
Abstract
Although Duchenne muscular dystrophy (DMD) primarily affects muscle tissues, the alterations to systemic metabolism manifested in DMD patients contribute to the severe phenotype of this fatal disorder. We propose that microRNA-378a (miR-378) alters carbohydrate and lipid metabolism in dystrophic mdx mice. In our study, we utilized double knockout animals which lacked both dystrophin and miR-378 (mdx/miR-378−/−). RNA sequencing of the liver identified 561 and 194 differentially expressed genes that distinguished mdx versus wild-type (WT) and mdx/miR-378−/− versus mdx counterparts, respectively. Bioinformatics analysis predicted, among others, carbohydrate metabolism disorder in dystrophic mice, as functionally proven by impaired glucose tolerance and insulin sensitivity. The lack of miR-378 in mdx animals mitigated those effects with a faster glucose clearance in a glucose tolerance test (GTT) and normalization of liver glycogen levels. The absence of miR-378 also restored the expression of genes regulating lipid homeostasis, such as Acly, Fasn, Gpam, Pnpla3, and Scd1. In conclusion, we report for the first time that miR-378 loss results in increased systemic metabolism of mdx mice. Together with our previous finding, demonstrating alleviation of the muscle-related symptoms of DMD, we propose that the inhibition of miR-378 may represent a new strategy to attenuate the multifaceted symptoms of DMD.
Similar content being viewed by others


Introduction
The lack of functional dystrophin is the primary cause of Duchenne muscular dystrophy (DMD) and is responsible for irreversible muscle weakening, with cardio-respiratory failure being the most common cause of premature death. However, the secondary consequences of dystrophin loss, though much less understood, significantly contribute to the severity of the disease. Accordingly, DMD should be recognized as a multi-systemic disorder and interdisciplinary care is recommended for the optimal management of DMD-associated complications1.
Disturbances in angiogenesis2,3 and brain4, renal, and bladder dysfunctions5,6, as well as bone-health issues7 and gastrointestinal problems8, reflect the range of pathological changes observed in DMD. Finally, systemic alterations in the metabolism of carbohydrates and lipids accompanied by cases of impaired glucose tolerance and insulin sensitivity are manifested by DMD patients and mouse models of the disease, including mdx mice and golden retriever muscular dystrophy (GRMD) dogs9,10,11,12,13.
Though a few mutation-specific compounds are used in clinical settings and other cell and gene therapy-based approaches are under extensive investigation, drugs that act mostly as anti-inflammatory agents, such as glucocorticoids (GCs), still serve as the gold standard of care for all patients suffering from DMD14. Apart from their indisputable beneficial effects in prolonging ambulation and improving the quality of the patient’s life, they, unfortunately, entail a list of multi-organ side effects15. This emphasizes the persistent need to evaluate novel targets to ameliorate disease symptoms, which could also potentially be applicable in the emerging concept of combined therapies for DMD patients16.
Given all of the above, factors that influence both muscular and systemic perturbations might be of special interest. One candidate might be short, noncoding microRNAs (miRs), due to their potential to modulate a wide range of physiological and pathological processes17. We have recently studied miR-378a (miR-378), embedded in the first intron of the peroxisome proliferator-activated receptor-gamma coactivator 1β (Ppargc1b) gene encoding PGC1β, a factor that has been implicated in energy metabolism18. miR-378 plays a multifaceted role in metabolism, angiogenesis, and muscle biology18. In the context of the latter, we and others have demonstrated that miR-378 is abundantly expressed in skeletal and cardiac tissues19,20 and that its deficiency influences vascularization of skeletal muscles19 as well as the functional properties and size of cardiomyocytes21, whereas overexpression of miR-378 attenuates muscle regeneration22. Notably, we previously demonstrated that the lack of miR-378 mitigates muscle dystrophy by reducing inflammation, decreasing fibrosis, and affecting the properties of satellite cells, the bona fide muscle stem cells. Most importantly, this was associated with increased physical performance and enhanced contractile properties of a single muscle. Surprisingly, RNA sequencing (RNA-seq) analysis of the gastrocnemius muscle revealed no profound transcriptional changes between mdx/miR-378−/− and mdx mice. On the other hand, the analysis of muscle fiber type composition pointed toward the influence of miR-378 on metabolism23. Accordingly, as miR-378 has been recognized as a potent modulator of lipid and carbohydrate homeostasis19 and was proposed as a biomarker of insulin resistance24, we examined whether the higher muscle functionality of mdx mice lacking miR-378 might be accompanied by improved systemic metabolism. Carrer et al. demonstrated previously that the loss of miR-378 diminished liver fat deposition while facilitating the response to glucose administration during a glucose tolerance test (GTT) in mice fed a high-fat diet25. Others reported that miR-378 regulates hepatic insulin signaling by targeting the p110α catalytic subunit of PI3K26,27, providing evidence for a hepatic-related function of miR-378. Though the incidence of hepatitis28, non-alcoholic fatty liver disease29, drug-induced hepatotoxicity30, or even acute liver failure secondary to dilated cardiomyopathy31 have been observed in DMD patients, these hepatic alterations are not well understood. Murphy et al.32 evaluated the liver proteome of the mdx-4Cv model of DMD and found dysregulated levels of the factors implicated in fatty acid and carbohydrate metabolism, among other findings. However, no studies have examined any candidate factors that would mediate those effects.
Higher levels of miR-378 in the serum/plasma of dystrophic animals and DMD patients, most likely a result of its leaking from damaged muscles, was reported previously by us23 and others33. Circulating miRNAs are often bound to lipoproteins, especially high-density lipoprotein (HDL) and low-density lipoprotein (LDL) cholesterol. In such complexes, they might be delivered to the liver34, which could also be the case regarding miR-378. In light of the above, in the current study, we sought to uncover liver pathology driven by dystrophin loss in an mdx model of DMD and to verify the hypothesis that a loss of miR-378 improves disturbed carbohydrate and lipid homeostasis in dystrophic animals. This, together with our previous findings23, sheds light on how the absence of miR-378 improves the dystrophic phenotype.
Results
RNA-seq revealed notable alterations in the hepatic transcriptome of mdx and mdx/miR-378−/− mice
Undetectable levels of mature strands of miR-378, miR-378-3p, and miR-378-5p, were confirmed in both the liver (Fig. 1A) and isolated hepatocytes (Supplementary Fig. 1A) of mdx/miR-378−/− mice, along with no apparent differences in miR-378 expression between the mdx and WT cohorts.

The global lack of miR-378 in mdx mice (mdx/miR-378−/−) does not profoundly affect liver histology and injury markers. (A) The level of both mature strands of miR-378, namely miR-378-3p and miR-378-5p assessed with the use of locked nucleic acid (LNA) technology; n = 5–8/group. (B) The liver weight was presented as g/kg BW; n = 15–16/group. (C) Bilirubin concentration determined in the serum by biochemical analyzer SPOTCHEM; n = 14–15/group. The level of (D) aspartate transaminase (AST) and (E) alanine transaminase (ALT) together with (F) calculated De Ritis ratio of AST/ALT in the serum collected from mice under non-fasting conditions (fed) and after overnight fasting (fasted); measurements performed with the use of biochemical analyzer SPOTCHEM; n = 5–6/group. (G) Liver histology was assessed by hematoxylin and eosin (H&E) and Masson’s Trichrome staining on paraffin-embedded sections, visualized with the use of Leica DMi8 microscope with CMOS Leica MC170 HD camera, and analyzed semiquantitatively (H&E) and quantitatively (Masson’s Trichrome); n = 4–6/group. Scale bars represent 1 mm (black) and 100 μm (white). Data are presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by one-way ANOVA with Tukey’s post-hoc test.
Higher relative liver weight was noticed in dystrophic mice without further changes driven by miR-378 loss (Fig. 1B), whereas the serum level of a typical marker of liver injury, bilirubin, did not differ between genotypes (Fig. 1C). On the contrary, both aspartate aminotransferase (AST, Fig. 1D) and alanine aminotransferase (ALT, Fig. 1E), whose levels increase as a result of both liver and muscle damage—as in the case of DMD—were markedly elevated in the mdx mice under normal fed conditions and after fasting overnight. This was also reflected by the substantially higher de Ritis ratio, which was not further escalated in the mdx/miR-378−/− mice (Fig. 1F).
