
Abstract
Tensor decomposition- and principal component analysis-based unsupervised feature extraction were proposed almost 5 and 10 years ago, respectively; although these methods have been successfully applied to a wide range of genome analyses, including drug repositioning, biomarker identification, and disease-causing genes’ identification, some fundamental problems have been identified: the number of genes identified was too small to assume that there were no false negatives, and the histogram of P values derived was not fully coincident with the null hypothesis that principal component and singular value vectors follow the Gaussian distribution. Optimizing the standard deviation such that the histogram of P values is as much as possible coincident with the null hypothesis results in an increase in the number and biological reliability of the selected genes. Our contribution was that we improved these methods so as to be able to select biologically more reasonable differentially expressed genes than the state of art methods that must empirically assume negative binomial distributions and dispersion relation, which is required for the selecting more expressed genes than less expressed ones, which can be achieved by the proposed methods that do not have to assume these.
Similar content being viewed by others

Introduction
Identifying differentially expressed genes (DEGs) on the basis of comparative analyses1,2 has always been difficult. This challenge is attributable to multiple reasons; however, the primary reason is being a large p small n problem. In a large p small n problem, it is difficult to select features based on statistical criteria because a small number of samples (\(=n\)) have a tendency to lead to low significance; in reality, the obtained P values must be heavily corrected by considering a large number of features (\(=p\)). This makes it difficult to find features with significance. To resolve this difficulty, many methods specific to gene expression analysis have been proposed. For example, significant analysis microarray (SAM)3 adds a small amount of constancy to gene expression, thereby avoiding the misidentification of low expressed genes as DEGs. Limma4 applied a Bayesian strategy to logarithmic gene expression. After high-throughput sequencing (HTS) became popular, P values are attributed to individual genes, assuming that gene expression follows a negative binomial (NB) distribution5,6, which is one of the simplest positively valued distributions with a tunable mean and variance. In addition to this, the so-called dispersion relation5,6,
has also been assumed, where \(\mu\) and \(\alpha\) are the mean and variance, respectively, and \(\alpha _0\) and \(\alpha _1\) are regression coefficients; to our knowledge, Eq. (1) is purely empirical and lacks rationalization. Despite these difficulties, many proposed state-of-art methods5,6,7,8,9 have been widely employed and used in various studies.
Contrary to these empirical methods, we proposed tensor decomposition (TD)- and principal component analysis (PCA)-based unsupervised feature extraction (FE)10 that only assumes that principal component (PC) and singular value vectors (SVVs) obey Gaussian distribution. Despite this simplicity, TD- and PCA-based unsupervised FE have been successfully applied to a wide range of genomic analyses. However, there have been two problems: 1. The histogram of the P values is not fully coincident with the null hypothesis that PC and SVV obey Gaussian distribution and 2. The number of genes selected is too small to have no false negatives. In this paper, we have shown that the optimization of standard deviation (SD) in Gaussian distribution can resolve these problems.
We tried optimizing SD for PCA-based unsupervised FE and applied this to two highly curated data sets–MAQC and SEQC. Then, we tested the optimization of SD for TD-based unsupervised FE and applied it to two more realistic problems: (1) drug repositioning for SARS-CoV-2 and (2) the analysis of gene expression of multiple organs treated with multiple drugs, to which TD-based unsupervised FE without SD optimization was already applied.
Our contributions are as follows. First, our methods allow more expressed genes to be more selected as DEGs without empirical dispersion relation, Eq. (1). Second, our methods can select significant DEGs without assuming not rationalized negative binomial distribution for individual gene expression. Third, our selected DEGs are much more biologically reasonable than those selected by other state of art methods.
Results
Outlines of TD and PCA based unsupervised FE
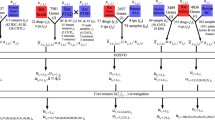
In this section, we have briefly explained the algorithm of PCA- and TD-based unsupervised FE (Fig. 1) before explaining how we could improve them.

