
Abstract
Primary ciliary dyskinesia (PCD) is a rare autosomal recessive condition often presenting with chronic respiratory infections in early life. Transmission electron microscopy (TEM) is used to detect ciliary ultrastructural defects. In this study, we aimed to assess ciliary ultrastructural defects using quantitative methods on TEM to identify its diagnostic role in confirming PCD. Nasal samples of 67 patients, including 37 females and 30 males (20.3 ± 10.7 years old), with suspected PCD symptoms were examined by TEM. The most common presentations were bronchiectasis: 26 (38.8%), chronic sinusitis: 23 (34.3%), and recurrent lower respiratory infections: 21 (31.3%). Secondary ciliary dyskinesia, including compound cilia (41.4%) and extra-tubules (44.3%), were the most prevalent TEM finding. Twelve patients (17.9%) had hallmark diagnostic criteria for PCD (class 1) consisting of 11 (16.4%) outer and inner dynein arm (ODA and IDA) defects and only one concurrent IDA defect and microtubular disorganization. Also, 11 patients (16.4%) had probable criteria for PCD (class 2), 26 (38.8%) had other defects, and 18 (26.9%) had normal ciliary ultrastructure. Among our suspected PCD patients, the most common ultrastructural ciliary defects were extra-tubules and compound cilia. However, the most prevalent hallmark diagnostic defect confirming PCD was simultaneous defects of IDA and ODA.
Similar content being viewed by others


Introduction
Chronic or recurrent infections of the respiratory tract are common among children. These infections may result from compromised mucociliary clearance. The interaction between ciliary and respiratory mucus is essential in the clearance of the respiratory tract, and any dysfunction in this system results in chronic or recurrent respiratory infections. A clear example is cystic fibrosis (CF)1,2. A normal ciliary ultrastructure was firstly introduced by Afzelius, consisting of a central pair of microtubules surrounded by nine peripheral microtubule doublets (known as 9 + 2) bonded to the center by several supporting radial spokes. Each doublet possesses outer and inner dynein arms (ODA and IDA). ODAs are molecular motors, which “walk” along axoneme microtubules and enable bending of the cilium (Fig. 1)3. This complex structure is not only found in the upper and lower airways (mucus‐propelling cilia) but is also seen in the middle ears, paranasal sinuses, ependymal lining of the brain ventricles, efferent ducts of the testis, and the oviduct (water‐propelling cilia)4.

Normal ciliary ultrastructure. (A) Tip: cross-sections from the tip of the cilium which consists of single microtubules. It should not be considered a defect. (B) Axoneme: it consists of a central pair of microtubules surrounded by nine peripheral microtubule doublets bond to the center pair by supporting radial spokes (known as the 9 + 2 arrangement) Each doublet possesses outer and inner dynein arms. (C) Base: cross-section at the base of the cilium, where the central pair is not present. Y-shaped linkers bond microtubule doublets together.
The ultrastructural ciliary impairments include the defect of the outer and/or inner dynein arms, the microtubular disorganization, and the rare absence of the central pair of microtubules (Fig. 2)5. These defects could be a hereditary defect called primary ciliary dyskinesia (PCD) or a post-infectious defect called secondary ciliary dyskinesia (SCD)6,7. On-time diagnosis of ultrastructural ciliary defects plays a key role in making clinical decisions and preventing harmful results such as hearing loss and speech impairment caused by recurrent otitis media and irreversible lung damage like bronchiectasis as a result of chronic pulmonary infections8.

Class 1 (hallmark) defects. (A) Outer dynein arm (ODA) defect: the absence of at least 7 out of 9 arms like this schematic cross-section in more than 50% of cilia. (B) ODA defect with inner dynein arm (IDA) defect which is defined as the absence of at least 5 out of 9 arms like here in more than 50% of cilia. (C) Microtubular disorganization (MTD) and IDA defect: the presence of IDA defect with disruption of 9 + 2 symmetry in more than 25% of cilia.