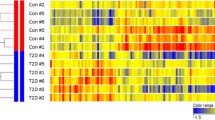
Simultaneously, the histological assessment did not reveal severe liver injury, collagen deposition abnormalities (Fig. 1G), or lipid deposition (Supplementary Fig. 2) in any of the cohorts. Despite that, RNA-seq analysis showed profound changes in the liver transcriptome. The 1000 most variable genes separated the WT, mdx, and mdx/miR-378−/− mice by the first and second principal components (PC1 and PC2, respectively) (Fig. 2A). Accordingly, the mdx and mdx/miR-378−/− mice clustered together, while the WT mice formed the second main cluster, indicating that the lack of dystrophin had the strongest impact on the gene expression profile in the liver (Fig. 2A). Of note, DESeq2 analysis identified 561 differentially expressed genes (DEGs) that distinguished mdx against WT and 194 DEGs that differentiated mdx/miR-378−/− from mdx livers (Fig. 2B, C), by hierarchical clustering and principal component analysis (PCA).

Alterations in the liver transcriptome of mdx mice are affected by the additional lack of miR-378. (A) Transcriptome profiling by RNA-seq in the mdx vs. WT mice and mice lacking both dystrophin and miR-378 (mdx/miR-378−/−) vs. mdx segregates the investigated groups into 3 distinct clusters based on 1000 most variable genes, n = 4/group. (B) RNA-seq analysis revealed 561 DEGs between mdx vs. WT livers, and (C) 194 DEGs between mdx/miR-378−/− vs. mdx that distinguish the analyzed groups by principal component analysis (PCA) and hierarchical clustering.
Gene ontology (GO) analysis pointed toward metabolism-related pathways, which were placed among the most significantly affected GO terms (Fig. 3A). Reduced basal respiration of dystrophic hepatocytes was revealed, without any apparent effect of miR-378 loss (Supplementary Fig. 1B), which is in line with the metabolic impairment observed in dystrophic liver mitochondria35. Moreover, the altered expression of cytochrome P450 (CYP450) enzymes—key players in xenobiotic and steroid metabolism36—was observed as a result of dystrophin deficiency and in mdx/miR-378−/− mice (Fig. 3B). The molecular and cellular functions identified by Ingenuity Pathway Analysis (IPA) also confirmed that DEGs were associated with metabolic processes, particularly related to lipids and carbohydrates (Fig. 3C).

Metabolic pathways are predominantly changed in the livers of both mdx and mdx/miR-378−/− mice. (A) Analysis of gene ontology (GO) terms of affected pathways. (B) The graph showing higher and lower expression of cytochrome P450 (CYP450) isoforms based on RNA-seq results, presented as the fold change. (C) Affected pathways associated with molecular and cellular functions were determined by ingenuity pathway analysis (IPA) and arranged according to the p-value range.
The impaired glucose tolerance and insulin sensitivity displayed by mdx mice are affected by the loss of miR-378
Further analysis revealed that the vast majority of genes regulating glucose metabolism (Fig. 4A) were elevated in the dystrophic mice (except for Adipor2), whereas factors related to lipid metabolism disorder displayed both upregulated and downregulated patterns (Fig. 4B). Additionally, the serum profile of miRNAs known predominantly as muscle-specific miRs (myomiR)37—but also reported as potential biomarkers of pre-diabetic/diabetic states and associated with glucose intolerance and insulin resistance38,39—was altered in the mdx and mdx/miR-378−/− animals. Accordingly, miR-1 (Fig. 4C) and miR-133 (Fig. 4D) levels were elevated in the mdx mice in comparison with the WT mice and were lower as a result of the additional lack of miR-378.

Disorder of glucose and lipid metabolism in the mdx mice is accompanied by higher expression of miR-1 and miR-133, further diminished as the result of the miR-378 loss. (A, B) Heat maps were created based on the list of genes indicated by ingenuity pathway analysis (IPA) that were attributed to the disorder of glucose (p-value = 1.04 × 10–12) and lipid metabolism (p-value = 1.31 × 10–6) in the liver of mdx mice. Blue color indicates downregulation, whereas orange upregulation. The expression of (C) miR-1-3p, (D) miR-133-3p, and miR-133-5p in the serum as assessed by locked nucleic acid (LNA) technology; n = 5–7/group. Data are presented as mean ± SEM. *p < 0.05; **p < 0.01; ****p < 0.0001 by one-way ANOVA with Tukey’s post-hoc test.
To further study the possible role of miR-378 in disturbed carbohydrate homeostasis in dystrophic mice, we utilized the functional GTT and the insulin tolerance test (ITT). A slightly lower glucose level was observed among the mdx/miR-378−/− mice even under fed conditions (random glucose), without apparent changes in the dystrophic animals (Fig. 5A). When the mice underwent a GTT, mild glucose intolerance was found in the mdx individuals, as indicated by an elevated glucose level 120 min after i.p. injection of glucose (Fig. 5B). Importantly, the mdx/miR-378−/− mice rapidly cleared the glucose, since diminished concentrations were visible even 30 min after injection and were sustained at a lower level than in the dystrophic animals in each subsequent time-point (Fig. 5B). This was accompanied by markedly reduced glycogen content in the mdx livers than in WT livers, restored to the level of the WT groups in mdx/miR-378−/− mice (Fig. 5C). Additionally, the lack of miR-378 decreased the level of hepatic interleukin 6 (IL6, Supplementary Fig. 3A), tumor necrosis factor (TNFα, Supplementary Fig. 3B), and its receptor (TNFR1, Supplementary Fig. 3C); these factors are positively correlated with glycogen degradation and insulin resistance40,41,42.

Disturbed systemic glucose homeostasis in the mdx mice is affected by the miR-378 loss. (A) The glucose level under non-fasting conditions was measured with a glucometer in the blood from the tail tip; n = 7–8/group. (B) A glucose tolerance test (GTT) was performed after overnight fasting. Glucose concentration was assessed in the blood from the tail tip before (0 min) and 15, 30, 60, and 120 min after i.p. injection of glucose solution; n = 7–8/group. (C) Liver glycogen content was evaluated after overnight fasting with the use of a biochemical assay; n = 4–6/group. (D) Serum insulin concentration was determined both under non-fasting conditions (fed) and after overnight fasting (fasted) by ELISA; n = 4–7/group. (E) Insulin tolerance test (ITT) was performed after 4 h of fasting. Glucose concentration was measured in the blood from the tail tip before (0 min) and 15, 30, 60, 90 min after intraperitoneal injection of insulin; n = 7–8/group. Data are presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 by (A, C, D) one-way ANOVA with Tukey’s post-hoc test or (B, E) by two-way ANOVA for repeated measures with Tukey’s post-hoc test. (A) #p < 0.05 additionally tested by Student’s t-test for comparison of mdx/miR-378−/− vs. mdx only.
Insulin concentration in the serum was higher in the mdx/miR-378−/− mice under fed conditions with no changes between the groups after overnight fasting (Fig. 5D). The dystrophic animals failed to respond to insulin in the ITT test, which resulted in markedly higher glucose levels in comparison to the WT mice 90 min after insulin administration, suggesting impaired insulin sensitivity in those animals (Fig. 5E). On the contrary, glucose concentration in the mdx/miR-378−/− animals was already at a low level before insulin injection; thus, the further drop following insulin administration was rather subtle (Fig. 5E). Starting from 15 min, the glucose concentration was similar to that of the WT mice, remaining markedly lower in comparison to the dystrophic animals following the next measurements (Fig. 5E). Taking all of the above into consideration, we suspected an altered expression of glucose transporter type 4 (GLUT4) in the muscles as the cause of impaired glucose uptake from the bloodstream. Lower levels of Glut4 mRNA were observed in the mdx animals as compared to the WT ones with no effect of miR-378 loss (Supplementary Fig. 4A). At the protein level, no alterations between genotypes were noticed in Western Blot (Supplementary Fig. 4B), whereas immunofluorescent staining revealed the presence of cytoplasmic aggregates of GLUT4 in the mdx animals and not the WT ones, without further changes resulting from an additional lack of miR-378 (Supplementary Fig. 4C).