Schematic figure of TD- and PCA-based unsupervised FE with optimized SD.
When a gene expression profile is formatted as a matrix, \(x_{ij} \in \mathbb {R}^{N \times M}\), which represents the gene expression of the ith gene of the jth sample, we use PCA-based unsupervised FE. After standardizing \(x_{ij}\) as
a gram matrix \(\sum _j x_{ij}x_{i'j} \in \mathbb {R}^{N \times N}\) was diagonalized as
where \(u_{\ell i} \in \mathbb {R}^{N \times N}\) is the \(\ell\)th PC score attributed to gene i. The \(\ell\)th PC loading attributed to the jth sample can be computed as
After identifying \(v_{\ell j}\), which is associated with a desired property, e.g., the district between control and treated samples, we attributed the P values to the gene i using the corresponding PC score, \(u_{\ell i}\), as
assuming that \(u_{\ell i}\) obeys the Gaussian distribution, where \(P_{\chi ^2} [ >x]\) is cumulative \(\chi ^2\) distribution when an argument larger than x and \(\sigma _\ell\) is the SD,
When we have gene expression that is formatted as a tensor, \(x_{ijk} \in \mathbb {R}^{N \times M \times K}\), for the expression of the ith gene at jth sample with the kth condition, we used TD-based unsupervised FE. After standardizing \(x_{ijk}\) as
Tucker decomposition of \(x_{ijk}\)
can be computed with a higher order singular value decomposition (HOSVD)10. After identifying which \(u_{\ell _2 j} \in \mathbb {R}^{M \times M}\) and \(u_{\ell _3 k} \in \mathbb {R}^{K \times K}\) are coincident with the target property, e.g., distinction between control and treated samples specifically under kth experimental condition, we try to find \(u_{\ell i} \in \mathbb {R}^{N \times N}\) associated with \(G(\ell _1 \ell _2 \ell _3) \in \mathbb {R}^{N \times M \times K}\) having the largest absolute value. Then, the P value is attributed to the ith gene as
by also assuming that \(u_{\ell _1 i}\) obeys the Gaussian distribution and
For both PCA- and TD-based unsupervised FE, \(P_i\) is corrected with the Benjamini-Hochberg (BH) criterion10; further, the ith genes associated with adjusted \(P_i\) less than the threshold value, which is usually 0.01, are selected.
Although PCA- as well as TD-based unsupervised FE were successfully applied to a wide range of genomic analyses, there were two weak points:
-
Too small a number of genes were selected to have no false negatives.
-
The histogram of \(P_i\) did not fully obey the null assumption that \(u_{\ell i}\) and \(u_{\ell _1 i}\) obey the Gaussian distribution.
In this paper, by fixing these two problems, we have tried to establish a new method at least comparable to or even superior to state-of-art methods.
Trials using highly curated data sets
Application to MAQC dataset
Initially, to assess what the problem is, we compared the performance of PCA-based unsupervised FE with DESeq2, a state-of-art method, using the MAQC11 data set, which has been carefully curated and frequently used for benchmark studies.
Figure 2C shows a scatter plot of genes using \(u_{1i}\) and \(u_{2i}\). Figure 2A,B show the PC loading \(v_{1j}\) and \(v_{2j}\); \(v_{1j}\) represents the mean gene expression and \(v_{2j}\) represents the differential expression between universal human reference (UHR) and brain. Occasionally, this reminds us of the horizontal and vertical axes of an MAPlot; the horizontal axis of an MAPlot represents the mean expression of individual genes, typically the mean logarithmic expression,
whereas the vertical axis of an MAPlot represents the differential expression between the two classes, typically the mean logarithmic fold change (LFC),
where \(M_A\) and \(M_B (=M-M_A)\) are sample numbers within one of the two classes, A and B, respectively, and summations are taken within individual classes. As can be seen in Fig. 2D, which represents the contribution of PC loading, \(x_{ij}\) can be expressed almost fully in the 2-dimensional space spanned by the first two PCs. Thus, PCA can derive, in a fully unsupervised manner, something that qualitatively corresponds to an MAPlot (Fig. 8), which is usually drawn artificially. In spite of that, unfortunately, the genes selected by the adjusted \(P_i\) are too small to have no false negatives (Table 3) and an histogram of \(P_i\) is hardly regarded to obey the null hypothesis;

PCA applied to MAQC data (A) \(v_{1j}\) (B) \(v_{2j}\) (C) Scatter plot of \(u_{1i}\) and \(u_{2i}\) (D) Contributions of individual PCs.
the left panel of Fig. 3 shows the histogram of \(1-P_i\), where \(P_i\)s were computed from \(u_{2i}\) by Eq. (6) using \(\sigma _2\) defined as
If \(1-P_i\) is coincident with the null hypothesis; the histogram of \(1-P_i < 1\) should have a flat distribution and that of \(1-P_i \sim 1\) should have a sharp peak.

Histogram of \(1-P_i\) of the MAQC data set with PCA-based unsupervised FE Left: \(P_i\)s by Eq. (6) using SD \(\sigma _2\) directly computed from \(u_{2i}\), right: using SD optimized to obey the Gaussian distribution as much as possible.
Top ranked genes are coincident with DESeq2
To understand the problem of \(P_i\)s computed by PCA-based unsupervsied FE, we compared \(P_i\)s computed by PCA-based unsupervised FE with those computed by DESeq2, a state-of-art method. At first, AUC was computed to predict the top 1000 genes based on \(P_i\) derived with DESeq2 using \(P_i\)s computed by PCA-based unsupervised FE; the area under the curve (AUC) was 0.97. Next, in contrast, the AUC was computed to predict the top 1000 genes based on \(P_i\) derived with PCA-based unsupervised FE using \(P_i\)s computed using DESeq2; the AUC was 0.98. This indicated that the top-ranked genes were suitably shared between PCA-based unsupervised FE and DESeq2. Thus, the problem of PCA-based unsupervised FE is not the genes’ ranking but the absolute value of \(P_i\)s.
Optimization of SD
Based on the observations at the end of the previous subsubsection, we arrived at optimizing \(\sigma _\ell\) such that \(u_{\ell i}\) and \(u_{\ell _1 i}\) obeyed the Gaussian distribution. Generally, optimizing SD to be fitted to the null hypothesis is not easy. For example, Mudge et al12 had to assume the equivalence between Type I and II errors, which we cannot assume because of an imbalance of numbers between DEGs and the other genes; typically, DEGs are expected to be minorities. Next, we decided to employ an alternative and more empirical approach. To visualize the idea, we have shown some illustrative examples.
Figure 4 shows a historgam of the variable \(x_i\) derived from the Gaussian distribution and outliers. If we attribute the P values to the ith variable with \(x_i\)
using the SD, \(\sigma\), directly computed by all points
and select outliers associated with adjusted P values \(<0.01\), we cannot select any of the outliers (Table 1); this is because the SD computed, \(\sigma = \frac{1000 \times 1 +100 \times 5^2}{1000+100} = 1.75\), is larger than that of the Gaussian distribution, \(\sigma =1\), because of outliers. Because \(P_i\)s computed with \(\sigma =1.75\) is larger than that with \(\sigma =1\), it fails to recognize outliers correctly.

