
Brown solid;Yield:90%;MP = 185–191 °C; IR (KBr, vmax) 3420(NH2 ), 3150(NH), 3030(C-H Aromatic), 2980 (CH2 Aliphatic), 1680(C = O), 1530 (C = N) Cm-1; 1H NMR (400 MHz,DMSO-d6) δ 11.87 (s, 1H, NH Thio amid), 10.60 (s, 1H, CH Hydrazone), 8.58 (s, 1H, NH Amid), 8.84, 8.45 (s, 2H, NH2), 7.95 (s, 1H, H4), 7.91(d, J = 7.77 Hz, 1H, H5), 7.80 (d, J = 8.50 Hz 1H, H8), 7.72 (t, J = 8.30 Hz, 1H, H7), 7.67 (d, 2H, J = 8.90 Hz,H2, , H6,), 7.52 (t, J = 8.00 Hz, 1H, H6)7.36 (d, J = 8.90 Hz, 2H, H3, , H5, ), 4.25 (s, 2H, CH2) ppm. 13C NMR (101 MHz, DMSO-d6): δ 178.72, 167.48, 156.60, 147.24, 138.65, 137.96, 135.30, 131.39, 129.18, 128.85, 127.56, 127.56, 127.26, 126.69, 126.09, 125.78, 121.04, 35.72 ppm; ESI–MS (C19H16ClN5OS2): calculated m/z 439.10 ,432.5[M + H]+and [M + H + 2]+,observed m/z 429.8,432.6 [M + H]+ and [M + H + 2]+, Anal. Calcd : C19H16ClN5OS2 ; C, 53.07; H, 3.75; N, 16.29; Found C, 53.19; H, 3.64; N,16.12C19H16N5OS2.
(E)-2-((3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)thio)-N-(2,4-dichlorophenyl)acetamide (10h)
Brown solid;Yield:86%;MP = 188–190 °C; IR (KBr, vmax) 3440(NH2 ), 3160(NH), 3035 (C-H Aromatic), 2960 (CH2 Aliphatic), 1675 (C = O), 1540 (C = N) Cm-1;1H NMR (400 MHz,DMSO-d6) δ 11.88 (s, 1H, NH Thio amid), 9.96 (s, 1H, CH Hydrazone), 8.87 (s, 1H, NH Amid), 8.58,7.94 (s, 2H, NH2), 8.45 (s, 1H, H4), 7.88 (d, J = 8.40 Hz, 1H, H5), 7.81 (d, J = 8.80 Hz, 1H, H6,), 7.75 (d, J = 8.30 Hz, 1H, H8), 7.72 (t, J = 8.10 Hz, 1H, H7), 7.63 (d,4 J = 2.4 Hz, 1H, H3,), 7.54 (t, J = 7.80 Hz, 1H, H6), 7.38 (dd, 3JC-H = 8.80 Hz,4JC-H = 2.50 Hz, 1H, H5,), 4.33 (s, 2H, CH2) ppm. 13C NMR (101 MHz, DMSO-d6): δ 178.71, 168.03, 156.30, 147.27, 137.90, 135.49, 134.58, 131.39, 129.45, 129.39, 129.33, 128.84, 128.08, 127.71, 126.79, 126.73, 126.48, 126.16, 125.87, 34.91 ppm;Anal. Calcd for C19H15Cl2N5OS2 ; C, 49.14; H, 3.26; N, 15.08; Found ; C, 49.34; H, 3.45; N, 15.30.
(E)-2-((3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)thio)-N-(4-fluorobenzyl)acetamide (10i)
Brown solid;Yield:89%;MP = 186–188 °C; IR (KBr, vmax) 3410(NH2 ), 3130(NH), 3060(C-H Aromatic), 2975 (CH2 Aliphatic), 1670(C = O), 1520 (C = N) Cm-1;1H NMR (400 MHz,DMSO-d6) δ 11.86 (s, 1H, NH Thio amid), 8.86 (s, 1H, CH Hydrazone), 8.76 (t, J = 6.10 Hz, 1H, NH Amid), 8.44 (s, 1H, H4), 8.56, 7.99 (s, 2H, NH2), 7.91(d, J = 8.00 Hz, 1H, H5), 7.78–7.68 (m, 2H, H6 , H8), 7.55 (t, J = 8,10 Hz, 1H, H7), 7.25–7.20 (m, 2H, H2, , H6,), 6.96 (t, J = 8,90 Hz, 2H, H3, , H5,), 4.11 (s, 2H, CH2) ppm. 13C NMR (101 MHz, DMSO-d6): δ 178.70, 168.21, 161.48(d, 1JCF=241), 156.59, 147.31, 137.85, 135.98, 135.94, 134.93, 131.19, 129.49, 129.41, 128.78, 127.80, 126.64, 126.22, 125.81, 115.33, 115.12, 42.24, 34.25 ppm;Anal. Calcd for C20H18FN5OS2: C, 56.19; H, 4.24; N, 16.38;Found; C, C, 56.40; H, 4.44; N, 16.59.