Complex regulation of factors influencing glucose and lipid homeostasis in the liver of mdx and mdx/miR-378−/− mice
Given that miR-378 was already reported to affect energy metabolism, for example, by targeting p110α25,26,27 and insulin-like growth factor 1 receptor (IGF1R)20, we aimed to check the levels of those markers in our experimental settings, together with glycogen synthase kinase 3 beta (GSK-3β) and pAKT/AKT kinase, due to their well-established role in glucose homeostasis and insulin action43. Nonetheless, the hepatic level of p110α was even lower in the mdx/miR-378−/− mice (Supplementary Fig. 5A,B). GSK-3β appeared to be potently upregulated in the mdx animals, without any further changes driven by the lack of miR-378 (Supplementary Fig. 5A,C), whereas IGF1R (Supplementary Fig. 5D) and pAKT/AKT (Supplementary Fig. 5E) displayed no differences between genotypes. We also analyzed the level of peroxisome proliferator-activated receptor alpha (PPARα), which plays a role not only in carbohydrate homeostasis44,45 but predominantly in fatty acid metabolism as well46. Despite the lack of changes in mRNA (Supplementary Fig. 5F), strong upregulation in the hepatic protein level of PPARα in the dystrophic mice with a concomitant decrease caused by miR-378 deficiency (Supplementary Fig. 5G) indicated altered lipid homeostasis.
Factors related to lipid homeostasis as the candidate mediators of miR-378 action in the liver
The IPA-based analysis predicted the opposite regulation of lipolysis in mdx and mdx/miR-378−/− livers – upregulation in the dystrophic animals and downregulation in the mice which additionally lacked miR-378 (Fig. 6A). Simultaneously, the incorporation of lipids was lower in the mdx mice, whereas the concentration of lipids was elevated in the mdx/miR-378−/− animals (Fig. 6B). Since triglycerides are hydrolyzed in the process of lipolysis, we aimed to measure their levels in the serum and liver. Despite the lack of changes in the serum (Fig. 6C), hepatic triglycerides appeared to accumulate under fasting conditions in the WT and mdx/miR-378−/− mice, which was not observed in the case of the dystrophic animals (Fig. 6D). No increase was noticed in the serum levels of total cholesterol (Supplementary Fig. 6A) and HDL (Supplementary Fig. 6B), under either fed or fasting conditions in any of the groups.

Disturbances in lipid metabolism in the liver of the mdx mice are affected by the additional lack of miR-378. (A) Ingenuity pathway analysis (IPA) revealed increased and decreased lipolysis in mdx vs. WT (p-value = 1.54 × 10–4) and mdx/miR-378−/− vs. mdx (p-value = 1.28 × 10−5) livers, respectively. (B) Incorporation of lipids was shown to be lower in the liver of mdx mice (p-value = 6.33 × 10–5) whereas the concentration of lipids was higher in mdx/miR-378−/− vs. mdx livers (p-value = 6.44 × 10–5). Triglyceride levels were assessed under non-fasting (fed) conditions and after overnight fasting (fasted) in (C) the serum; n = 5–6/group and (D) in the liver; 4–6/group. Data are presented as mean ± SEM. ***p < 0.001; ****p < 0.0001 by one-way ANOVA with Tukey’s post-hoc test.
To indicate potential mediators of miR-378 action in the liver, we examined the expression pattern of all 561 DEGs in mdx vs. WT and 194 DEGs in mdx/miR-378−/− vs. mdx mice (Fig. 7). Among them, 67 DEGs were identified as being changed in both comparisons, with 53 DEGs exhibiting the opposite expression pattern (Fig. 7). Accordingly, 20 of these DEGs had lower expression in mdx vs. WT mice and concomitantly higher expression in the mdx/miR-378−/− vs. mdx animals, pointing toward those factors being directly regulated by miR-378. More than half of them (11 out of 20) were found to be predicted targets of miR-378 with the miRmap database47 (Fig. 7). On the other hand, an indirect regulation might be proposed in the case of 33 DEGs which had higher expression in mdx vs. WT and lower expression in mdx/miR-378−/− vs. mdx livers, with 12 DEGs being indicated as predicted targets of miR-378 (Fig. 7). When we analyzed 20 DEGs that were downregulated in mdx in comparison with WT and were upregulated in mdx/miR-378−/− over mdx using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING), 7 of them were found to have multi-level interaction (Fig. 8A). We further validated the lower level of all of these DEGs in the mdx when compared with the WT mice and higher expression in mdx/miR-378−/− than the mdx animals (though to a lesser extent in the case of perilipin 2; Plin2 and glycerol-3-phosphate acyltransferase 3; Agpat9) by qRT-PCR analysis (Fig. 8B). Five out of 7 DEGs were identified as predicted targets of miR-378: ATP citrate synthase (Acly), glycerol-3-phosphate acyltransferase, mitochondrial (Gpam), Patatin-like phospholipase domain-containing protein 3 (Pnpla3), stearoyl-CoA desaturase-1 (Scd1), and fatty acid synthase (Fasn) (Figs. 7, 8A). GO term analysis indicated that all interacting genes were implicated in processes related to lipid homeostasis (Fig. 8C). On the other hand, only 2 DEGs, complement factor properdin (Cfp) and hemolytic complement (Hc)—related to complement/protein activation pathways, elevated in dystrophic animals, and downregulated by the additional lack of miR-378 (Fig. 7, Fig. 8D)—were revealed by STRING to interact with each other (Fig. 8E). Notably, IPA indicated activation of the complement system in mdx vs. WT counterparts (data not shown).

The analysis of oppositely expressed genes in the liver of the mdx vs. WT and mdx/miR-378−/− vs. mdx mice. Based on RNA-seq analysis, 67 genes were found to be differentially expressed in both comparisons. 53 of them exhibited opposite expression patterns: 20 genes were downregulated in mdx and upregulated in mdx/miR-378−/− livers, whereas 33 were elevated in mdx and downregulated in mdx/miR-378−/− livers. The expression pattern and gene names are presented on heat maps. Orange color indicates upregulation whereas blue color downregulation. Asterisks indicate genes predicted to be miR-378 targets according to the miRmap database.

Search Tool for the Retrieval of Interacting Genes/Proteins (STRING)—based analysis uncovered possible mediators of miR-378 action in the liver. The STRING analysis was performed on differentially expressed genes that were oppositely regulated in mdx vs. WT and mdx/miR-378−/− vs. mdx livers. (A) 7 out of 20 genes that were downregulated in mdx vs. WT and upregulated in mdx/miR-378−/− vs. mdx mice were found by STRING to interact with each other and exhibit co-expression patterns. Five of them indicated by asterisks were predicted by miRmap database to be direct targets of miR-378. (B) Verification of RNA sequencing results by qRT-PCR analysis of ATP citrate synthase (Acly), fatty acid synthase (Fasn), glycerol-3-phosphate acyltransferase, mitochondrial (Gpam), patatin-like phospholipase domain-containing protein 3 (Pnpla3), and stearoyl-CoA desaturase-1 (Scd1) predicted as miR-378 targets along with perilipin 2 (Plin2) and glycerol-3-phosphate acyltransferase 3 (Agpat9); n = 4–8/group, together with (C) gene ontology (GO) terms indicating pathways affected by interacting genes. (D) The expression of 2 out of 22 genes, hemolytic complement (Hc) and complement factor properdin (Cfp), that were higher in mdx vs. WT and lower in mdx/miR-378−/− vs. mdx liver based on RNA-seq data, was verified by qRT-PCR analysis; n = 4–8/group. (E) Those genes, presented together with the affected GO terms, were shown by STRING to interact with each other. Data are presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 by one-way ANOVA with Tukey’s post-hoc test; #p < 0.05 additionally tested by Student’s t-test for comparison of mdx vs. WT and mdx/miR-378−/− vs. mdx only.
Implications for metabolic alterations associated with by-product generation in mdx and mdx/miR-378−/− mice
Additionally, we implemented the metabolic cages approach to examine potential changes in the total energy expenditure48. After a 3 day acclimation period, mice were kept in metabolic cages for an additional 24 h to assess food and water consumption, as well as feces and urine excretion. Though no differences in food intake were noticed (Supplementary Fig. 7A), the dystrophic animals displayed intensified defecation (Supplementary Fig. 7B) and mdx/miR-378−/− mice showed increased water consumption (Supplementary Fig. 7C). Furthermore, it appeared that the urine volume excreted by the WT and dystrophic mice lacking miR-378−/− was at a comparable level with significantly lower amounts in the mdx animals (Supplementary Fig. 7D). These effects were also associated with changes in urinary uric acid concentration, which was elevated in the dystrophic animals and diminished as a consequence of miR-378 loss (Supplementary Fig. 7E). Interestingly, based on the RNA-seq, IPA suggested increased levels of blood urea nitrogen and creatinine, indicators of kidney injury (Supplementary Fig. 7F). All of the above strengthens multi-organ perturbations in dystrophic animals, modeling human pathology which is not solely restricted to muscle tissues.