PCD is an inherited genetic disorder that results from impaired ciliary beating caused mainly by an abnormal ciliary ultrastructure such as the ODA defect with or without the IDA defect4. These cilia also play a pivotal role during the embryonic period by determining the laterality of organs; therefore, Situs Inversus is a common finding among PCD patients. PCD cases develop into chronic upper and lower respiratory infections (such as recurrent otitis media, hearing impairment, and recurrent sinusitis), often starting in the early years of life, followed by inevitable adult bronchiectasis9. PCD may also manifest as male infertility, as cilia are also present in other organs4,6. The prevalence of PCD is estimated to be 1 in 10,000–20,000 newborns and is also more prevalent among consanguineous married family clusters4,6. Due to the limited epidemiological data and phenotypic heterogeneity, PCD is rather an unfamiliar disease for most general practitioners. Therefore, these patients are mostly visited more than 50 times without having a definite diagnosis10. The lack of accurate and sensitive diagnostic methods has also contributed to the delayed and often incorrect diagnosis of PCD6.
This cross-sectional study aimed to assess ciliary ultrastructural defects in terms of demographic and clinical characteristics using quantitative methods on transmission electron microscopy (TEM) to assess the findings of 87 suspected patients. This survey can contribute to a more comprehensive look at the role of EM in evaluating these patients and help improve diagnostic algorithms in PCD diagnosis.
Results
Initially, 87 participants were enrolled in the study. Of the samples, 11 had metaplasia of squamous cells, eight were poorly preserved, and one did not have adequate cilia for examination. Therefore, 20 patients were excluded, and the final study population consisted of 67 participants (Fig. 3).

Inclusion and exclusion criteria.
Among the patients, 30 (42.9%) were male, and 37 (52.9%) were female. They were between 4 and 47 years of age (mean age = 20.3 ± 10.7 years). Recurrent lower respiratory infections were seen in 21 (31.3%) of the patients. Also, 26 (38.8%) and 23 (34.3%) of the cases had bronchiectasis and recurrent sinusitis, respectively. Infertility was reported in two cases, and nasal polyposis was reported in only one case. Based on TEM analysis, our samples consisted of 12 (17%) hallmark defects, 11 (15.7%) class 2 defects, 26 (37.1%) other defects, and 18 (25.7%) normal TEM findings. Moreover, SCD was also seen: 31 (44.3%) of the patients had extra-tubule, and 29 (41.4%) of them had compound cilia (Fig. 4).

Compound cilia as an SCD. It should be mentioned that some primary defects like ODA defects are also evident in this picture (arrows). (Original magnification: ×140,000). ODA outer dynein arm.
The demographic characteristics and ciliary ultrastructural defects of the cases are shown in Table 1.
Situs Inversus was only seen in the hallmark defects group (three patients), and auditory symptoms were reported mostly in the hallmark defects and class 2 groups (four of the five cases presenting with these symptoms).
Table 2 shows the clinical presentations of the cases with a definite diagnosis of PCD and their ciliary ultrastructural defects detected by TEM.
Figure 5 shows some examples of microtubular disorganization and Fig. 6 shows some examples of IDA and ODA defects in our patients.

Microtubular disorganization (arrows) (Original magnification: ×50,000).

IDA and ODA defects. (a) ODA defects (arrows). (b, c, d) Some absence of IDA (I) and ODA (O) are marked. (Original magnification: a, c & d: ×50,000, b: ×140,000).
Discussion
This survey was the first study conducted in Iran to evaluate ciliary ultrastructural defects among patients presenting with symptoms attributable to PCD. In the current study, 12 patients had a definite diagnosis of PCD. Eleven patients were indicated as PCD cases, as they had class 2 defects with compatible symptoms and other diagnoses were ruled out. Therefore, they benefited from the early diagnosis of PCDs, which is important because the earlier onset of clinical intervention prevents permanent bronchiectasis and other insidious lung tissue damages11. We also reported 18 cases with normal patterns in TEM. However, 30% of the PCD patients might present normal TEM findings, such as those with large dynein heavy chain Dynein Axonemal Heavy Chain 11 (DNAH11) and defects in HYDIN12.