A histogram of Gaussian distribution with outliers.
We computed the histogram of \(1-P_i\), Fig. 5A, which is far being idealized, Fig. 5C, that should have a constant histogram \(h(1-P_i)\) up to \(1-P_i\) very close to 1 and has one with a narrow peak near \(1-P_i \sim 1\). To optimize the SD, we tried to find an optimal SD such that the histogram for those not recognized as outliers was as flat as possible, i.e, obeying the null hypothesis of the Gaussian distribution; we decided to find the optimal SD that results in the most flat \(h(1-P_i)\) for \(1-{\text{adjusted}} \; P_i\) less than threshold value \(1-{\text{adjusted}} \; P_0\) (\({\text{adjusted}} \; P_0\) should be small enough). To minimize the SD of binned \(h_i=h(1-P_i)\), \(\sigma _h\),
with respect to \(\sigma\), where \(N({\text{adjusted}} \; P_0)\) is the number of his associated with \({\text{adjusted}} \; P_i >{\text{adjusted}} \; P_0\), i.e., not recognized as outliers and recognized as a part of the Gaussian distribution. After optimizing \(\sigma _\ell\), we recomputed \(P_i\). Figure 5A,B show the histogram of \(1-P_i\) using \(\sigma =1.75\) and optimized SD, respectively; the latter is closer to an idealized histogram of \(P_i\), Fig. 5C, than the former.

To validate the effectiveness of the optimization of SD, we repeated this procedure 100 times.
Figure 6 shows the dependence of \(\sigma _h\) on SD (upper panel) and the comparison between SD in Eq. (20), optimized SD, and SD computed using is for \({\text{adjusted}} \; P_i < {\text{adjusted}} \; P_0\) (lower panel). In the lower panel, the optimized SD was approximately 1.2, which is much closer to 1 than 1.75, computed by Eq. (20). In addition, the fact that SD computed using is for \({\text{adjusted}} \; P_i < {\text{adjusted}} \; P_0\), which is expected to correspond to the Gaussian distribution part in Fig. 4, is almost 1 helps justify our optimization procedure (Fig. 6, lower panel). The reason why SD = 0 with \(\sigma _h=0\) in the upper panel of Fig. 6 was not selected as optimal (as having the smallest \(\sigma _h\)) is because \(\sigma =0\) corresponds to nothing selected and is thus meaningless. Using \(P_i\) computed by optimized SD, we can discriminate the outliers almost perfectly (Table 2).

Next, we applied this strategy to the MAQC data set. Figure 7 shows \(\sigma _h\), defined in Eq. (22), as a function of SD to compute \(P_i\) in Eq. (19) using the MAQC data set; the optimal SD was 0.05557979. It is close to the SD recomputed using is with \({\text{adjusted}} \; P_i < {\text{adjusted}} \; P_0\), 0.03871846; moreover, \(h(1-P_i)\) derived from optimal SD looks more idealized (the right panel of Fig. 3). Thus, the optimal SD improved PCA-based unsupervised FE.

Table 3 shows the number of genes selected using DESeq2 (list of genes available as Data S1), the original PCA-based unsupervised FE, than by using optimal SD (list of genes available as Data S2). Although the number of genes selected by original PCA-based unsupervised FE, 344, is too small to regard no false negatives, that of genes selected by PCA-based unsupervised FE with optimal SD, 12252, is large enough to regard no false negatives. Furthermore, that of DESeq2, 20546, seems to be too large to have no false positives, because it is unlikely true that more than half the genes (40933) are distinctly expressed between the brain and controls.
Less expressed genes are less likely to be DEGs
Figure 8 shows the selected genes in MAPlot. Although we assumed neither NB distribution nor dispersion relation, Eq. (1), the distribution of selected genes in the MAPlot is reasonable; genes with the same LFC (vertical axis) are less likely selected when associated with smaller mean expression (horizontal axis). Although this property is explicitly assumed in DESeq2 with dispersion relation, Eq. (1), PCA-based unsupervised FE seems to possess the property without assuming dispersion relation explicitly (see the “Discussion” section). On the other hand, DESeq2 selects too many genes and is less likely reasonable. This suggests that PCA-based unsupervised FE with optimized \(\sigma _\ell\) is a promising method.

MAPlot with selected genes colored in red Upper: PCA-based unsupervised FE with optimized SD, lower: DESeq2.
Confirmation using the SEQC dataset
To see if it occurs only occasionally, we repeated all computations on as many as 13 data sets in SEQC13, which is yet another curated data set. Coincidence between DESeq2 and PCA-based unsupervised FE (Fig. 9), a reasonable number of selected genes (\(\sim 10^3\), Fig. 10), and a lower opportunity of less expressed genes to be DEGs (Fig. 11) are also observed, as in the case of MAQC. In addition to this, although the number of genes selected by DESeq2 are too large (\(\sim 10^4\)) and heavily dependent upon sample numbers (\(\sim 10^3\) for the smallest sample number \(\sim 10^0\)), that by PCA-based unsupervised FE is not and is always \(\sim 10^3\), regardless of sample numbers. Thus, PCA-based unsupervised FE is seemingly superior to DESeq2.

Coincidence of top-ranked genes between DESEq2 and PCA-based unsupervised FE using the SEQC data set. Open circles: AUC when P values computed by PCA-based unsupervised FE with optimized SD discriminates top 1000 genes ranked by P values computed by DESeq2. Open red triangles: AUC when P values computed by DESeq2 discriminating top 1000 genes ranked by P values computed by PCA-based unsupervised FE with optimized SD.

Dependence of the number of DEGs on sample numbers using the SEQC data set. Open circles: the number of genes selected by PCA-based unsupervised FE with optimized SD. Open red triangles:the number of genes selected by DESeq2.