(E)-N-(2-bromophenyl)-2-((3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)thio)acetamide (10j)
Brown solid; Yield: 93%; MP = 181–183° C; IR (KBr, vmax) 3380(NH2), 3140(NH), 3030 (C-H Aromatic), 2965(CH2 Aliphatic), 1655(C = O), 1540(C = N) Cm-1; 1H NMR (400 MHz,DMSO-d6) δ 11.85 (s, 1H, NH Thio amid), 10.52 (s, 1H, CH Hydrazone), 8.86 (t, J = 6.10 Hz, 1H, NH Amid), ), 8.57, 7.95 (s, 2H, NH2), 8.45 (s, 1H, H4), 7.91(d, J = 7.70 Hz, 1H, H5), 7.82 (d, J = 8,40 Hz, 1H, H8), 7.72 (t, J = 8,30 Hz, 1H, H7), 7.67–7.61 (m, 2H, H2, , H6,), 7.53 (t, J = 7.50 Hz, 1H, H6), 7.15 (t, J = 8,90 Hz, 2H, H3, , H5,), 4.20 (s, 2H, CH2), ppm. 13C NMR (101 MHz, DMSO-d6): δ 178.71, 167.64, 156.25, 147.32, 137.87, 136.62, 135.39, 133.10, 131.40, 128.84, 128.54, 127.83, 127.16, 126.80, 126.32, 126.20, 125.89, 117.12, 34.86 ppm; Anal. Calcd : C19H16BrN5OS2 ; C, 48.10; H, 3.40; N, 14.76; Found C, 48.30; H, 3.60; N, 14.95.
(E)-N-benzyl-2-((3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)thio)acetamide (10k)
Brown solid;Yield:94%;MP = 183–185 °C; IR (KBr, vmax) 3410(NH2 ), 3160(NH) , 3045(C-H Aromatic), 2975 (CH2 Aliphatic) 1655 (C = O), 1520 (C = N) Cm-1;1H NMR (400 MHz,DMSO-d6) δ 11.84 (s, 1H, NH Thio amid), 8.88 (s, 1H, CH Hydrazone), 8.75 (t, J = 5.80 Hz, 1H, NH Amid), 8.54, 7.98 (s, 2H, NH2), 8.45(s, 1H, H4), 7.92(d, J = 8.00 Hz, 1H, H5), 7.82 (d, J = 8.30 Hz 1H, H8), 7.74 (t, J = 8,00 Hz, 1H, H7), 7.57 (d, J = 7.20 Hz, 1H, H6), 7.23–7.13 (m, 5H, H phenyl), 4.11 (s, 2H, CH2), ppm. 13C NMR (101 MHz, DMSO-d6): δ 178.72, 168.15, 156.64, 147.36, 139.75, 137.81, 134.87, 131.24, 128.80, 128.57, 127.86, 127.45, 127.08, 126.66, 126.25, 125.84, 42.92, 34.22 ppm; Anal. Calcd : C20H19N5OS2 ; C, 58.66; H, 4.68; N, 17.10; Found C, 58.54; H, 4.57; N, 17.24.