Discussion
In this study, we comprehensively described the secondary consequences of dystrophin loss associated with alterations in systemic metabolism and we proposed the inhibition of miR-378 as a novel approach to mitigating those abnormalities, at least partially.
We functionally proved disturbed systemic carbohydrate metabolism by demonstrating a delayed glucose clearance from the bloodstream after a GTT and a completely blunted response following insulin administration in ITT in mdx mice, constituting the most commonly utilized model in DMD research. To the best of our knowledge, impaired insulin sensitivity in dystrophic males has not yet been reported, although insulin resistance has been revealed in female mdx mice49. Similar observations to our results regarding GTT in male dystrophic mice have been published by Stapleton et al.9 and Swiderski et al.50, although contradictory reports also exist10,51. It is known that the status of carbohydrate homeostasis may largely depend on the disease stage, as impaired glucose tolerance was observed only in severely immobilized DMD patients13, though the molecular mechanisms responsible for such abnormalities are not well understood. The vast majority of studies focused on skeletal muscles as the major players in glucose transport and homeostasis52. In our analysis, the protein level of GLUT4, a pivotal glucose transporter, was not affected in the dystrophic gastrocnemius muscle. However, Rodríguez-Cruz et al.11 found that GLUT4 forms cytoplasmic aggregates in muscle biopsies from DMD patients, which we also noticed in mdx mice, suggesting diminished glucose incorporation into the muscles. Moreover, altered insulin receptor affinity13,53 and GCs treatment54 were thought to trigger those complications. On the other hand, impaired insulin sensitivity, independent of GCs administration, was also observed in DMD boys11 and by us in non-treated mdx mice, pointing toward rather intrinsic causes of carbohydrate abnormalities in DMD. Accordingly, it appears that alterations in the dystrophin-glycoprotein complex (DGC) may facilitate the development of insulin resistance due to the impaired assembly of insulin receptors via DGC components55. DGC also binds neuronal nitric oxide synthase (nNOS), which facilitates glucose uptake into the muscles56. Thus, it might be suspected that the reduction of the nNOS pool in the absence of dystrophin contributes, at least partially, to the dysfunctional carbohydrate metabolism in DMD57. Importantly, apart from changes within dystrophic muscles, we and others9 found lower glycogen content and altered expression of genes implicated in glucose homeostasis in the liver. In the latter case, delta 4-desaturase, sphingolipid 1 (Degs1)—the most significantly upregulated factor in mdx livers compared with WT livers (padj = 5.68 × 10–171, fold change = 30.51)—could be proposed as one of the key players of the effects observed in dystrophic animals. Its deletion in the liver was reported to resolve insulin resistance in mice caused by leptin deficiency or an obesogenic diet58, among other effects.
Likewise, we provided some important insight into abnormalities in lipid homeostasis mediators which could be important for the development of dyslipidemia, recently described in DMD patients and animal models of the disease59. In the livers of mdx mice, we observed a dysregulated expression of genes affecting the level of circulating triglycerides, cholesterol, LDL, and HDL—such as Apoa1, Abca1, Lpl (normally not expressed in the liver under physiological conditions), Pltp, and Pparg60. Nonetheless, despite this molecular signature, we did not notice any significant abnormalities in the levels of triglycerides, total cholesterol, or HDL in the serum, under either fed or fasting conditions. No pathological accumulation of lipids in the liver was found either, in contrast to Murphy et al.32, who observed hepatic fat deposition in older, 6-month-old mdx-4Cv animals. On the other hand, Milad et al.61 did not find differences in the levels of total cholesterol, HDL, and triglycerides in the same model of the disease, namely 7-month-old mdx-4Cv mice fed a normal chow diet. However, because White et al.59 suggested that the elevation of lipids in DMD dogs progresses with age, it would be of particular importance for future studies to further investigate dyslipidemia features in older dystrophic mice, not only under normal conditions but also when animals are kept on an obesogenic diet. Importantly, lipid-lowering agents such as statins, have already been proposed to exert beneficial effects in DMD62,63; however, our64 and other studies65 did not confirm the rationale to utilize simvastatin, at least, to ameliorate muscle-related DMD symptoms. Our study indicated reduced hepatic expression of the key lipogenic factors, Scd1 and Fasn, similarly to the results obtained in skeletal muscles of mdx animals66. In the latter study, Paran et al. reported that a reduction of approx. 50% in de novo lipogenesis contributes to the dysfunction of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) activity, whereas the recovery of this pathway through the overexpression of lipogenic factors mitigates SERCA function by affecting the composition of the sarcoplasmic reticulum membrane66. Of note, pharmacological activation of SERCA was recently demonstrated to provide a promising therapeutic strategy for DMD67. Moreover, apart from lipogenic genes, we observed decreased expression of the gene encoding lipid droplet coat protein perilipin 2 (Plin2) in dystrophic livers, similarly to the findings reported by Murphy et al.32 on protein level, further emphasizing the perturbations in lipid homeostasis displayed by dystrophic animals.
Finally, the most important finding is that we identified miR-378 as a factor that regulates not only muscle-related dysfunctions, as we showed previously23, but also metabolic disturbances in the mdx model of DMD, as reported in the current study. Though miR-378 is a well-documented mediator of metabolism18, this is the first report on its relevance to metabolic alterations under dystrophic conditions. Mdx mice devoid of miR-378 exhibited improved glucose tolerance, which is in line with studies performed not only in miR-378 knockout25, 26,68 but also in miR-378 overexpressing animals, which manifest impaired glucose and/or insulin tolerance26,27,69.
At the same time, we did not observe higher levels of the verified targets of miR-378, p110α26 and IGF1R in the livers of dystrophic mice lacking miR-378. This indicates that the molecular outcome of miR-378 loss might largely depend on the pathological conditions and the existence of factors that mask miR-378-specific effects. The analysis of potential mediators in the whole tissue is also associated with limitations, as the relevance of cell type-specific alterations might be overlooked in such experimental settings. On the other hand, we unravel the restoration of hepatic glycogen content in dystrophic mice devoid of miR-378. Importantly, in agreement with our findings, Liu et al.26 reported diminished glycogen content in the livers of miR-378 overexpressing mice.
miR-378 loss restored the expression of genes implicated in lipogenesis and lipid storage, such as Fasn and Scd1, which were diminished in the dystrophic mice. Of note, they were already reported to have higher levels in miR-378 knockout mice26, whereas Scd1 was additionally verified as a direct target of miR-37869. Interestingly, bioinformatics analysis predicted intensified and decreased lipolysis in mdx and mdx/miR-378−/− mice, respectively. We link this with the pattern of hepatic triglyceride content after a fasting period. However, no accumulation of triglycerides in mdx livers under food deprivation could be also explained by reduced lipogenesis, whereas their elevation in mdx/miR-378−/− animals would reflect restored lipogenesis. In line with such an assumption, we propose that Gpam, a factor involved in the glycerophosphate pathway of de novo triglyceride synthesis in mammals, might serve as an important player in the observed effects. Although further studies are warranted, it was reported that knockout of Gpam results in a profound reduction of triglyceride biosynthesis70, among other things.
Lastly, PPARα was validated as a target of miR-37868; therefore, the drastic reduction of PPARα levels in the mdx/miR-378−/− mice observed in the current study is likely the result of indirect regulation of this transcription factor by miR-378 loss under dystrophic conditions. We previously made similar findings concerning fibroblast growth factor 1 (FGF1), a predicted target of miR-378, which was significantly lower in mdx/miR-378−/− than mdx mice23. Although the role of PPARα in DMD is poorly understood, its agonist, fenofibrate, is known to exert beneficial effects in mdx mice related to muscle pathology and fatty acid metabolism71. Moreover, PPARα inducers were proposed as factors that prevent insulin resistance72, but other results showed that PPARα knockout mice were protected from insulin resistance induced by a high-fat diet73. In light of the above, the use of PPARα antagonist would be important to resolve its potential to mimic the effect of miR-378 loss in dystrophic animals. Also, the evaluation of the potential link between PPARα, increased liver mass, and cardiac complications74,75 in dystrophic mice seems to be an interesting aspect of further studies.
In conclusion, we provided an insightful report on metabolic disturbances accompanied by the global, hepatic-specific changes displayed by the mdx model of DMD. Importantly, we uncovered for the first time that the lack of miR-378 in dystrophic mice can, at least partially, reverse those alterations. Although more mechanistic studies are warranted, we believe that the downregulation of miR-378 might serve as a promising therapeutic approach to mitigate the multifaceted symptoms of DMD.