MAPlot for SEQC PCA-based unsupervised FE with optimized SD: the first, third, and fifth columns, DESeq2: the second, forth, and sixth columns. Three character IDs represent platform and sites. Blue: genes associated with adjusted P values less than 0.1 but greater than 0.01. Red: genes associated with adjusted P values less than 0.01.
Biological validation
Based on the above results, PCA-based unsupervised FE is seemingly better than DESeq2. Nonetheless, PCA-based unsupervised FE can select a reasonable number of genes regardless of sample numbers (Fig. 10), and less expressed genes are unlikely to be DEGs when genes are selected by PCA-based unsupervised FE with optimized SD (Figs. 8, 11), even without assuming NB distribution and dispersion relations, Eq. (1), which DESeq2 requires, if the selected genes are not biological, it is meaningless. To evaluate the selected genes biologically, we uploaded the genes selected using MAQC to Enrichr. As can be seen in Fig. 12, the genes selected by PCA-based unsupervised FE were better than those selected by DESeq2 (Full list of enrichment analysis is available in Data S1 and S2).

Enrichment analysis of the selected genes, whose numbers in Table 3P values are adjusted P values (based upon “Jensen Tissues” category in Enrichr). Seven terms associated with \(- \log _{10} P =350\) are linked with \(\infty\), since \(P=0\).
One may still wonder the other state-of-art methods might be better than PCA-based unsupervised FE. To deny this possibility, we biologically evaluated the genes selected for MAQC using edgeR6 (full list of enrichment analysis available in Data S3), voom8 (full list of enrichment analysis available in Data S4), and NOISeq9 (full list of enrichment analysis available in Data S5); it is obvious that these three methods are even inferior to DESeq2 biologically (Fig. 13).

Enrichment analysis for MAQC with other methods in Enrichr (A) KEGG (B) GO BP (C) Human gene atlas. Numbers in (B) correspond to 1. “axonogenesis,” 2. “axon guidance,” 3. “axon development,” 4.“regulation of axonogenesis,” 5. “synapse organization,” 6. “modulation of chemical synaptic transmission,” 7. “positive regulation of axonogenesis,” 8.“modulation of excitatory postsynaptic potential,” 9. “regulation of axon extension,” 10. “positive regulation of synaptic transmission,” 11. “axon extension,” 12. “negative regulation of axonogenesis,” 13. “chemical synaptic transmission,” 14. “signal release from synapse,” 15. “synapse assembly,” 16. “regulation of neuronal synaptic plasticity,” 17. “positive regulation of axonextension,” 18. “regulation of trans-synaptic signaling,” 19. “positive regulation of excitatory postsynaptic potential,” 20. “negative regulation of axon extension,” 21. “regulation of synapse assembly,” 22.“retrograde axonal transport,” 23. “synaptic vesicle endocytosis,” 24.“synaptic transmission, GABAergic,” 25. “synaptic transmission, glutamatergic,” 26.“regulation of long-term synaptic potentiation,” 27. “regulation of axon extension involved in axon guidance,” 28. “synaptic membrane adhesion,” 29. “regulation of synaptic transmission, glutamatergic,” 30. “regulation of postsynaptic neurotransmitter receptor activity.” P values are adjusted P values.
Drug discovery for SARS-CoV-2
Although we have demonstrated that PCA-based unsupervised FE with optimized SD can outperform other state-of-art methods in highly curated data, one might wonder that it is not the case for a realistic and more noisy case. To check if PCA-based unsupervised FE with optimized SD can outperform DESeq2 in more realistic data sets, we considered the drug repositioning of SARS-CoV-2, to which we applied TD-based unsupervised FE14 and its kernelized version15.
In our implementation, we employed HOSVD to obtain the tensor decomposition, Eq. (11); because HOSVD is equivalent to SVD applied to a matrix obtained by unfolding a tensor, we can obtain the identical \(u_{\ell i}\) independent of which of PCA or HOSVD is used; SD used in Eq. (12) can be optimized too. Next, we applied the optimization of SD and could select 3627 genes associated with adjusted P values of less than 0.1 (list of genes available as Data S6), which is a much higher number of genes than 163 genes than that selected in previous studies14,15.
Overlap with human genes known to interact with SARS-CoV-2 protein
We evaluated the selected 3627 genes based on the overlap with the human genes known to interact with SARS-CoV-2, as has been done in previous studies14,15 (Fig. 14). It is obvious that TD-based unsupervised FE with an optimized SD can outperform kernel TD-based unsupervised FE, original (without optimized SD) TD-based unsupervised FE as well as DESeq2 (list of overlap available in Data S7). Thus, it is indeed an outstanding method.