(E)-2-((3-((2-carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)-N-(4-methylbenzyl)acetamide (10l)
Brown solid; Yield: 93%; MP = 180–182° C; IR (KBr, vmax) 3370 (NH2 ), 3120 (NH), 3025 (C-H Aromatic), 2970(CH2 Aliphatic), 1675(C = O),1510 (C = N) Cm-1; 1H NMR (400 MHz,DMSO-d6) δ 11.85 (s, 1H, NH Thio amid), 8.87 (s, 1H, CH Hydrazone), 8.69 (t, J = 5.90 Hz, 1H, NH Amid), 8.54, 7.98 (s, 2H, NH2), 8.44 (s, 1H, H4), 7.92(d, J = 8.00 Hz, 1H, H5), 7.78 (d, J = 8.20 Hz 1H, H8), 7.77–7.69 (m, 1H, H7), 7.56 (t, J = 7.90 Hz, 1H, H6), 7.09 (d, J = 7.80 Hz, 2H, H2, , H6,), 6.98 (d, J = 7.80 Hz, 2H, H3, , H5,), 4.26 (d, J = 5.90 Hz, 2H, CH2-NH), 4.11 (s, 2H, CH2), 2.22(s, 3H, CH3) ppm. 13C NMR (101 MHz, DMSO-d6): δ 178.70, 171.11, 168.05, 156.64, 147.35, 137.79, 136.69, 136.10, 134.85, 131.19, 129.13, 128.78, 127.86, 127.50, 126.64, 126.22, 125.82, 42.69, 34.24, 21.13 ppm; Anal. Calcd : C21H21N5OS2 ; C, 59.55; H, 5.00; N, 16.54; Found C, 59.75; H, 5.20; N, 16.74.
(E)-2-((3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)thio)-N-(4-fluorobenzyl)acetamide (10m)
Brown solid;Yield:89%;MP = 186–188 °C; IR (KBr, vmax) 3410(NH2 ), 3130(NH), 3060(C-H Aromatic), 2975 (CH2 Aliphatic), 1670(C = O), 1520 (C = N) Cm-1;1H NMR (400 MHz,DMSO-d6) δ 11.86 (s, 1H, NH Thio amid), 8.86 (s, 1H, CH Hydrazone), 8.76 (t, J = 6.10 Hz, 1H, NH Amid), 8.44 (s, 1H, H4), 8.56, 7.99 (s, 2H, NH2), 7.91(d, J = 8.00 Hz, 1H, H5), 7.78–7.68 (m, 2H, H6 , H8), 7.55 (t, J = 8,10 Hz, 1H, H7), 7.25–7.20 (m, 2H, H2, , H6,), 6.96 (t, J = 8,90 Hz, 2H, H3, , H5,), 4.11 (s, 2H, CH2) ppm. 13C NMR (101 MHz, DMSO-d6): δ 178.70, 168.21, 161.48(d, 1JCF=241), 156.59, 147.31, 137.85, 135.98, 135.94, 134.93, 131.19, 129.49, 129.41, 128.78, 127.80, 126.64, 126.22, 125.81, 115.33, 115.12, 42.24, 34.25 ppm;Anal. Calcd for C20H18FN5OS2: C, 56.19; H, 4.24; N, 16.38;Found; C, C, 56.40; H, 4.44; N, 16.59.
Tyrosinase assay
Mushroom tyrosinase (EC 1.14.18.1) (Sigma Chemical Co.) was assayed using L-Dopa as the substrate as reported in our previous studies with some modifications30,31,32. The enzyme diphenolase activity was monitored spectrophotometrically by observing dopachrome formation at 490 nm. All the test samples were first dissolved in DMSO at 10 mM and diluted to the required concentrations. Initially, in a 96-well microplate, 10 µl of test samples were added to 160 µl of 50 mM phosphate buffer (pH = 6.8) and then 10 µl tyrosinase (500 U mL−1) was added. After the mixture was pre-incubated at 28 °C for 20 min, 20 µl of L-Dopa solution (7 mM) was added to the mixture. After 10 min incubation absorbance of samples was measured. DMSO without test compounds was used as the control, and kojic acid was used as a positive control. Each assay was conducted as three separate replicates. The inhibitory activity of the tested compounds was expressed as the concentration that inhibited 50% of the enzyme activity (IC50). The percentage inhibition ratio was calculated according to the following equation:
Enzyme kinetic studies
The kinetic study for the inhibition of tyrosinase by compound 10g was carried out using four different concentrations of inhibitor (1, 10, 20, and 40 µM) against tyrosinase with different concentrations of L-Dopa (0.25, 0.5, 0.75, and 1 mM) as the substrate. The Lineweaver–Burk reciprocal plot was provided by plotting 1/V against 1/[S] at variable concentrations of the substrate L-Dopa (0.25, 0.5, 0.75, and 1 mM) The inhibition constant Ki was achieved by the plot of slopes versus the corresponding concentrations of the compound 10g24.