Materials and methods
Animals
Animal procedures were performed after approval by the 2nd Institutional Animal Care and Use Committee (IACUC) in Kraków, Poland (approval numbers: 230/2018, 200/2019, 237/2019), following Polish and European legislation and according to the ARRIVE guidelines. Mice were housed under specific pathogen-free (SPF) conditions in individually ventilated cages with a 14 h/10 h light/dark cycle and were kept on a normal, chow diet with water and food available ad libitum (fed conditions). Under fasting conditions, mice were placed in a new cage with no food for 4 (in the morning) or 16 (overnight) hours, water ad libitum. Control (C57BL/10ScSnJ) and mdx (C57BL/10ScSn-Dmdmdx/J) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA, stock nos. 000476 and 001,801, respectively), whereas miR-378−/− mice (129SvEv/C57BL/6) were kindly provided by Dr. Eric Olson (Department of Molecular Biology, University of Texas Southwestern Medical Center, Dallas, TX, USA)25. Mice globally lacking both dystrophin and miR-378 (mdx/miR-378−/−) were generated as described previously23. Wild-type (WT), mdx, and mdx/miR-378−/− mice were bred at mixed C57BL/10ScSn x 129SvEv/C57BL/6 background23. For all experiments only male, 3-month-old animals were used.
Tolerance tests
GTT and ITT were conducted according to the published protocols with minor modifications76. Briefly, GTT and ITT were performed in mice after an overnight (16 h) or 4 h fasting period, respectively. At first, fasting blood glucose was measured (indicated as 0 min timepoint) followed by intraperitoneal (i.p.) administration of 20% D-( +)-Glucose solution (Sigma-Aldrich, St. Louis, MO, USA) of a final dose of 2 g/kg body weight (BW) (GTT) or 1 U/mL insulin (Humulin® R, Lilly, Warsaw, Poland) in a dose of 0.5 U/kg BW (ITT). Next, the measurement of blood glucose concentration was repeated after 15, 30, 60, and 120 min (GTT) or after 15, 30, 60, and 90 min (ITT). Additionally, the random glucose level under non-fasting conditions (random glucose) and insulin under both non-fasting (random insulin) and after overnight (16 h) fasting conditions were measured. In any case, the blood glucose level was estimated from the tail tip using a glucometer (Diagnosis S.A., Białystok, Poland), whereas insulin level was assessed by Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem, Elk Grove Village, IL, USA) in serum prepared from the blood collected from vena cava, after allowing the blood to clot at room temperature for 30 min and centrifugation at 2000 × g for 10 min at 4 °C.
Metabolic cages
For the assessment of total energy expenditure, we utilized metabolic cages. Mice were housed individually in cages and acclimatized to standard animal house conditions for 3 days. Water and food were precisely measured to enable the determination of the food and water intake, feces, and urine excretion after consecutive 24 h. Upon collection, feces were dried for 1 h at 37 °C, whereas urine was centrifuged (2000 × g for 10 min at 4 °C). All results were calculated per kg BW.
RNA isolation, reverse transcription, and quantitative real-time PCR (qRT-PCR)
For RNA isolation, tissues (a piece of a right lobe of the liver and gastrocnemius muscle) were collected, preserved in RNAlater solution (Sigma-Aldrich, St. Louis, MO, USA), snap-frozen in liquid nitrogen, and stored at −80 °C. Tissues were homogenized using TissueLyser in 1 mL of QIAzol Lysis Reagent (all from QIAGEN, Hilden, Germany) followed by RNA isolation that was performed according to our previous expertise using the standard Chomczynski-Sacchi method77. For the reverse transcription, 1 µg of total RNA was transcribed to cDNA in a mix containing recombinant M-MuLV reverse transcriptase (Thermo Fisher Scientific, Waltham, MA, USA), dNTPs, oligodT (both from Genomed, Warsaw, Poland), and water, or was conducted according to the miRCURY LNA RT Kit (QIAGEN, Hilden, Germany) in the case of miRNAs expression analysis. qRT-PCR was performed using StepOne Plus Real-Time PCR (Applied Biosystems—Thermo Fisher Scientific, Waltham, MA, USA) on obtained cDNA with SYBR Green PCR Master Mix, and specific primers (all from Sigma-Aldrich, St. Louis, MO, USA) which sequences are listed in Table 1. Eukaryotic elongation factor 2 (Eef2), with a stable expression between analyzed groups, served as a housekeeping gene. In the case of miRNA level assessment, qRT-PCR was conducted according to the miRCURY LNA SYBR PCR Kit with specific primers (miRCURY LNA™ miRNA PCR Assays, all from QIAGEN, Hilden, Germany). Normalization of miRNAs expression was done based on the small nucleolar RNA, C/D box 68 (Snord68) which level was not affected by mice genotype. Relative gene expression was calculated based on the comparative Ct method according to the 2−ΔCt formula, where (ΔCt = Ct gene of interest − Ct Eef2/Snord68). Primer specificity was monitored based on the melting curves.
Liver transcriptome sequencing
For RNA-seq, mice were euthanized, perfused immediately with saline containing 0.5 U/ml heparin (Polfa, Warsaw, Poland) through the left ventricle, and a piece of the liver (left lateral lobe) was collected to tubes containing RNAlater (Sigma-Aldrich, St. Louis, MO, USA). Samples were immediately snap-frozen in liquid nitrogen and stored at -80 °C before RNA isolation which was performed as described above according to the standard Chomczynski-Sacchi method78.
High-throughput gene expression profiling was conducted using next-generation sequencing and a highly multiplexed amplification method provided by Ion AmpliSeq™ technology and Ion Proton™ machine (Thermo Fisher Scientific, Waltham, MA, USA). Libraries for 16 samples were prepared using Ion AmpliSeq™ Transcriptome Mouse Gene Expression Kit covering > 20,000 mouse RefSeq genes in a single assay. Before the library preparation step, integrity and concentrations of RNA samples were determined using an Agilent 2100 Bioanalyzer with RNA 6000 Nano Kit (Agilent, Santa Clara, CA, USA). Libraries were prepared according to the manufacturer's protocol using 100 ng of total RNA as the input material. Emulsion PCR, templating and chip loading was conducted manually. Sequencing was performed using Ion PI Hi-Q Sequencing 200 chemistry. Two Ion PI Chip v3 chips were used with 8 barcoded and pooled in equimolar concentrations samples sequenced per single chip. The primary bioinformatics analyses including mapping were carried out using Torrent Suite Server v5.10.0. Transcripts were counted with the HTSeq Python package (https://htseq.readthedocs.io/en/release_0.10.0/) while read-count normalization and differential gene expression analysis (DGE) was carried out using DESeq2 package implemented in R version 3.3.3 software. P-values for differentially expressed genes were corrected for multiple comparisons using the Benjamini–Hochberg approach and the results with the corrected P-adjusted < 0.05 were considered significant if not otherwise stated. Heatmaps were generated using ggplot2 R package79 or were created in GraphPad Prism 8 Software. Gene set enrichment analysis (GSEA) was conducted with the GSEA method using the Java GSEA implementation. GSEA pre-ranked module and log2 fold change values were used as the input values (http://software.broadinstitute.org/gsea/index.jsp). Mapping to Gene Ontology terms (GO) was conducted using the “GOstats” package implemented in R version 3.3.3 software80. Data were analyzed through the use of IPA (QIAGEN, Hidden, Germany, https://www.QIAGENbioinformatics.com/products/ingenuitypathway-analysis)81 and STRING database for known and predicted interactions82. miRmap database with the default parameters was recruited to predict miR-378 targets47.
Biochemical assays
The measurements of ALT, AST, total cholesterol, HDL, triglycerides, and bilirubin level in serum were performed using biochemical analyzer SPOTCHEM EZ SP-4430 (ARKRAY, Kyoto, Japan) according to the vendor’s instructions. Triglyceride content in the liver was assessed by Triglyceride Quantification Colorimetric/Fluorometric Kit (Sigma-Aldrich, St. Louis, MO, USA), whereas glycogen level was analyzed using Glycogen Colorimetric/Fluorometric Assay Kit (BioVision, Milpitas, CA, USA). The urinary uric acid concentration was tested according to the vendor’s instruction (CORMAY, Warsaw, Poland).