P values computed by Fishers’ exact test to evaluate the overlap between human genes known to interact with SARS-Cov-2 proteins and genes selected by various methods. DESeq2 is only for A549 cell lines.
Drug repositioning
We also tried drug discovery using the genes selected by TD-based unsupervised FE with optimized SD. See Table 4 (Full list of drug repositioning available as Data S6). The first one, imatinib, was once identified as a promising drug toward COVID-19, although it was rejected later16. The second one, apratoxin A, was reported to be a promising compound based on its protein binding affinity17. The third and fourth one, doxycycline, was supposed to be a promising drug toward COVID-1918. The seventh one, trovafloxacin, was reported to be a promising compound based on its protein binding affinity19. The eighth one, doxorubicin, was also reported to be a promising compound based on its protein binding affinity20. The ninth one, cisplatin, and the tenth one, carboplatin, were proposed as a result of drug repositioning21. Seven of the nine compounds identified as the top 10 compounds have been previously reported as drugs toward SARS-CoV-2.
See Table 5. The first, fourth, and tenth one, estradiol, was reported as a promising compound22. The second one, tamoxifen, was reported to inhibit SARS-CoV-2 infection by suppressing viral entry23. The third one, apratoxin A, has been listed in Table 4, too. The fifth one, MK-886, was reported to be an inhibitor of 3CL protease24, although its efficiency was limited to 40 %. The sixth one, IFN-alphacon1, was reported to be an inhibitor of SARS-CoV 25 but not for SARS-CoV-2. The seventh one, arachidonic acid, was generally expected to inhibit SARS-CoV-2 infection26. The eighth one, arsenic, was also generally expected to act against the RdRp of coronavirus27. The ninth one, metoprolo, was reported to be a promising drug toward COVID-1928. Thus, all the top 10 compounds were reported to be promising.
On the other hand, for DESeq2, see Table 6 (full list of drug repositioning is available in Data S8), The use of the second and third one, dexamethasone, resulted in lower 28-day mortality among those who received either invasive mechanical ventilation or oxygen alone at randomization but not among those receiving no respiratory support.29, The seventh one, metformin, suppressed SARS-CoV-2 in cell culture30. The eighth one, etanercept, significantly decreased the risk of developing COVID-19 in patients with rheumatoid arthritis or spondyloarthropathies31. The tenth one, lipopolysaccharide, is not a compound but a bacterial protein reported to bind to the SARS-CoV-2 spike protein 32.
See Table 7. The first and fourth one, resveratrol, inhibits HCoV-229E and SARS-CoV-2 coronavirus replication in vitro33. The second, third, and fifth one, carboplatin, was proposed as a result of drug repositioning21. The seventh one, lipopolysaccharide, is listed in Table 6, too.
The proposed method can predict effective drugs for COVID-19 based on gene expression analysis, at least, comparatively to DESeq2. Nevertheless, DESeq2 has less significance and has a tendency to list the same compounds multiple times. The proposed method can identify more convincing and diverse candidate compounds than DESeq2.
Based on the overlap between human genes known to interact with SARS-CoV-2 proteins and selected genes (Fig. 14) and from the point of drug repositioning, TD-based unsupervised FE with optimized SD is, at least, competitive with DESeq2.
Comparison of methods using multi-organ measurements with multiple drug treatments
One might wonder if the proposed methods, TD- and PCA-based unsupervised FE with optimized SD, are applicable to a more complicated set-up. To investigate this point, we checked the case where multiple drugs are applied to mice whose gene expression of multiple tissues are measured, to which we applied TD-based unsupervised FE34.
Enrichment of tissue-specific genes
In the previous study34, although we applied TD-based unsupervised FE to gene expression profiles, there existed some problems. First of all, the number of genes selected was too small to have no false negatives.
Using the optimized SD, the number of selected genes increased (Table 8; for more details, e.g., the definition of the four gene sets, neurons and testis, muscle, gastrointestine 1 and 2, see the previous study34. This topic has not been discussed herein as it is not directly related to the comparison of the performance between the original TD-based unsupervised FE and that with the optimised SD. The full list of the selected genes is available in Data S9). Although an increased number of genes is meaningless if the biological reliability is less, the biological reliability of selected genes is also improved (lower panel of Fig. 15, which corresponds to a present study and is associated with a greater number of cell lines and tissue specificity than that in the upper panel of Fig. 15, which corresponds to a previous study).

Enrichment analysis of cell and tissue specificity with Metascape35. Upper: original TD-based unsupervised FE (using genes with adjusted \(P \le 0.01\) in Table 8), lower; the present study with optimized SD (using genes with adjusted \(P \le 0.1\) in Table 8). Metascape 3.5, https://metascape.org/.
Thus, the employment of optimized SD is also effective to a more complicated data set than simple pairwise comparisons between the treated and control samples investigated in the previous sections.
Coincidence with drug treatment
We have also performed additional validation of the genes selected by TD-based unsupervised FE with optimized SD associated with adjusted P values less than 0.1 (Table 8, full list is available in Data S10–S13). We have uploaded selected genes to Enrichr36 and evaluated the overlaps between the genes selected and those whose expression was altered with the treatment of the 15 drugs used in this study. Then, we found that all four gene sets in Table 8 had a significant overlap with the genes whose expression was altered with the treatment of 5 of the drugs (acetaminophen, cisplatin, clozapine, doxycycline, and olanzapine) in DrugMatrix, which does not include other drug treatments (Supplementary material). This suggests that TD-based unsupervised FE with optimal SD can correctly recognize drug treatments based on gene expression; this was impossible in the previous study34 because of the very small number of genes selected (Table 8). Thus, considering the optimization of SD enables TD-based unsupervised FE to recognize a greater number of biologically reliable genes than the original TD-based unsupervised FE, which did not include the optimization of SD.
Discussion
In this study, we have introduced the optimization of SD to TD- and PCA-based unsupervised FE and have improved their performance by increasing the identified DEGs associated with greater biological reliability. One of the striking features is that DEGs with lesser gene expression are less likely recognized even with the same LFC, if the genes are selected by TD- and PCA-based unsupervised FE with optimized SD. In DESeq2, the tendency that less expressed genes are hardly recognized as DEGs is artificially introduced by assuming dispersion relation, Eq. (1). Nevertheless, in PCA- and TD-based unsupervised FE, it is automatically introduced. Generally, there exists a relationship between difference, \(\Delta\) of two variables, x and y, and LFC as
Then
Because \(v_{2j}\) (Fig. 2B) corresponds to \(\Delta\), if DEGs are identified using \(u_{2i}\) that corresponds to \(v_{2j}\) as in TD- and PCA-based unsupervised FE (see Eqs. (6) and (12)), DEGs associated with the same LFC are less likely selected for the smaller y that corresponds to \(\mu\). This results in the distribution of DEGs in MAPlot (Fig. 8), where genes with the same LFC (vertical axis) are less likely identified as DEGs with smaller gene expression (horizontal axis). Figure 16 shows the MAPlot drawn using two independent random variables obeying the same positive uniform distribution; the red colored region associated with \(|\Delta |\) larger than some threshold values qualitatively represents the tendency that indicates that a smaller \(x+y\) is less likely selected even with the same LFC, \(\log _2 \frac{x}{y}\). Thus, TD- and PCA-based unsupervised FE can introduce the tendency that genes with less expression are less likely to be DEGs, even with the same amount of LFC more naturally than DESeq2, which has to manually introduce a dispersion relation, Eq. (1).