According to the European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia, it is highly recommended to screen patients with recurrent productive cough, chronic rhinitis, persistent middle ear disease, congenital heart disease, and neonatal respiratory infections for PCD. Also, PCD patients' siblings should undergo PCD investigations13,14. In our cases, the most prominent symptoms among the patients were bronchiectasis (39%) and persistent sinusitis (34%). However, recurrent lower respiratory tract infections were even more common in the hallmark defects group than in all subjects (50% compared to 31%). It is pivotal to note that five patients in the hallmark defects group had bronchiectasis; however, they did not report productive cough. The probable reason for this discrepancy can be the strict criteria used for productive cough's definition in this study. These patients might suffer from ongoing non-productive coughing or ineffective coughing as a result of a defect in mucociliary clearance15.
PCD and SCD may have similar symptoms. However, in PCD, TEM findings are permanent and seen in most cilia. Secondary changes, however, could be the result of former infections and would recover over time or by cell culture7,16,17. In our study, the most common ultrastructural ciliary defects were extra-tubules (44.3%) and compound cilia (41.4%), and examples of SCD were identified in almost half of the sample population (Fig. 4).
Furthermore, changes such as microtubular disorganization and central pair abnormalities may be common between PCD and SCD. Therefore, to confirm PCD diagnosis, other specific diagnostic methods should be used. Repeating the biopsy and considering a sufficient healthy interval before re-sampling also help to differentiate between PCD and SCD more precisely12. Almost 30% of our samples had IDA absence, overlapping between PCD and non-PCD cases; the probable reason may be that it is difficult to see IDA by TEM18,19,20. Therefore, it is highly suggested to repeat the biopsy, especially for this group21. Besides, in the event of an isolated IDA defect, the cultivation may result in a reversal and can exclude the diagnosis of PCD7. To avoid misinterpretation as an ultrastructural defect, great attention should be paid to the difference between the normal shape of the tip and the base of the cilia and its body, because the diameter of the cilium varies along its length, it is wider at the base and narrower at the tip12 (Figs. 1 and S1).
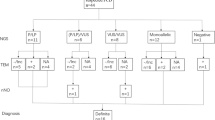
To date, PCD diagnosis has remained a significant medical challenge in resource-limited regions, and a variety of diagnostic approaches are used in different countries based on their local guidelines. Saccharine testing was once used for PCD screening but is no longer considered a reliable method22. Although TEM was previously thought to be a gold-standard test, there is currently no stand-alone diagnostic test for PCD, and a combination of several techniques is required to confirm the disease23. To date, nasal nitric oxide (nNO) and high-speed video microscopy analysis (HSVA) have been used in combination with TEM to confirm a PCD diagnosis based on local diagnostic algorithms. However, the European Respiratory Society guideline13 labels nNO for patients under the age of 6 years and HSVA as a "weak recommendation" and still strongly advocates TEM. The limitation of TEM in PCD diagnosis, particularly in class 2 defects and SCD changes, prompts researchers to use other diagnostic tests or repeat sampling to reach the definite diagnosis (Fig. 7).

Challenges of PCD diagnosis and possible solutions. TEM transmission electron microscopy, PCD primary ciliary dyskinesia, SCD secondary ciliary dyskinesia, PICADAR PrImary CiliAry DyskinesiA Rule, IF immunofluorescence, nNO nasal nitric oxide, HSVA high-speed video analysis.
Reviewing the literature13,22,23 and considering the cost of TEM analysis, we think nNO in combination with the primary ciliary dyskinesia rule (PICADAR) score24 is a proper initial screening test for an adult. However, further studies are needed in this regard. From our experience and due to its high specificity, TEM is a reliable tool for diagnosing PCD. However, up to 30% of PCD patients have normal TEM results12. Furthermore, there is no confirmed diagnosis for patients with class 2 defects by using TEM alone. Therefore, we suggest performing other tests for these groups of patients, along with re-sampling or cell culture, to make further decisions12.