Protein isolation
For protein isolation, collected tissues were homogenized using TissueLyser (QIAGEN, Hilden, Germany) in PBS containing 1% Triton X-100 (BioShop, Burlington, ON, Canada) and protease inhibitors (cOmplete™, Mini Protease Inhibitor Cocktail; Roche Diagnostic, Basel, Switzerland). After centrifugation (14,000 × g for 10 min at 4 °C), supernatants were collected and total protein content was assessed by the bicinchoninic acid assay (BCA) method (Sigma-Aldrich, Saint Louis, MO, USA).
Western Blot
For the assessment of protein level of pAKT, AKT, p110α, GSK-3β, PPARα, GLUT4, GAPDH, and VINCULIN, 25 µg of protein lysates were subjected to SDS-PAGE electrophoresis, transferred to nitrocellulose membrane (wet transfer, 100 V/1.5 h, 4 °C), blocked in 5% non-fat milk (in the case of p110α, GSK-3β, PPARα, GLUT4, GAPDH) or BSA (for pAKT, AKT, and VINCULIN) in TBS containing 0.1% Tween20 (1 h, room temperature), and incubated overnight with primary antibodies diluted in the appropriate blocking solution (Table 2). If needed, the membranes were cut before the incubation with the desired antibody. The next day, membranes were washed, incubated with secondary antibodies (diluted in the appropriate blocking solution) conjugated to horseradish peroxidase (HRP) (Table 2) for 1 h at room temperature, washed again, and the chemiluminescence was measured by ChemiDoc™ MP Imaging System (Bio-Rad, Hercules, CA, USA) after addition of the Immobilon® Western Chemiluminescent HRP Substrate (Millipore, Burlington, MA, USA). For stripping and reprobing, the membranes were washed thoroughly with TBS containing 0.1% Tween20 followed by the incubation with 0.5 M glycine solution (pH = 2.6), twice for 30 min with washing steps after each incubation (3 times per 5 min). Afterward, the membrane was blocked and subsequent steps were done as described above. Densitometric analysis was performed with the use of ImageJ software. Unedited Western Blot results from all experiments subjected to densitometric analysis are presented in Supplementary Fig. 8.
ELISA
IL6, TNFα, TNFR1, and IGF1R levels in the liver were assessed by ELISA assay (R&D, Minneapolis, MN, USA, and Abbexa, Cambridge, UK, respectively) according to the vendor’s instruction using 100 µg of protein lysates as determined by the BCA method. The results were calculated per mg of the total protein.
Histological analyses
A piece of the liver (right lobe) was collected directly to 10% formalin in PBS for 48 h. Afterward, tissues were transferred to 70% ethanol and stored at 4 °C before further processing. Tissues were dehydrated, embedded in paraffin, and cut using a microtome (Thermo Fisher Scientific, Waltham, MA, USA) on 4.5 µm sections. After deparaffinization, sections were subjected to hematoxylin and eosin (H&E) and Masson’s Trichrome staining according to the vendor’s instructions (Sigma-Aldrich, St. Louis, MO, USA). The pictures were taken using the Leica DMi8 microscope with the CMOS Leica MC170 HD camera. The assessment of liver injury was conducted according to previous reports83 with modifications. Briefly, semiquantitative total scoring of necrosis, vacuolization, congestion, and leukocyte infiltration was performed, based on the following scale (for each parameter): 0—none, 1—minimal, 2—mild, 3—moderate, 4—severe. The liver fibrosis was calculated using ImageJ software and presented as the percentage of Masson’s trichrome positive area of the whole liver biopsy/scan84.
Oil Red O (ORO) staining for the determination of lipid accumulation was performed on 5 μm thick frozen sections, which were cut after embedding the liver in an OCT medium and freezing in an isopentane-cooled liquid nitrogen bath. The sections were fixed in formalin, briefly washed with running tap water, rinsed with 60% isopropanol, and stained for 15 min with freshly prepared ORO (ICN Biomedicals, Inc., USA) working solution (0.5% of the stock solution prepared in isopropanol was diluted 1.6 times in water). Afterward, the sections were rinsed with 60% isopropanol, distilled water, and mounted with an aqueous mounting medium. The pictures were taken using the Nikon Eclipse Ti microscope.
Immunofluorescent staining of GLUT4 was performed on frozen sections according to the protocol described previously19, with the use of rabbit anti-rat GLUT4 antibody (Abcam, Cambridge, UK, ab654, 1:200) and Alexa Fluor 488 donkey anti-rabbit IgG (H + L) secondary antibody (Thermo Fisher Scientific, Waltham, MA, USA, A21206, 1:500). The pictures were taken using the Leica DMi8 microscope with Leica DFC7000 GT fluorescent camera.
Isolation of primary hepatocytes
Isolation of primary hepatocytes was performed according to the established protocol85 with a modified digestion step in which 200 U/mL of collagenase IV (Thermo Fisher Scientific, Waltham, MA, USA) was used. For RNA isolation, cells were seeded at a density of 2 × 105 cells/well of 12-well plates. 24 h after isolation, the medium was removed, cells were washed with PBS, lysed with Fenozol (A&A Biotechnology, Gdańsk, Poland), and subjected to RNA isolation and miRNA level determination as described in “RNA isolation, reverse transcription (RT), and quantitative real-time PCR (qRT-PCR)” section of the manuscript.
Seahorse analyses
Oxygen consumption rate (OCR) in primary hepatocytes was measured using Seahorse Bioscience XFe96 Analyzer (Agilent Technologies, Santa Clara, CA, USA) according to our protocol described previously86, 87. Briefly, 1 × 105 of live hepatocytes (determined by the trypan blue staining) were plated into Seahorse XFe96-well plates directly after isolation. The next day, the medium was switched for low-buffered assay medium (8.3 g/L DMEM, 2 mM L-Glutamine, 1 mM sodium pyruvate, and 0.5% phenol red (all from Sigma-Aldrich, St. Louis, MO, USA ) at pH = 7.4, and the cells were incubated at 37 °C, 20% O2, without CO2 for ~ 1 h. OCR was assessed at a basal level and after consecutive injections of 1.5 µg/mL oligomycin, 0.4 µM FCCP, and 0.5 µM rotenone + antimycin A (all from Sigma-Aldrich, St. Louis, MO, USA). All parameters were optimized before the test. Basal respiration was calculated as the last rate measurement before oligomycin injection minus non-mitochondrial respiration (minimum rate measurement after antimycin A and rotenone injection). The results were normalized to the protein content using the BCA method.
Statistical analysis
The results are presented as mean ± SEM. In the figure legends, the “n” number indicates biological replicates equal to the number of mice used in each experiment. Differences between groups were tested for statistical significance using one-way ANOVA with Tukey’s posthoc test for multiple comparisons or two-way ANOVA for repeated measures with Tukey’s posthoc tests for multiple comparisons. When indicated, the unpaired 2-tailed Student’s t-test was applied test for the comparison of two groups. p < 0.05 was considered significant, whereas p = 0.05–0.1 was described as a tendency. Grubb’s test was used to identify outliers. Graphs and statistical analyses were performed with the use of GraphPad Prism 8 Software.
References
Bushby, K. et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurol. 9, 177–189 (2010).
Podkalicka, P. et al. Age-dependent dysregulation of muscle vasculature and blood flow recovery after hindlimb ischemia in the mdx model of duchenne muscular dystrophy. Biomedicines 9, 481 (2021).
Podkalicka, P., Mucha, O., Dulak, J. & Loboda, A. Targeting angiogenesis in Duchenne muscular dystrophy. Cell. Mol. Life Sci. 76, 1507–1528 (2019).
Rae, M. G. & O’Malley, D. Cognitive dysfunction in Duchenne muscular dystrophy: a possible role for neuromodulatory immune molecules. J. Neurophysiol. 116, 1304–1315 (2016).
Motoki, T. et al. Renal dysfunction can occur in advanced-stage Duchenne muscular dystrophy. Muscle Nerve 61, 192–197 (2020).
MacLeod, M., Kelly, R., Robb, S. A. & Borzyskowski, M. Bladder dysfunction in Duchenne muscular dystrophy. Arch. Dis. Child 88, 347–349 (2003).
Ward, L. M., Hadjiyannakis, S., McMillan, H. J., Noritz, G. & Weber, D. R. Bone health and osteoporosis management of the patient with duchenne muscular dystrophy. Pediatrics 142, S34–S42 (2018).
Lo Cascio, C. M. et al. Gastrointestinal Dysfunction in Patients with Duchenne Muscular Dystrophy. PLoS One 11, e0163779 (2016).