“MAPlot” using two independent variables, x and y, drawn from uniform distribution \(\in [0,1]\). Red dots are associated with \(|x-y|>0.5\).
In addition to this, although DESeq2 assumes NB distribution that does not have any rationalization other than that it takes only positive values and has a tunable mean as well as variance simultaneously, TD- and PCA-based unsupervised FE assume only that \(u_{\ell i}\) obeys the Gaussian distribution (Eqs. (6) and (12)), which is more reasonable because Gaussian distributions can generally appear when independent random variables are summed up. Actually, NOISeq does not assume NB distribution as well but achieves comparative performance with DESeq2 (Fig. 13). In this sense, TD- and PCA-based unsupervised FE can realize DEG distribution in an MAPlot more naturally than DESeq2.
Another remarkable point of TD- and PCA-based unsupervised FE with optimized SD is that it does not have to screen for selected genes by LFC after the genes are selected using P values. As can be seen in Fig. 10, state-of-art methods, including DESeq2, often identify too many DEGs. In these circumstances, LFC is often used to reduce the number of DEGs. Nevertheless, Stupnikov et al37 found that the coincidence of the selected genes among the various state-of-art methods drastically decreases if the genes selected based on P values are further screened with LFC. In this sense, TD- and PCA-based unsupervised FE with optimized SD are more promising methods than state-of-art methods that need screening by LFC to yield a reasonable number of DEGs.
Yet another advantage is that TD- and PCA-based unsupervised FE have already been applied to a wide range of problems. Not only can optimized SD improve the performance of PCA- and TD-based unsupervised FE, as can be seen in Figs. 14 and 15, but also the alteration is limited to the last stage, i.e., P value computation, Eqs. (6) and (12). Thus, the optimized SD is expected to improve the performance in a wide range of problems, to which TD- and PCA-based unsupervised FE have been applied.
One might wonder if the validation should be based upon ground truth. Nevertheless, we do not think that there are ground truth for DEGs; DEGs are depend upon the definition of DEGs since the amount of differential expression is not discrete variable but continuous one. We need to decide threshold values for DEGs which affects which genes are DEGs. In contrast, biological significance is more trustable. In addition to this, the purpose of identification of DEGs is to further make use of them as biological studies. Thus, we believe that the proposed methods that can select biologically more reasonable genes than stat of art methods is worthwhile publishing.
Conclusions
In this study, we optimized SD to improve TD- and PCA-based unsupervised FE. As a result, not only the obtained DEGs increased and became reasonable in number but also the histogram of 1-P became more reliable, i.e., more coincident with the null hypothesis that SVV and PC obey Gaussian distribution. In addition to this, TD- and PCA-based unsupervised FE provide reliable distribution of DEGs in MAPlot, i.e., less expressed genes are less likely selected as DEGs even if they are associated with the same LFC; this property was implemented manually by assuming dispersion relation, Eq.(1), in DESeq2. The biological reliability of the selected genes is also much better by this method than by other state-of-art methods. These points suggest that TD- and PCA-based unsupervised FE are superior than state-of-art methods in terms of achieving better performance with less assumption.
Methods
Sample R code to perform analyses in this study is available as Data S14.
Gene expression profiles
MAQC
Seven human brain expression profiles were downloaded from SRA38 (ID SRX016359), and seven UHR expression profiles were downloaded from SRA (ID SRX016367). Fourteen FASTQ files were mapped to the hg38 human genome using rapmap39. htseq-count40 was used to convert the obtained bam files to count data files using the gtf file taken from ftp://ftp.ensembl.org/pub/release-105/gtf/ homo_sapiens/Homo_sapiens.GRCh38.105.gtf.gz.
SEQC
SEQC13 were obtained from bioconductor41 as an experimental package, seqc. It includes thirteen profiles shown in Fig. 11. For more details, see Vignettes in the seqc experimental package.
The histogram composed of Gaussian distribution and outliers in Fig. 4
The Gaussian part is one thousand values drawn from Gaussian distribution with zero mean and an SD of one. Outliers are 100 values, which are equal to 5.
PCA-based unsupervised FE applied
MAQC
Genes not expressed in any of the 14 samples have been excluded. Four rows having annotations “__no_feature”, “__ambiguous”, “__not_aligned”, and “__alignment_not_unique” have also been excluded. As a result, we got \(x_{ij} \in \mathbb {R}^{40933 \times 14}\). The \(x_{ij}\) was processed as described in the main text.
SEQC
Regardless of which of the 13 data sets was considered, only those genes expressed in all samples were considered. An individual data set has a distinct number of rows (genes) and columns (samples). The \(x_{ij}\) obtained from an individual data set was processed as described in the main text.
SARS-CoV-2
All processes used were exactly the same as those described in the previous study14. After obtaining \(u_{5i}\), the SD was optimized as described in the main text.
Multi-organ