Fluorescence spectroscopy
The fluorescent measurements were carried out in each well of the black 96 well plate by adding various concentrations of inhibitor 10g, (Q = 5, 10, 20, 40, and 100 µM) to a constant concentration of tyrosinase (273 IU/mL). Next, phosphate buffer solution (0.05 M, pH = 6.8) was used to bring the capacity of each well to 200 µL. Before measurements, all solutions were properly mixed and allowed to stand for 20 min at 298 K and 304 K. A fluorescence spectrometer (BioTek’s multi-mode plate reader, USA) was used to measure the fluorescence spectra. The bandwidths of excitation and emission were both set at 5 nm, and fluorescence emission spectra were captured spanning the wavelength range of 310–400 nm at an excitation wavelength of 280 nm.
The linear Stern–Volmer Eq. (1) was used to examine the fluorescence quenching:
where, F and F0 represent the fluorescence intensity in the presence or absence of inhibitors, respectively. The bimolecular quenching constant is denoted by the symbol kq, whereas the average lifespan of a biomolecule without a quencher is denoted by the symbol τ0, and its value is 10−8 s. [Q] refers to the inhibitor concentration (µM). KSV which is calculated by kq . τ0. is the fluorescence quenching constant.
In order to calculate the apparent binding constant and binding fraction of fluorophore group of tyrosinase accessible to inhibitor 10g, Stern–Volmer Eq. (1) should be modified to Eq. (2):
In this equation, KA indicates either the modified apparent binding constant or the Stern–Volmer quenching constant. n represents the fraction of fluorophore accessible to the quencher. The intercept of the curve that is obtained by plotting log (F0-F/F) versus log [Q] is log KA, and the slope of the curve is n. Therefore, the intercept and slope data may be used to calculate the values of KA and n.
In silico studies
The molecular docking studies of the most potent inhibitor were performed against tyrosinase (PDB code: 2Y9X) to observe the binding orientation and interactions using the MolDock program. The 3D crystal structure of tyrosinase was retrieved from Protein Data Bank. Water molecules and the cognate ligand (tropolone) were removed from the receptor and the hydrogen atoms were added and non-polar hydrogens were merged into related atoms of the receptor via protein preparation of MolDock software. Moldock scoring function and MolDock SE algorithm of the program were used for re-dock tropolone inside the enzyme with a binding site radius of 9 Å. All other options were set as default. 10g were drawn using Hyperchem and subjected to energy minimization using MM+ and AM1 algorithms. Similarly, the MolDock program was applied for doing docking analyses of 10g. The top-score binding pose was analyzed with Discovery Studio Visualizer18,21,31.
MTT assay for cell viability
The cytotoxic activity of all derivatives against the HEK 293 cell line and 10g against A375 (at a density of 5 × 103 cells/ml) using 3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide (MTT) assay were performed. Cells were grown at 37 ºC in the presence of CO2 5% in DMEM (Gibco BRL, Grand Island, NY, USA), 10% fetal bovine serum (FBS, Gibco BRL), and penicillin/streptomycin (100 IU/mL and 100 µg/mL, respectively). Next cells were seeded in a 96-well plate and incubated at 37 \(^\circ{\rm C}\) with derivatives at different concentrations for 48 h. Following the treatment, cells were incubated with MTT (0.5 mg/ml) at 37 \(\mathrm{^\circ{\rm C} }\) for 3 h. The MTT-containing medium was then removed, and 100 μl of DMSO was added to each well, mixed thoroughly with a 10 min shake to dissolve formazan crystals. The absorbance of each well was measured at 540 nm33.
Determination of melanin content
The assay was performed according to previously reported procedures. In detail, A375 cells were seeded in six-well plates (2.0 × 105 cells/well). After 24 h, the medium was replaced by a fresh one and treated with 10g at different concentrations and the plate was incubated for an extra 48 h. After that cells were washed twice with PBS and harvested using 0.25 M trypsin, then dissolved in 300 μl of 1 N NaOH/10% DMSO buffer and boiled for 2 h at 80 ºC to solubilize the melanin. The absorbance of the supernatant was measured at 470 nm in a microplate reader. The obtained results were normalized using total protein content. Kojic acid was used as a positive30,34.
Data availability
The datasets generated and/or analysed during the current study are available in the Worldwide Protein Data Bank (wwPDB) repository. (http://www.rcsb.org).
References
Costin, G.-E. & Hearing, V. J. Human skin pigmentation: melanocytes modulate skin color in response to stress. FASEB J. 21(4), 976–994 (2007).
Solano, F. Photoprotection and skin pigmentation: Melanin-related molecules and some other new agents obtained from natural sources. Molecules 25(7), 1537 (2020).
Lee, S.-E. et al. Deoxyvasicinone with Anti-Melanogenic Activity from Marine-Derived Streptomyces sp. CNQ-617. Marine Drugs 20(2), 155 (2022).