Stapleton, D. I. et al. Dysfunctional muscle and liver glycogen metabolism in mdx dystrophic mice. PLoS One 9, e91514 (2014).
Strakova, J. et al. Integrative effects of dystrophin loss on metabolic function of the mdx mouse. Sci. Rep. 8, 13624 (2018).
Rodríguez-Cruz, M. et al. Evidence of insulin resistance and other metabolic alterations in boys with duchenne or becker muscular dystrophy. Int J Endocrinol 2015, 867273 (2015).
Bostock, E. L. et al. Impaired glucose tolerance in adults with duchenne and becker muscular dystrophy. Nutrients 10, E1947 (2018).
Freidenberg, G. R. & Olefsky, J. M. Dissociation of insulin resistance and decreased insulin receptor binding in Duchenne muscular dystrophy. J. Clin. Endocrinol. Metab. 60, 320–327 (1985).
Łoboda, A. & Dulak, J. Muscle and cardiac therapeutic strategies for Duchenne muscular dystrophy: past, present, and future. Pharmacol. Rep. 72, 1227–1263 (2020).
Kourakis, S. et al. Standard of care versus new-wave corticosteroids in the treatment of Duchenne muscular dystrophy: Can we do better?. Orphanet J. Rare Dis. 16, 117 (2021).
Cordova, G., Negroni, E., Cabello-Verrugio, C., Mouly, V. & Trollet, C. Combined therapies for duchenne muscular dystrophy to optimize treatment efficacy. Front. Genet. 9, 114 (2018).
Friedman, R. C., Farh, K.K.-H., Burge, C. B. & Bartel, D. P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105 (2009).
Krist, B., Florczyk, U., Pietraszek-Gremplewicz, K., Józkowicz, A. & Dulak, J. The Role of miR-378a in Metabolism, Angiogenesis, and Muscle Biology. Int J Endocrinol 2015, 281756 (2015).
Krist, B. et al. miR-378a influences vascularization in skeletal muscles. Cardiovasc. Res. 116, 1386–1397 (2020).
Knezevic, I. et al. A novel cardiomyocyte-enriched microRNA, miR-378, targets insulin-like growth factor 1 receptor: Implications in postnatal cardiac remodeling and cell survival. J. Biol. Chem. 287, 12913–12926 (2012).
Martyniak, A. et al. Generation of microRNA-378a-deficient hiPSC as a novel tool to study its role in human cardiomyocytes. J. Mol. Cell Cardiol. 160, 128–141 (2021).
Zeng, P. et al. miR-378 attenuates muscle regeneration by delaying satellite cell activation and differentiation in mice. Acta Biochim. Biophys. Sin. (Shanghai) 48, 833–839 (2016).
Podkalicka, P. et al. Lack of miR-378 attenuates muscular dystrophy in mdx mice. JCI Insight 5, 135576 (2020).
Jones, A. et al. miRNA signatures of insulin resistance in obesity. Obesity (Silver Spring) 25, 1734–1744 (2017).
Carrer, M. et al. Control of mitochondrial metabolism and systemic energy homeostasis by microRNAs 378 and 378*. Proc. Natl. Acad. Sci. USA 109, 15330–15335 (2012).
Liu, W. et al. Hepatic miR-378 targets p110α and controls glucose and lipid homeostasis by modulating hepatic insulin signalling. Nat. Commun. 5, 5684 (2014).
Li, Y.-Y. et al. miR-378b regulates insulin sensitivity by targeting insulin receptor and p110α in alcohol-induced hepatic steatosis. Front. Pharmacol. 11, 717 (2020).
Facco, F., Navone, C., Bado, M. & Giacchino, R. The role of liver in muscular dystrophy. Minerva Pediatr. 45, 15–19 (1993).
Veropalumbo, C. et al. Duchenne and Becker muscular dystrophy presenting as nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 53, 463–464 (2011).
Lao, Y. E. et al. Fatal liver failure after therapeutic doses of paracetamol in a patient with Duchenne muscular dystrophy and atypical pharmacogenetic profile of drug-metabolizing enzymes. Basic Clin. Pharmacol. Toxicol. 127, 47–51 (2020).
Janowski, K. et al. Acute liver failure due to previously unrecognised dilated cardiomyopathy in a 17-year-old boy with Duchenne muscular dystrophy. Pediatr. Pol. 95, 178–183 (2020).
Murphy, S. et al. Proteomic profiling of liver tissue from the mdx-4cv mouse model of Duchenne muscular dystrophy. Clin. Proteomics 15, 34 (2018).
Vignier, N. et al. Distinctive serum miRNA profile in mouse models of striated muscular pathologies. PLoS One 8, e55281 (2013).
Michell, D. L. & Vickers, K. C. Lipoprotein carriers of microRNAs. Biochim Biophys Acta 1861, 2069–2074 (2016).
Howland, J. L. & Challberg, M. D. Altered respiration and proton permeability in liver mitochondria from genetically dystrophic mice. Biochem. Biophys. Res. Commun. 50, 574–580 (1973).
Manikandan, P. & Nagini, S. Cytochrome P450 structure, function and clinical significance: A review. Curr. Drug Targets 19, 38–54 (2018).
Horak, M., Novak, J. & Bienertova-Vasku, J. Muscle-specific microRNAs in skeletal muscle development. Dev. Biol. 410, 1–13 (2016).
Vasu, S. et al. MicroRNA signatures as future biomarkers for diagnosis of diabetes states. Cells 8, E1533 (2019).
Al-Kafaji, G., Al-Muhtaresh, H. A. & Salem, A. H. Expression and clinical significance of miR-1 and miR-133 in pre-diabetes. Biomed. Rep. 14, 33 (2021).
Lienenlüke, B. & Christ, B. Impact of interleukin-6 on the glucose metabolic capacity in rat liver. Histochem. Cell Biol. 128, 371–377 (2007).
Krogh-Madsen, R., Plomgaard, P., Møller, K., Mittendorfer, B. & Pedersen, B. K. Influence of TNF-alpha and IL-6 infusions on insulin sensitivity and expression of IL-18 in humans. Am. J. Physiol. Endocrinol. Metab 291, E108-114 (2006).
Steinberg, G. R. et al. Tumor necrosis factor α-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 4, 465–474 (2006).
Patel, S. et al. Tissue-specific role of glycogen synthase kinase 3β in glucose homeostasis and insulin action. Mol. Cell. Biol. 28, 6314–6328 (2008).
Tordjman, K. et al. PPARα deficiency reduces insulin resistance and atherosclerosis in apoE-null mice. J. Clin. Invest. 107, 1025–1034 (2001).
Haluzík, M. M. & Haluzík, M. PPAR-alpha and insulin sensitivity. Physiol. Res. 55, 115–122 (2006).
Burri, L., Thoresen, G. H. & Berge, R. K. The Role of PPAR Activation in Liver and Muscle. PPAR Research 2010, e542359 (2010).
Vejnar, C. E. & Zdobnov, E. M. MiRmap: Comprehensive prediction of microRNA target repression strength. Nucl. Acids Res. 40, 11673–11683 (2012).
Kennedy, B. W. Metabolic caging for animal research. Lab. Anim. 41, 171–176 (2012).
Moore, T. M. et al. Mitochondrial dysfunction is an early consequence of partial or complete dystrophin loss in mdx mice. Front. Physiol. 11, 690 (2020).
Swiderski, K. et al. Tranilast administration reduces fibrosis and improves fatigue resistance in muscles of mdx dystrophic mice. Fibrogenes. Tiss. Repair. 7, 1 (2014).
Mokhtarian, A. & Even, P. C. Effect of intraperitoneal injection of glucose on glucose oxidation and energy expenditure in the mdx mouse model of Duchenne muscular dystrophy. Pflügers Arch. Eur. J. Physiol. 432, 379 (1996).
Koistinen, H. A. & Zierath, J. R. Regulation of glucose transport in human skeletal muscle. Ann. Med. 34, 410–418 (2002).
DePirro, R. et al. Decreased insulin receptors but normal glucose metabolism in Duchenne muscular dystrophy. Science 216, 311–313 (1982).
Hwang, J. L. & Weiss, R. E. Steroid-induced diabetes: a clinical and molecular approach to understanding and treatment. Diabet. Metab. Res. Rev. 30, 96–102 (2014).
Eid Mutlak, Y. et al. A signaling hub of insulin receptor, dystrophin glycoprotein complex and plakoglobin regulates muscle size. Nat. Commun. 11, 1381 (2020).