All processes used were exactly the same as those described in the previous study34. After getting \(u_{\ell i}\), the SD was optimized as described in the main text.
Optimization of SD
At first, a histogram of \(1-P_i\) was computed using hclust function in R with the “break=100” option. Then, an SD of the binned histogram, hc$count associated with hc$breaks less than 1-P whose adjusted P value was less than threshold value \(P_0\), was minimized using optim function in R. The R code has been provided in Data S14 to show how to optimize SD in an individual data set.
Coincidence between PCA-based unsupervised FE and DESeq2
The coincidence between PCA-based unsupervised FE and DESeq2 was evaluated by AUC (Fig. 9) as follows. At first, the top 1000 genes based on P values computed by DESeq2 were regarded positive and the remaining genes were regarded negative. Then, P values computed by PCA-based unsupervised FE were used to predict positive genes. Using this result, AUC was computed. Next, on the contrary, the top 1000 genes based on P values computed by PCA-based unsupervised FE were regarded positive and the remaining genes were negatives. Then, P values computed by DESeq2 were used to predict positive genes. Using this result, AUC was computed.
Enrichment analyses
Enrichment analyses were performed using either Metascape35 or Enrichr36 by uploading gene symbols. If the gene ID was not a gene symbol in individual data sets, the gene ID conversion tool in Database for Annotation, Visualization, and Integrated Discovery (DAVID)42,43 was used for conversion.
DEG identification of SARS-CoV-2 data by DESeq2
We used author-provided adjusted P values and LFC (in supplementary data in their paper) to identify DEGs. If we considered only adjusted P values to identify DEGs, DESeq2 would identify too many genes (Table 9). Thus, we had to consider LFC as well. Table 9 shows the number of DEGs used in this study.
The evaluation of the overlap with human genes known to interact with SARS-CoV-2 proteins is available in Supplementary materials. The best one, that for the ACE2-expressed A549 cell line, is also included in the main text as Fig. 14.
Data availability
The sequencing datasets are available via the NIH/NCBI Sequence Read Archive (SRA) repository using accession number SRX016359 and SRX016367, via biocondutor with the package of seqc [https://doi.org/doi:10.18129/B9.bioc.seqc, accessed 10th July 2022], via the NIH/NCBI Gene Expression Omnibus (GEO) repository using accession number GSE147507 and GSE142068.
References
Taguchi, Y-h. Comparative transcriptomics analysis. In Encyclopedia of Bioinformatics and Computational Biology (eds Ranganathan, S. et al.) 814–818 (Academic Press, 2019). https://doi.org/10.1016/B978-0-12-809633-8.20163-5.
Rapaport, F. et al. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 14, 3158. https://doi.org/10.1186/gb-2013-14-9-r95 (2013).
Tusher, V. G., Tibshirani, R. & Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. 98, 5116–5121. https://doi.org/10.1073/pnas.091062498 (2001).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. https://doi.org/10.1093/bioinformatics/btp616 (2009).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297. https://doi.org/10.1093/nar/gks042 (2012).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. https://doi.org/10.1186/gb-2014-15-2-r29 (2014).
Tarazona, S., García, F., Ferrer, A., Dopazo, J. & Conesa, A. NOIseq: a RNA-seq differential expression method robust for sequencing depth biases. EMBnet.journal 17, 18–19. https://doi.org/10.14806/ej.17.B.265
Taguchi, Y-h. Unsupervised Feature Extraction Applied to Bioinformatics (Springer International Publishing, 2020).
Shi, L. et al. The MicroArray quality control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 24, 1151–1161. https://doi.org/10.1038/nbt1239 (2006).
Mudge, J. F., Baker, L. F., Edge, C. B. & Houlahan, J. E. Setting an optimal \(\alpha\) that minimizes errors in null hypothesis significance tests. PLoS ONE 7, 1–7. https://doi.org/10.1371/journal.pone.0032734 (2012).
SEQC/MAQC-III Consortium, A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the sequencing quality control consortium. Nature Biotechnology 32, 903–914. https://doi.org/10.1038/nbt.2957 (2014).
Taguchi, Y.-H. & Turki, T. A new advanced in silico drug discovery method for novel coronavirus (SARS-CoV-2) with tensor decomposition-based unsupervised feature extraction. PLoS ONE 15, 1–16. https://doi.org/10.1371/journal.pone.0238907 (2020).
Taguchi, Y.-H. & Turki, T. Application of tensor decomposition to gene expression of infection of mouse hepatitis virus can identify critical human genes and efffective drugs for SARS-CoV-2 infection. IEEE J. Sel. Top. Signal Process. 15, 746–758. https://doi.org/10.1109/JSTSP.2021.3061251 (2021).
Zhao, H., Mendenhall, M. & Deininger, M. W. Imatinib is not a potent anti-SARS-CoV-2 drug. Leukemia 34, 3085–3087. https://doi.org/10.1038/s41375-020-01045-9 (2020).
Naidoo, D., Roy, A., Kar, P., Mutanda, T. & Anandraj, A. Cyanobacterial metabolites as promising drug leads against the mpro and plpro of SARS-CoV-2: An in silico analysis. J. Biomol. Struct. Dyn. 39, 6218–6230. https://doi.org/10.1080/07391102.2020.1794972 (2021).
Dorobisz, K., Dorobisz, T., Janczak, D. & Zatoński, T. Doxycycline in the coronavirus disease 2019 therapy. Ther. Clin. Risk Manag. 17, 1023–1026. https://doi.org/10.2147/tcrm.s314923 (2021).
Gimeno, A. et al. Prediction of novel inhibitors of the main protease (M-pro) of SARS-CoV-2 through consensus docking and drug reposition. Int. J. Mol. Sci. 21, 3793. https://doi.org/10.3390/ijms21113793 (2020).