These patients would have a regular joyful life only if diagnosed on time and given proper antibiotic treatment for each respiratory infection. Regular physiotherapy is helpful, as well25. On the other hand, recurrent infections will result in respiratory complications, including recurrent pneumonia, bronchiectasis, parenchymal fibrosis, and hypoxemia induced by ventilation/perfusion mismatch. If hypoxemia persists for a long time, it may lead to right ventricular failure. Hence, early diagnosis would prevent irreversible consequent complications occurred during the clinical course and reduce the burden of disease and its great cost for the health system26.
Our work has led us to conclude that the most prevalent hallmark diagnostic defect among suspected PCD patients was simultaneous lack of IDA and ODA. Further studies with other PCD diagnostic methods and their comparison with TEM are needed to establish a local guideline in western Asian countries.
Methods
Study design and patient enrollment
This prospective cohort study was conducted on referral patients from several affiliated centers to the TEM of the central laboratory, School of Medicine, the Shahid Beheshti University of Medical Sciences, Tehran, Iran, from 2017 to 2019. The patients had at least one of the suspected symptoms of PCD listed in Table S1 or positive history of PCD in the family (particularly in siblings and consanguineous marriages).
The patients were previously evaluated for probable differential diagnoses, including asthma (using both salbutamol and methacholine challenge tests), cystic fibrosis, immunological disorders, and chronic gastroesophageal reflux (depending on their past medical history). If one of the above diagnoses was confirmed and the patient's symptoms were justified with this diagnosis, these patients would be excluded.
Overall, 87 patients were enrolled in the study and tested for ciliary ultrastructural defects. Informed consent was taken from the participants or their parents in the case of children. The study was approved by the Ethics Committee of the Medical School, the Shahid Beheshti University of Medical Sciences (Code = IR.SBMU.MSP.REC.1397.382). All methods were carried out following relevant guidelines and regulations.
All the participants filled out a questionnaire designed by the expert medical team. The questionnaire provided demographic data (age and sex) and their clinical presentations, including chronic productive cough, recurrent sinusitis, recurrent lower respiratory infections, auditory symptoms, Situs Inversus, and infertility in both genders. The study defined the chronic productive cough as a purulent cough lasting more than 8 weeks15. Also, recurrent sinusitis was defined as four or more episodes of sinusitis with symptomless intervals each year27 and chronic sinusitis as a condition in which the symptoms last for more than 12 weeks28; we considered both of these definitions. Auditory symptoms included recurrent, chronic otitis and hearing loss. Infertility was defined based on past medical history.
TEM analysis
An expert otolaryngologist collected ciliary respiratory mucosa. It is important to note that sampling was avoided in patients with upper or lower respiratory tract infections, and they were asked to return without any respiratory symptoms after 6 weeks. Nasal samples were fixed by glutaraldehyde 2.5%, and then the second fixation was performed using osmium tetroxide 1% in 0.1 M veronal acetate buffer. The samples were dehydrated by passing through the ethanol series and then embedded in the epoxy-containing resin. The ultrathin sections of lead citrate and uranyl acetate were stained to provide an appropriate ultrastructural view. These sections were evaluated using a ZEISS EM900 transmission electron microscope to detect ciliary ultrastructural defects. At least 50 intact cilia from healthy areas with minimal artifacts were examined for each case12.
In addition, poorly preserved samples, samples with squamous cell metaplasia resulting in non-detectable cilia in the light-microscope or samples with an insufficient number of cilia (less than 50 sections for each patient) were excluded (Fig. 3).
Ultrastructural defects are classified into two main classes, according to the International Consensus guideline for reporting transmission electron microscopy results in the diagnosis of PCD12.
Class 1 defects include:
-
1.
ODA defects: It is defined as the absence of more than seven out of nine arms in each cilium that must be seen in more than 50% of cilia.
-
2.
Both ODA and IDA defects: ODA defects with IDA defects are defined as the absence of more than five out of nine arms in each cilium that must be seen in more than 50% of cilia (Fig. S2).
-
3.
Microtubular disorganization and IDA defects: Microtubular disorganization is defined as the disruption of 9 + 2 symmetry in more than 25% of cilia (Fig. 5).
Class 2 defects include:
-
1.