Slominski, R. M., Zmijewski, M. A. & Slominski, A. T. The role of melanin pigment in melanoma. Exp. Dermatol. 24(4), 258–259 (2015).
Kim, Y. J., Kang, K. S. & Yokozawa, T. The anti-melanogenic effect of pycnogenol by its anti-oxidative actions. Food Chem. Toxicol. 46(7), 2466–2471 (2008).
Bose, A., Petsko, G. A. & Eliezer, D. Parkinson’s disease and melanoma: Co-occurrence and mechanisms. J. Parkinsons Dis. 8(3), 385–398 (2018).
Chen, C.-Y., Lin, L.-C., Yang, W.-F., Bordon, J. & Wang, H.-M.D. An updated organic classification of tyrosinase inhibitors on melanin biosynthesis. Curr. Org. Chem. 19(1), 4–18 (2015).
Sánchez-Ferrer, Á., Rodríguez-López, J. N., García-Cánovas, F. & García-Carmona, F. Tyrosinase: A comprehensive review of its mechanism. Biochimica et Biophysica Acta (BBA) Protein Struct. Mol. Enzymol. 1247(1), 1–11 (1995).
Strothkamp, K., Jolley, R. & Mason, H. Quaternary structure of mushroom tyrosinase. Biochem. Biophys. Res. Commun. 70(2), 519–524 (1976).
Ismaya, W. T. et al. Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry 50(24), 5477–5486 (2011).
Goldfeder, M., Kanteev, M., Isaschar-Ovdat, S., Adir, N. & Fishman, A. Determination of tyrosinase substrate-binding modes reveals mechanistic differences between type-3 copper proteins. Nat. Commun. 5(1), 4505 (2014).
Bukhari, S. N. A. et al. Biological activity and molecular docking studies of curcumin-related α, β-unsaturated carbonyl-based synthetic compounds as anticancer agents and mushroom tyrosinase inhibitors. J. Agric. Food Chem. 62(24), 5538–5547 (2014).
Dettori, M. A. et al. Synthesis and studies of the inhibitory effect of hydroxylated phenylpropanoids and biphenols derivatives on tyrosinase and laccase enzymes. Mol. (Basel, Switzerland) 25(11), 2709 (2020).
Musiol, R. et al. Investigating biological activity spectrum for novel quinoline analogues. Bioorg. Med. Chem. 15(3), 1280–1288 (2007).
Chu, X.-M. et al. Quinoline and quinolone dimers and their biological activities: An overview. Eur. J. Med. Chem. 161, 101–117 (2019).
Alam, M. B. et al. Inhibition of melanogenesis by jineol from Scolopendra subspinipes mutilans via MAP-Kinase mediated MITF downregulation and the proteasomal degradation of tyrosinase. Sci. Rep. 7(1), 45858 (2017).
Mahajan, P. G. et al. Facile synthesis of new quinazolinone benzamides as potent tyrosinase inhibitors: Comparative spectroscopic and molecular docking studies. J. Mol. Struct. 1198, 126915 (2019).
Sepehri, N. et al. The natural-based optimization of kojic acid conjugated to different thio-quinazolinones as potential anti-melanogenesis agents with tyrosinase inhibitory activity. Bioorg. Med. Chem. 36, 116044 (2021).
Pillaiyar, T., Namasivayam, V., Manickam, M. & Jung, S.-H. Inhibitors of melanogenesis: An updated review. J. Med. Chem. 61(17), 7395–7418 (2018).
Soares, M. A. et al. Thiosemicarbazones as inhibitors of tyrosinase enzyme. Bioorg. Med. Chem. Lett. 27(15), 3546–3550 (2017).
Hosseinpoor, H. et al. A series of benzylidenes linked to hydrazine-1-carbothioamide as tyrosinase inhibitors: Synthesis, biological evaluation and structure−activity relationship. Chem. Biodivers. 17(8), e2000285 (2020).
Iraji, A. et al. Synthesis, biological evaluation and molecular docking analysis of vaniline–benzylidenehydrazine hybrids as potent tyrosinase inhibitors. BMC Chem. 14(1), 28 (2020).
Iraji, A., Khoshneviszadeh, M., Bakhshizadeh, P., Edraki, N. & Khoshneviszadeh, M. Structure-based design, synthesis, biological evaluation and molecular docking study of 4-Hydroxy-N’-methylenebenzohydrazide derivatives acting as tyrosinase inhibitors with potentiate anti-melanogenesis activities. Med. Chem. 16(7), 892–902 (2020).