Kellogg, D. L., McCammon, K. M., Hinchee-Rodriguez, K. S., Adamo, M. L. & Roman, L. J. Neuronal nitric oxide synthase mediates insulin- and oxidative stress-induced glucose uptake in skeletal muscle myotubes. Free Radic Biol. Med. 110, 261–269 (2017).
Brenman, J. E., Chao, D. S., Xia, H., Aldape, K. & Bredt, D. S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 82, 743–752 (1995).
Chaurasia, B. et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 365, 386–392 (2019).
White, Z. et al. High prevalence of plasma lipid abnormalities in human and canine Duchenne and Becker muscular dystrophies depicts a new type of primary genetic dyslipidemia. J. Clin. Lipidol. 14, 459-469.e0 (2020).
García-Giustiniani, D. & Stein, R. Genetics of dyslipidemia. Arq. Bras. Cardiol. 106, 434–438 (2016).
Milad, N. et al. Increased plasma lipid levels exacerbate muscle pathology in the mdx mouse model of Duchenne muscular dystrophy. Skelet. Muscle. 7, 19 (2017).
Amor, F. et al. Cholesterol metabolism is a potential therapeutic target in Duchenne muscular dystrophy. J. Cachexia Sarcopenia Muscle 12, 677–693 (2021).
Whitehead, N. P., Kim, M. J., Bible, K. L., Adams, M. E. & Froehner, S. C. A new therapeutic effect of simvastatin revealed by functional improvement in muscular dystrophy. Proc. Natl. Acad. Sci. USA 112, 12864–12869 (2015).
Mucha, O. et al. Simvastatin does not alleviate muscle pathology in a mouse model of Duchenne muscular dystrophy. Skelet Muscle 11, 21 (2021).
Verhaart, I. E. C. et al. Simvastatin treatment does not ameliorate muscle pathophysiology in a mouse model for duchenne muscular dystrophy. J. Neuromuscul. Dis. 8, 845–863 (2021).
Paran, C. W. et al. Lipogenesis mitigates dysregulated sarcoplasmic reticulum calcium uptake in muscular dystrophy. Biochim. Biophys. Acta. 1851, 1530–1538 (2015).
Nogami, K. et al. Pharmacological activation of SERCA ameliorates dystrophic phenotypes in dystrophin-deficient mdx mice. Hum. Mol. Genet. 30, 1006–1019 (2021).
Wang, H. et al. Activation of dsRNA-dependent protein kinase R by miR-378 sustains metabolic inflammation in hepatic insulin resistance. Diabetes 70, 710–719 (2021).
Zhang, Y. et al. miR-378 activates the pyruvate-PEP futile cycle and enhances lipolysis to ameliorate obesity in mice. EBioMedicine 5, 93–104 (2016).
Yu, H. et al. Bovine lipid metabolism related gene GPAM: Molecular characterization, function identification, and association analysis with fat deposition traits. Gene 609, 9–18 (2017).
Sun, C. et al. Duchenne muscular dystrophy hiPSC-derived myoblast drug screen identifies compounds that ameliorate disease in mdx mice. JCI Insight 5, 134287 (2020).
Cho, Y., Hazen, B. C., Russell, A. P. & Kralli, A. Peroxisome proliferator-activated receptor γ coactivator 1 (PGC-1)- and estrogen-related receptor (ERR)-induced regulator in muscle 1 (Perm1) is a tissue-specific regulator of oxidative capacity in skeletal muscle cells. J. Biol. Chem. 288, 25207–25218 (2013).
Guerre-Millo, M. et al. PPAR-alpha-null mice are protected from high-fat diet-induced insulin resistance. Diabetes 50, 2809–2814 (2001).
Kaimoto, S. et al. Activation of PPAR-α in the early stage of heart failure maintained myocardial function and energetics in pressure-overload heart failure. Am. J. Physiol. Heart Circ. Physiol. 312, H305–H313 (2017).
Hall, A. P. et al. Liver hypertrophy: A review of adaptive (adverse and non-adverse) changes–conclusions from the 3rd International ESTP Expert Workshop. Toxicol Pathol 40, 971–994 (2012).
Vinué, Á. & González-Navarro, H. Glucose and insulin tolerance tests in the mouse. Methods Mol. Biol. 1339, 247–254 (2015).
Pietraszek-Gremplewicz, K. et al. Heme oxygenase-1 influences satellite cells and progression of duchenne muscular dystrophy in mice. Antioxid. Redox Signal. 29, 128–148 (2018).
Chomczynski, P. & Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–159 (1987).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. (Springer International Publishing, 2016). https://doi.org/10.1007/978-3-319-24277-4.
Falcon, S. & Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 23, 257–258 (2007).
Krämer, A., Green, J., Pollard, J. & Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530 (2014).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucl. Acids Res. 47, D607–D613 (2019).
Suzuki, S., Toledo-Pereyra, L. H., Rodriguez, F. J. & Cejalvo, D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation 55, 1265–1272 (1993).
Hirao, H. et al. Donor hepatic occult collagen deposition predisposes to peritransplant stress and impacts human liver transplantation. Hepatology 74, 2759–2773 (2021).
Charni-Natan, M. & Goldstein, I. Protocol for primary mouse hepatocyte isolation. STAR Protoc 1, 100086 (2020).
Jeż, M. et al. Role of heme-oxygenase-1 in biology of cardiomyocytes derived from human induced pluripotent stem cells. Cells 10, 522 (2021).
Podkalicka, P. et al. Synthetically lethal interactions of heme oxygenase-1 and fumarate hydratase genes. Biomolecules 10, E143 (2020).
Acknowledgements
We would like to acknowledge colleagues from the Faculty of Biochemistry, Biophysics, and Biotechnology of the Jagiellonian University in Kraków: Ewa Werner, Karolina Hajduk, Łukasz Szczepanik, and Janusz Drebot from the Animal Facility for the mice care and breeding; Katarzyna Polak and Joanna Strzęp-Knapiak for the help in mice genotyping; Dr. Sławomir Lasota for the assistance in microscopic imaging; Dr. Monika Jakubowska for consulting liver histology; Dr. Natalia Pydyn and Dr. Jerzy Kotlinowski for technical hints regarding primary hepatocyte isolation; Barbara Bober-Sowa and Dr. Paweł Koteja for providing us with metabolic cages; Dr. Iwona Bronisz-Budzyńska and Dr. Magdalena Kozakowska for valuable advice and discussions; Katarzyna Sarad for providing us with liver tissue from the mouse fed on high-fat which served as a positive control for ORO staining. We would like to thank the administrative staff (Agnieszka Andrychowicz-Róg and Joanna Uchto-Bajołek) for their assistance. This work was supported by grant PRELUDIUM 16 #2018/31/N/NZ4/02500 from the National Science Centre awarded to P. Podkalicka who is a recipient of the START 2021 scholarship for outstanding young scientists funded by the Foundation for Polish Science. The work on miR-378 in DMD is supported by the MAESTRO 10 grant (2018/30/A/NZ3/00412, JD) and was initiated in the MAESTRO 3 grant (2012/06/A/NZ1/00004, JD) from the National Science Centre. The open-access publication of this article was funded by the Priority Research Area BioS under the program “Excellence Initiative–Research University” at the Jagiellonian University in Kraków.
Author information
Authors and Affiliations
Contributions
P.P., J.D., and A.Ł. designed the study; P.P., O.M., K.K., J.S., L.I., and A.Ł. performed the experiments; P.P. and A.Ł. wrote the main manuscript text; P.P. prepared the figures and analyzed data; P.P., K.S., and E.P. analyzed R.N.A. sequencing data; A.J., J.W.K.-W., and H.H. contributed to the data analysis, interpretation of the results, and corrected the final version of the manuscript; A.Ł. and J.D. supervised the research. All authors approved and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Podkalicka, P., Mucha, O., Kaziród, K. et al. miR-378 affects metabolic disturbances in the mdx model of Duchenne muscular dystrophy. Sci Rep 12, 3945 (2022). https://doi.org/10.1038/s41598-022-07868-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-07868-z
- Springer Nature Limited
This article is cited by
-
miR-378 influences muscle satellite cells and enhances adipogenic potential of fibro-adipogenic progenitors but does not affect muscle regeneration in the glycerol-induced injury model
Scientific Reports (2023)
-
Hydrogen sulfide as a therapeutic option for the treatment of Duchenne muscular dystrophy and other muscle-related diseases
Cellular and Molecular Life Sciences (2022)