Central complex defect: It is defined as the absence of one or two central microtubules in more than 20% of examined cilia or lateral transposition of central pair. Also, abnormal counts like 8 + 1 are in this category (Fig. S3).
-
2.
Mislocalization of basal bodies with few or no cilia: It refers to very low count of cilia along with basal bodies failure to dock the apical surface of each cilium.
-
3.
Microtubular disorganization with IDA present: In this criterion, the disruption of 9 + 2 symmetry is seen in less than 25% of cilia.
-
4.
ODA absence from 25–50% of cilia cross-sections
-
5.
Combined IDA and ODA absence from 25–50% of cilia cross-sections
Therefore, based on these findings, the TEM findings of the patients were classified into four groups (Fig. 3) (Table S2).
-
1.
Hallmark defects defined as the existence of at least one of the class 1 defects
-
2.
Class 2 defects defined as the existence of at least one of the class 2 defects with other supporting evidence
-
3.
Other defects: having other ciliary ultrastructural defects that are not classified in class 1 or 2 defects.
-
4.
Normal: normal ciliary ultrastructure
Statistical analysis
The data were recorded in Microsoft Excel 2019 (Supplementary file 2) and then imported to the SPSS version 21 (the URL of SPSS version 21 was https://www.ibm.com/support/pages/spss-statistics-210-available-download). Data analysis was conducted using descriptive analysis of each patient's demographic and ciliary ultrastructural characteristics after splitting them into hallmark defects, class 2 defects, other defects, and normal groups.
Data availability
The materials and data of this study are available from the corresponding author and can be provided upon reasonable request.
References
Bergeron, C. & Cantin, A. M. (eds) Cystic Fibrosis: Pathophysiology of Lung Disease. Seminars in Respiratory and Critical Care Medicine (Thieme Medical Publishers, 2019).
de Iongh, R. U. & Rutland, J. Ciliary defects in healthy subjects, bronchiectasis, and primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 151(5), 1559–1567 (1995).
Afzelius, B. Cilia-related diseases. J. Pathol. J. Pathol. Soc. Great Britain Ireland 204(4), 470–477 (2004).
Lucas, J. S. et al. Diagnosis and management of primary ciliary dyskinesia. Arch. Dis. Child. 99(9), 850–856 (2014).
Ehouman, A., Pinchon, M., Escudier, E. & Bernaudin, J. Ultrastructural abnormalities of respiratory cilia. Virchows Archiv B 48(1), 87 (1985).
Abitbul, R. et al. Primary ciliary dyskinesia in Israel: Prevalence, clinical features, current diagnosis and management practices. Respir. Med. 119, 41–47 (2016).
Dixon, M. & Shoemark, A. Secondary defects detected by transmission electron microscopy in primary ciliary dyskinesia diagnostics. Ultrastruct. Pathol. 41(6), 390–398 (2017).
Rubbo, B. & Lucas, J. S. Clinical care for primary ciliary dyskinesia: Current challenges and future directions. Eur. Respir. Rev. 26(145), 170023 (2017).
Shapiro, A. J. et al. Laterality defects other than situs inversus totalis in primary ciliary dyskinesia: Insights into situs ambiguus and heterotaxy. Chest 146(5), 1176–1186 (2014).
Sommer, J. U. et al. ENT manifestations in patients with primary ciliary dyskinesia: Prevalence and significance of otorhinolaryngologic co-morbidities. Eur. Arch. Oto-Rhino-Laryngol. Off. J. Eur. Feder. Oto-Rhino-Laryngol. Soc. (EUFOS) Affiliated German Soc. Oto-Rhino-Laryngol. Head Neck Surg. 268(3), 383–388 (2011).
Karakoc, G. B., Yilmaz, M., Altintas, D. U. & Kendirli, S. G. Bronchiectasis: Still a problem. Pediatr. Pulmonol. 32(2), 175–178 (2001).
Shoemark, A. et al. International consensus guideline for reporting transmission electron microscopy results in the diagnosis of primary Ciliary dyskinesia (BEAT PCD TEM criteria). Eur. Respir. J. 55(4), 190072 (2020).