Jamal, Q. M. S., Alharbi, A. H. & Ahmad, V. Identification of doxorubicin as a potential therapeutic against SARS-CoV-2 (COVID-19) protease: a molecular docking and dynamics simulation studies. J. Biomol. Struct. Dyn. 40, 7960–7974. https://doi.org/10.1080/07391102.2021.1905551 (2021).
MotieGhader, H., Safavi, E., Rezapour, A., Amoodizaj, F. F. & asl Iranifam, R. Drug repurposing for coronavirus (SARS-CoV-2) based on gene co-expression network analysis. Sci. Rep. 11, 21872. https://doi.org/10.1038/s41598-021-01410-3 (2021).
Mansouri, A., Kowsar, R., Zakariazadeh, M., Hakimi, H. & Miyamoto, A. The impact of calcitriol and estradiol on the SARS-CoV-2 biological activity: A molecular modeling approach. Sci. Rep. 12, 717. https://doi.org/10.1038/s41598-022-04778-y (2022).
Zu, S. et al. Tamoxifen and clomiphene inhibit SARS-CoV-2 infection by suppressing viral entry. Signal Transduct. Targeted Therapy 6, 435. https://doi.org/10.1038/s41392-021-00853-4 (2021).
Zhu, W. et al. Identification of SARS-CoV-2 3cl protease inhibitors by a quantitative high-throughput screening. ACS Pharmacol. Transl. Sci. 3, 1008–1016. https://doi.org/10.1021/acsptsci.0c00108 (2020).
Paragas, J., Blatt, L. M., Hartmann, C., Huggins, J. W. & Endy, T. P. Interferon alfacon1 is an inhibitor of SARS-corona virus in cell-based models. Antiviral Res. 66, 99–102. https://doi.org/10.1016/j.antiviral.2005.01.002 (2005).
Ripon, M. A. R., Bhowmik, D. R., Amin, M. T. & Hossain, M. S. Role of arachidonic cascade in covid-19 infection: A review. Prostaglandins Other Lipid Mediators 154, 106539. https://doi.org/10.1016/j.prostaglandins.2021.106539 (2021).
Chowdhury, T., Roymahapatra, G. & Mandal, S. M. In silico identification of a potent arsenic based approved drug darinaparsin against sars-cov-2: Inhibitor of RNA dependent RNA polymerase (RdRp) and necessary proteases. ChemRxiv. https://doi.org/10.26434/chemrxiv.12200495.v1 (2020).
Clemente-Moragón, A. et al. Metoprolol in critically ill patients with COVID-19. J. Am. Coll. Cardiol. 78, 1001–1011. https://doi.org/10.1016/j.jacc.2021.07.003 (2021).
The RECOVERY Collaborative Group, Dexamethasone in hospitalized patients with covid-19. N. Engl. J. Med. 384, 693–704. https://doi.org/10.1056/nejmoa2021436 (2021).
Parthasarathy, H., Tandel, D. & Harshan, K. H. Metformin suppresses SARS-CoV-2 in cell culture. bioRxiv. https://doi.org/10.1101/2021.11.18.469078 (2021).
Salesi, M., Shojaie, B., Farajzadegan, Z., Salesi, N. & Mohammadi, E. TNF-\(\alpha\) blockers showed prophylactic effects in preventing COVID-19 in patients with rheumatoid arthritis and seronegative spondyloarthropathies: A case-control study. Rheumatol. Therapy 8, 1355–1370. https://doi.org/10.1007/s40744-021-00342-8 (2021).
Petruk, G. et al. SARS-CoV-2 spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J. Mol. Cell Biol. 12, 916–932. https://doi.org/10.1093/jmcb/mjaa067 (2020).
Pasquereau, S. et al. Resveratrol inhibits HCoV-229E and SARS-CoV-2 coronavirus replication in vitro. Viruses 13, 354. https://doi.org/10.3390/v13020354 (2021).
Taguchi, Y-h. & Turki, T. Universal nature of drug treatment responses in drug-tissue-wide model-animal experiments using tensor decomposition-based unsupervised feature extraction. Front. Genet. 11, 695. https://doi.org/10.3389/fgene.2020.00695 (2020).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523. https://doi.org/10.1038/s41467-019-09234-6 (2019).
Xie, Z. et al. Gene set knowledge discovery with Enrichr. Curr. Protocols 1, e90. https://doi.org/10.1002/cpz1.90 (2021).
Stupnikov, A. et al. Robustness of differential gene expression analysis of RNA-seq. Comput. Struct. Biotechnol. J. 19, 3470–3481. https://doi.org/10.1016/j.csbj.2021.05.040 (2021).
Leinonen, R., Sugawara, H. & Shumway, M. On behalf of the international nucleotide sequence database collaboration, the sequence read archive. Nucleic Acids Res. 39, D19–D21. https://doi.org/10.1093/nar/gkq1019 (2010).
Srivastava, A., Sarkar, H., Gupta, N. & Patro, R. RapMap: A rapid, sensitive and accurate tool for mapping RNA-seq reads to transcriptomes. Bioinformatics 32, i192–i200. https://doi.org/10.1093/bioinformatics/btw277 (2016).
Putri, G. H., Anders, S., Pyl, P. T., Pimanda, J. E. & Zanini, F. Analysing high-throughput sequencing data in python with htseq 2.0. Bioinformatics 38, 2943–2945. https://doi.org/10.1093/bioinformatics/btac166 (2022).
Huber, W. et al. Orchestrating high-throughput genomic analysis with bioconductor. Nat. Methods 12, 115–121. https://doi.org/10.1038/nmeth.3252 (2015).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. https://doi.org/10.1038/nprot.2008.211 (2008).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. https://doi.org/10.1093/nar/gkn923 (2008).
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science, KAKENHI [Grant numbers 19H05270, 20K12067, and 20H04848] to YHT.
Author information
Authors and Affiliations
Contributions
Y.H.T. planned the research and performed analyses. Y.H.T. and T.T. evaluated the results, discussions, and outcomes and drafted and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taguchi, Yh., Turki, T. Adapted tensor decomposition and PCA based unsupervised feature extraction select more biologically reasonable differentially expressed genes than conventional methods. Sci Rep 12, 17438 (2022). https://doi.org/10.1038/s41598-022-21474-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-21474-z
- Springer Nature Limited