Iraji, A., Panahi, Z., Edraki, N., Khoshneviszadeh, M. & Khoshneviszadeh, M. Design, synthesis, in vitro and in silico studies of novel Schiff base derivatives of 2-hydroxy-4-methoxybenzamide as tyrosinase inhibitors. Drug Dev. Res. 82(4), 533–542 (2021).
Mistry, B. M. & Jauhari, S. Synthesis and in vitro antimicrobial and anti-tubercular evaluation of some quinoline-based azitidinone and thiazolidinone analogues. Med. Chem. Res. 22(2), 635–646 (2013).
Kiran, B. M., Nandeshwarappa, B. P., Vaidya, V. P. & Mahadevan, K. M. Chemistry of substituted quinolines: Thieno [2, 3-b] and thiopyrano [2, 3-b] quinolines. Phosphorus Sulfur Silicon Relat. Elem. 182(5), 969–980 (2007).
Pires, D. E. V., Blundell, T. L. & Ascher, D. B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 58(9), 4066–4072 (2015).
Noori, M. et al. Design, synthesis, in vitro, and in silico enzymatic evaluations of thieno[2,3-b]quinoline-hydrazones as novel inhibitors for α-glucosidase. Bioorg. Chem. 127, 105996 (2022).
Mohammadi-Khanaposhtani, M. et al. New 4-phenylpiperazine-carbodithioate-N-phenylacetamide hybrids: Synthesis, in vitro and in silico evaluations against cholinesterase and α-glucosidase enzymes. Arch. Pharm. 355(5), 2100313 (2022).
Karimian, S. et al. Design, synthesis, and biological evaluation of symmetrical azine derivatives as novel tyrosinase inhibitors. BMC Chem. 15(1), 54 (2021).
Hosseinpoor, H. et al. Anti-melanogenesis and anti-tyrosinase properties of aryl-substituted acetamides of phenoxy methyl triazole conjugated with thiosemicarbazide: Design, synthesis and biological evaluations. Bioorganic Chem. 114, 104979 (2021).
Mahdavi, M. et al. Synthesis of new benzimidazole-1,2,3-triazole hybrids as tyrosinase inhibitors. Chem. Biodivers. 15(7), e1800120 (2018).
Edraki, N. et al. 2-Imino 2H-chromene and 2-(phenylimino) 2H-chromene 3-aryl carboxamide derivatives as novel cytotoxic agents: Synthesis, biological assay, and molecular docking study. J. Iran. Chem. Soc. 13(12), 2163–2171 (2016).
Hosseinpoor, H. et al. Anti-melanogenesis and anti-tyrosinase properties of aryl-substituted acetamides of phenoxy methyl triazole conjugated with thiosemicarbazide: Design, synthesis and biological evaluations. Bioorg Chem 114, 104979 (2021).
Author information
Authors and Affiliations
Contributions
M.N. synthesized compounds. R.S. performed the biological assay. A.T. synthesized compounds. M.S. performed the biological assay. A.I performed the biological assay and contributed to the preparation of the manuscript. M.K.G performed chemical analysis. N.D. contributed to the design and characterization of compounds. C.I performed in silico study. E.Z. performed the biological assay. S.M.M.P performed in silico study. F.R performed chemical analysis. B.L supervised the biological tests. M.A supervised the biological tests. M.H. synthesized compounds M.M. supervised all phases of the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Noori, M., Sabourian, R., Tasharoie, A. et al. Thioquinoline derivatives conjugated to thiosemicarbazide as potent tyrosinase inhibitors with anti-melanogenesis properties. Sci Rep 13, 2578 (2023). https://doi.org/10.1038/s41598-023-28852-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-28852-1
- Springer Nature Limited
This article is cited by
-
Structure-based development of 3,5-dihydroxybenzoyl-hydrazineylidene as tyrosinase inhibitor; in vitro and in silico study
Scientific Reports (2024)
-
Synthesis, in vitro potency of inhibition, enzyme kinetics and in silico studies of quinoline-based α-glucosidase inhibitors
Scientific Reports (2024)
-
Thiazolopyrimidine derivatives as novel class of small molecule tyrosinase inhibitor
BMC Chemistry (2023)
-
Synthesis, in vitro α-glucosidase inhibitory activities, and molecular dynamic simulations of novel 4-hydroxyquinolinone-hydrazones as potential antidiabetic agents
Scientific Reports (2023)