Lucas, J. S. et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur. Respir. J. 49(1), 1601090 (2017).
Pizzi, S., Cazzato, S., Bernardi, F., Mantovani, W. & Cenacchi, G. Clinico-pathological evaluation of ciliary dyskinesia: Diagnostic role of electron microscopy. Ultrastruct. Pathol. 27(4), 243–252 (2003).
McCallion, P. & De Soyza, A. Cough and bronchiectasis. Pulm. Pharmacol. Ther. 47, 77–83 (2017).
Mygind, N., Pedersen, M. & Nielsen, M. Primary and secundary ciliary dyskinesia. Acta Otolaryngol. 95(5–6), 688–694 (1983).
Al-Rawi, M. M., Edelstein, D. R. & Erlandson, R. A. Changes in nasal epithelium in patients with severe chronic sinusitis: A clinicopathologic and electron microscopic study. Laryngoscope 108(12), 1816–1823 (1998).
Escudier, E. et al. Computer-assisted analysis helps detect inner dynein arm abnormalities. Am. J. Respir. Crit. Care Med. 166(9), 1257–1262 (2002).
Keith Funkhouser, W. III. et al. A new tool improves diagnostic test performance for transmission em evaluation of axonemal dynein arms. Ultrastruct. Pathol. 38(4), 248–255 (2014).
Papon, J.-F. et al. Quantitative analysis of ciliary beating in primary ciliary dyskinesia: A pilot study. Orphanet J. Rare Dis. 7(1), 78 (2012).
Smith, C. M. et al. Cooling of cilia allows functional analysis of the beat pattern for diagnostic testing. Chest 140(1), 186–190 (2011).
Barbato, A. et al. Primary ciliary dyskinesia: A consensus statement on diagnostic and treatment approaches in children. Eur. Respir. J. 34(6), 1264–1276 (2009).
Rumman, N. et al. Diagnosis of primary ciliary dyskinesia: Potential options for resource-limited countries. Eur. Respir. Rev. 26(143), 160058 (2017).
Behan, L. et al. PICADAR: A diagnostic predictive tool for primary ciliary dyskinesia. Eur. Respir. J. 47(4), 1103–1112 (2016).
Bush, A. et al. Primary ciliary dyskinesia: Diagnosis and standards of care. Eur. Respir. J. 12(4), 982–988 (1998).
Rossman, C. M. & Newhouse, M. T. Primary ciliary dyskinesia: Evaluation and management. Pediatr. Pulmonol. 5(1), 36–50 (1988).
Sharma, G. K., Lofgren, D. H. & Taliaferro, H. G. Recurrent Acute Rhinosinusitis. StatPearls [Internet] (2020).
Melen, I. Chronic sinusitis: Clinical and pathophysiological aspects. Acta Otolaryngol. 114(sup515), 45–48 (1994).
Acknowledgements
We would like to express our sincere gratitude to Dr. Amir Human Hoveidaei for native grammar editing and Dr. Amirhossein Aghdaei for his conceptual preparation of schematic figures in this study.
Author information
Authors and Affiliations
Contributions
M.R., A.F., A.A.V., and H.R.J. conceived of the presented idea. S.A.Z. and M.R. supervised the project. J.G., M.K., and A.E. provided critical feedback and helped shape the research. M.R., S.A.M., and A.S. designed the study and along with H.T. and M.V. contributed to the final version of the manuscript. M.M., H.A.G., and S.M. were involved in the proofreading and sample provision. H.T., D.M., and M.H. helped in preparing the new figures and the new design of the study. M.P., S.R., and P.T. contributed to the re-evaluation and data analysis. M.M. revised and corrected the manuscript and re-designed the pictures and suplementary file. All authors discussed the results and commented on the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rezaei, M., Soheili, A., Ziai, S.A. et al. Transmission electron microscopy study of suspected primary ciliary dyskinesia patients. Sci Rep 12, 2375 (2022). https://doi.org/10.1038/s41598-022-06370-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-06370-w
- Springer Nature Limited